

Abstract
Influenza A virus (flu) is a respiratory tract pathogen causing high morbidity and mortality among the human population. Nitric oxide (NO) is a cellular mediator involved in tissue damage due to apoptosis of target cells and resulting enhancement of local inflammation. Inducible nitric oxide (iNOS) is involved in the production of NO following infection. Although NO is a key player in the development of exaggerated lung disease during flu infection, the underlying mechanism including the role of NO in apoptosis during infection has not been reported. Similarly, the mechanism of iNOS gene induction during flu infection is not well defined in terms of host trans-activator(s) required for iNOS gene expression. In the current study we have identified kruppel-like factor 6 (KLF6) as a critical transcription factor essential for iNOS gene expression during flu infection. We have also underscored the requirement of iNOS in inducing apoptosis during infection. KLF6 gene silencing in human lung epithelial cells resulted in drastic loss of NO production, iNOS-promoter specific luciferase activity and expression of iNOS mRNA following flu infection. Chromatin immuno-precipitation assay revealed a direct interaction of KLF6 with iNOS promoter during both in vitro and in vivo flu infection of human lung cells and mouse respiratory tract, respectively. Significant reduction in flu mediated apoptosis was noted in KLF6 silenced cells, cells treated with iNOS inhibitor and in primary murine macrophages derived from iNOS knock-out (KO) mice. A similar reduction in apoptosis was noted in the lungs following intra-tracheal flu infection of iNOS KO mice.
Introduction
Influenza A virus (flu) is a single-stranded RNA virus that causes severe respiratory diseases upon infection of the airway (1, 2). Flu infection among high risk individuals (e.g. elderly, immuno-compromised individuals) manifest in progression of inflammatory disease state like pneumonia (1, 2, 3).
Nitric oxide (NO) (4) plays an important role in innate immune response against pathogens (4, 5, 6, 7, 8, 9). Although it confers a protective role in the infected host, it can also exaggerate the disease pathology associated with infection. This detrimental effect of NO is due to its ability to launch a hyper-inflammatory response leading to tissue damage (10). Two mechanisms are primarily responsible for inducing NO mediated tissue damage – a) action of NO-induced inflammatory mediators, and b) apoptosis of NO targeted cells. In regard to host defense against viruses, NO could act as a “double-edged sword”. While functioning as an antiviral molecule NO can contribute to progression/exaggeration of virus-induced disease (11). Flu infection results in NO production due to induction of inducible nitric oxide synthase (iNOS) gene (12). Although a specific transcription factor(s) required for iNOS gene expression during flu infection is yet to be identified and characterized, one study suggested that NF-κB (an inflammatory transcription factor) may play a role in iNOS gene activation during flu infection (12). NO produced from the lungs during infection contributes to tissue damage and enhanced disease pathology associated with flu infection (13, 14, 15). NO mediated apoptosis represents one of the key factors contributing to enhanced inflammation and tissue damage during non-infectious respiratory diseases like asthma and chronic obstructive pulmonary disease (COPD) (10, 16). Although flu infection results in NO production, the contribution of NO in apoptosis during infection is not known. Furthermore, apart from NF-κB, the role of “non-inflammatory” transcription factor(s) in the regulation of iNOS gene expression during respiratory virus infection (especially during flu infection) has not been examined.
In the current study we have identified triple zinc finger containing DNA binding transcription factor Kruppel-like factor 6 (KLF6) (17, 18) as a transcription factor required for iNOS gene induction during flu infection. We have also demonstrated that induction of iNOS (leading to production of NO) during flu infection contributes to apoptosis of both epithelial and immune cells (i.e. macrophages). The in vivo physiological relevance of iNOS in contributing to apoptosis during flu infection was further documented by using wild-type and iNOS knock-out mice.
The family of Kruppel-like factor (KLF) transcription factors controls a wide spectrum of biological and physiological processes including cell growth, cell proliferation and differentiation (17, 18). Although KLF factors have been implicated in regulating normal cellular/tissue homeostasis, their role during infection has not been examined yet. In the present study we have identified KLF6 as a regulator of iNOS gene expression during flu infection and furthermore, we demonstrated the requirement of iNOS expression for apoptosis during flu infection.
Materials and Methods
Virus and Cell culture
Influenza A [A/PR/8/34 (H1N1)] virus (flu) was grown in theallantoic cavities of 10-day-old embryonated eggs (19). Virus was purified by centrifugation (two times) on discontinuous sucrose gradients (20). A549 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and glutamine (21). Bone marrow-derived macrophages (BMDMs) were obtained from femurs and tibiasof wild-type (WT) and iNOS knock-out (KO) mice and were cultured for 6–8 days as described earlier (19, 22). Cells were plated on 6-well plates containing RPMI, 10% FBS, 100 IU/mL Penicillin, 100 μg/mL Streptomycin and 20 ng/ml GM-CSF. The flu titer was monitored by plaque assay analysis with MDCK cells.
Generation of stable shRNA expressing A549 cells
A549 cells transduced (in the presence of 5 μg/ml polybrene at 2 multiplicity of infection or MOI of lentivirus) with either scrambled shRNA (control) or KLF6-specific shRNA expressing lentivirus (Santa Cruz Biotechnology, CA, USA) were selected in the presence of puromycin dihydrochloride. Antibiotic resistant cells were expanded to generate a population of stable cells expressing either scrambled shRNA or KLF6-specific shRNA.
Reverse transcription-PCR (RT-PCR)
Total RNA was extracted from A549 cells using Tri Reagent (Invitrogen). cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed using 0.25 units of Taq polymerase, 10 pmol of each oligonucleotide primer, 1 mM MgCl2, and 100 μM deoxynucleotide triphosphates in a final reaction volume of 25 μl. Following amplification, the PCRproducts were analyzed on 1.5% agarose gel. Equal loading in each well was confirmed by analyzing expression of the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase(GAPDH). The primers used to detect the indicated genes by RT-PCR are shown below.
GAPDH forward, 5′-GTCAGTGGTGGACCTGACCT, GAPDH reverse, 5′-AGGGGTCTACATGGCAACTG,
Human iNOS forward, 5′- TCCGAGGCAAACAGCACATTCA, iNOS reverse, 5′-GGGTTGGGGGTGTGGTGATGT,
Human KLF6 forward, 5′- CTCTCAGCCTGGAAGCTTTTAGCCTAC, KLF6 reverse, 5′-ACAGCTCCGAGGAACTTTCTCCCA,
Chromatin immuno-precipitation (ChIP) assay
Standard ChIP assay protocol (23) was followed for examining binding of KLF6 to iNOS promoter in cells and lung tissue. Following chromatin isolation from infected (at 1 MOI) human A549 cells and mouse BMDMs (after cross-linking with formaldehyde), it was immuno-precipitated at 4°C with either human or mouse specific anti-KLF6 antibody or isotype matched control IgG (Santa Cruz Biotechnology, CA, USA). Human and mouse iNOS promoter-specific PCR primer pairs (encompassing the consensus CACCC KLF6 binding site located in the proximal region of iNOS promoter) were used to analyze the antibody precipitated DNA. As a negative control, PCR primer pairs corresponding to iNOS promoter region that does not possess consensus KLF6 binding site was used. Primers used for the ChIP assay are shown in Table-1. Lung tissue derived from WT and iNOS KO mice were also used for ChIP analysis as described previously (23). In order to demonstrate that amount of DNA used for immuno-precipitation (with either KLF6 antibody or control antibody) is equivalent for each experiment, an equal portion of input DNA was analyzed for each sample.
Table-1.
ChIP primers
| Encompassing the KLF6 binding site at the human iNOS promoter |
Human iNOS forward 5′ CAGAGAGCTCCCTGCTGAGGAAA 3′ Human iNOS reverse 5′ GAGAGTTGTTTTTGCATAAAGGTCTC 3′ |
Product: 321 bp (encompassing −45 to −366 region of human iNOS promoter) | Optimal annealing temp: 55.7°C |
| Encompassing the region in human iNOS promoter that does not bind to KLF6 (negative control) |
Human iNOS forward 5′ GGAGAACATTCAACCCCAGGTAATC 3′ Human iNOS forward 5′ ACCAATAAGGAAACCGAGACACAG 3′ |
Product: 249 bp (encompassing −4417 to −4666 region of human iNOS promoter) | Optimal annealing temp: 56.0 °C. |
| Encompassing the KLF6 binding site at the mouse iNOS promoter |
Mouse iNOS forward 5′ CCCAGTTTTGAAGTGACTACG 3′ Mouse iNOS reverse 5′ AAAGTTGTGACCCTGGCAGC 3′ |
Product: 229 bp (encompassing −12 to −241 region of mouse iNOS promoter) | Optimal annealing temp: 62.6°C. |
| Encompassing the region in mouse iNOS promoter that does not bind to KLF6 (negative control) |
Mouse iNOS forward 5′ GCTCTTGTTTCCCAGGTTAC 3′ Mouse iNOS forward 5′ GAGATGGCTCAGTTGGTAAAG 3′ |
Product: 320 bp (encompassing −1157 to −1477 region of mouse iNOS promoter) | Optimal annealing temp: 56.6 °C. |
Viral infection of cells
A549 cells were infected with purified flu in serum and antibiotic free OPTI-MEM medium (GIBCO). Following adsorption for 1.5h at 37°C, cells were washed twice with serum-containing DMEM and the infection was continued in the presence of serum-containing DMEM for the specified time points. BMDMs were grown in RPMI medium supplemented with 10% FBS, sodium pyruvate, L-glutamine, GM-CSF, Hepes buffer. BMDMs were infected with purified flu in serum and antibiotic free RPMI medium. Following adsorption for 1.5h at 37°C, cells were washed twice with serum-containing RPMI and the infection was continued in the presence of serum containing RPMI for the specified time points. For some experiments, A549 cells were pre-treated with either water (vehicle) or N6-(1-iminoethyl)-L-lysine dihydrochloride (L-NIL) (Cayman Chemical) (200 μM) for 2h before infection. Infection was continued in the presence of L-NIL.
Nitric oxide quantification
NO production from infected cells was determined by measuring nitrite levels in the medium supernatant as described previously (24). Medium supernatant collected from mock and flu infected cells were mixed with Griess reagent for 10 min at room temperature. The absorbance (at 543 nm) was measured using a micro-plate reader. Diluted sodium nitrite was used to derive a standard curve.
Luciferase assay
Luciferase assay was performed as described previously (19, 25). A549 cells were transfected (using Lipofectamine 2000 from Invitrogen) with pRL-null-renilla luciferase and luciferase (firefly) reporter gene fused to human iNOS promoter (iNOS-luc). At 24h post-transfection, cells were infected with flu. At 12h and 24h post-infection, cells were washed once with PBS and then lysed using passive lysis buffer (Promega). Luciferase activity was measured using Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. Transfection efficiency was normalized by measuring expression of renilla luciferase. Luciferase units were measured by standard methodology and presented as fold activation of luciferase activity.
Apoptosis assay
Flu-infected A549 cells and BMDMs were examined for apoptosis by annexin V labeling, using annexin V/propidium iodide (PI) apoptosis detection kit (BioVision, CA) (26, 27).
Flu infection of mice
6–8-week old pathogen-free WT C57BL/6 and iNOS KO C57BL/6 mice (Jackson laboratory) were anesthetized and inoculated (via intra-tracheal route) with flu (5X104 pfu/mouse) in 100 μl of Opti-MEM medium (Invitrogen) (19). Mock infected animals were sham inoculated with 100 μl of Opti-MEM. At 5d post-infection, whole lung was collected. Lungs were utilized for in situ TUNEL analysis (with lung tissue sections) and Western blot analysis (with lung homogenate) with anti-caspase-3 antibody (Cell Signaling Technology). Another set of WT mice were also infected with flu for 5d and the isolated lungs were used for tissue ChIP assay. The mouse experiments performed in the current study was reviewed and approved by the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee (IACUC).
in situ TUNEL assay
To evaluate apoptotic cell death in the respiratory tract TUNEL assay was performed. Formalin fixed lungs from flu infected WT and iNOS KO mice were utilized for in situ TUNEL assay using ApopTag Peroxidase In Situ Apoptosis Detection Kit (Milipore, MA, USA). Light microscopy was performed to capture digital images of the TUNEL stained lung sections. The digital images were used to count the number of TUNEL positive cells in lung sections by using Image J software from NIH (http://rsbweb.nih.gov/ij/) as described previously (28). For each analysis, an area of 858.95 μm (microns) X 646.74 μm of TUNEL stained lung section was scanned by Image J software. Gross apoptotic area was expressed as pixels/micron and this value was used to calculate percentage apoptotic area during each analysis. Three mice from each group were used to prepare lung sections (i.e. three flu infected WT mice and three flu infected iNOS KO mice). The data was collected from twenty seven areas (nine areas/mouse) of the lung sections from each experimental group (i.e. twenty seven areas for flu infected WT mice and twenty seven areas for flu infected iNOS KO mice). The values obtained from twenty seven lung section areas of each experimental group (WT and iNOS KO) were used for statistical analysis.
Results
KLF6 is required for nitric oxide (NO) production
In order to study the role of KLF6 during infection, we utilized KLF6-specific shRNA to silence KLF6 expression in human lung epithelial A549 cells. A549 cells are routinely used as model type-II human alveolar epithelial cells and alveolar cells are major target of flu during productive infection of human respiratory tract. Lentivirus mediated shRNA delivery system was used to generate stable A549 cells lacking KLF6 expression. Loss of KLF6 is evident in stable cells expressing KLF6-specific shRNA (Fig. 1A). Control cells represent stable cells derived after transduction with scrambled shRNA expressing lentivirus.
FIGURE 1.
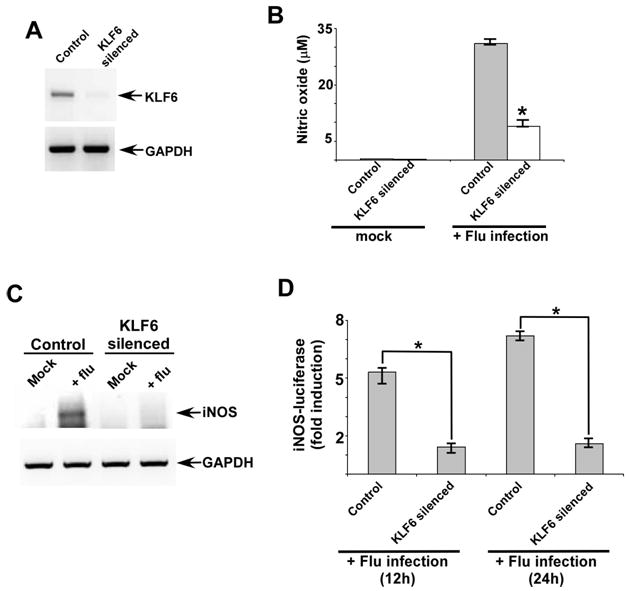
KLF6 is required for nitric oxide (NO) production and iNOS gene expression. (A) RT-PCR analysis of KLF6 expression in stable A549 cells generated following transduction with scrambled shRNA (control) and KLF6 specific shRNA (KLF6 silenced cells) expressing lentivirus. (B) NO production from mock and flu infected control and KLF6 silenced cells. NO was measured by monitoring nitrite production using Griess reagent. Each value represents the mean ± standard deviation from three independent experiments performed in triplicate. *p <0.05 using a Student’s t test. (C) RT-PCR analysis of iNOS expression in scrambled shRNA (control) and KLF6-specific shRNA expressing stable A549 cells infected with flu. RT-PCR data is representative of three independent experiments. (D) Activation of iNOS-luciferase reporter gene in control and KLF6 silenced cells infected with flu for 12h and 24h. The luciferase assay results are presented as mean ± standard deviation from three independent experiments performed in triplicate. *p <0.05 using a Student’s t test.
Control and KLF6 silenced cells were used subsequently to examine whether KLF6 plays any role in NO production and iNOS gene expression during flu infection. To assess the requirement of KLF6 for NO production, control and KLF6 silenced cells were infected with flu. At 12h post-infection, the medium supernatant was collected to measure nitrite (the end product of nitric oxide) by Griess reagent. Griess assay revealed significant reduction in NO production from KLF6 silenced cells compared to control cells (Fig. 1B). These results demonstrated that KLF6 is required for NO production during flu infection.
KLF6 is required for iNOS gene expression
To further evaluate the mechanism responsible for KLF6 dependent NO production, we investigated whether KLF6 regulates iNOS gene expression. Total RNA was isolated from control and KLF6 silenced cells infected with flu for 12h. The RNA was used to determine iNOS mRNA levels by RT-PCR. iNOS gene expression is regulated by KLF6 at the transcriptional level because low levels of iNOS transcripts were detected in flu infected KLF6 silenced cells compared to infected control cells (Fig. 1C).
The loss of iNOS mRNAs in KLF6 silenced cells suggested that KLF6 could positively transactivate the iNOS promoter. In order to test this possibility, we utilized luciferase reporter gene fused to human iNOS promoter (iNOS-luc). Control and KLF6 silenced cells transfected with luciferase expressing plasmids were infected with flu. Expression of luciferase gene was monitored at 12h and 24h post-infection. KLF6 directly regulates iNOS promoter during flu infection, since significant reduction in luciferase activity was observed in cells lacking KLF6 expression (Fig. 1D). These results showed that KLF6 is an important transcription factor regulating iNOS gene expression during flu infection.
Binding of KLF6 to iNOS promoter during infection of human lung epithelial cells
The role of KLF6 in transactivation of iNOS gene was further confirmed by performing chromatin immuno-precipitation (ChIP) assay to investigate direct association of KLF6 with iNOS promoter during infection. Human lung epithelial A549 cells were either mock infected or infected with flu for 6h, 12h and 24h. At various post-infection time-periods, chromatin from these cells was immuno-precipitated using either isotype matched control IgG (negative control) or anti-KLF6 antibody. The KLF6-DNA binding complex was analyzed by PCR using primer corresponding to KLF6 binding site on human iNOS promoter (29) (Table 1). A primer specific for non-KLF6 binding region on human iNOS promoter element (an upstream region in the human iNOS gene devoid of the KLF6 responsive region) (Table 1) was used as a negative control. Although KLF6 binding to human iNOS gene was not detected in mock infected cells, flu infection led to association of KLF6 with human iNOS promoter (Fig. 2). Interestingly, maximal KLF6 binding was detected at 12h post-infection, which corresponds to the time-frame at which iNOS induction and NO production was observed during infection of A549 cells (Fig. 1). The specificity of KLF6 binding to human iNOS promoter is borne out by the observation that immuno-precipitation with control IgG did not generate any amplified product. Moreover, amplification with primers that do not correspond to KLF6 binding site on human iNOS promoter did not yield any amplified product. These results demonstrated that during flu infection KLF6 directly transactivates human iNOS gene expression following its association with human iNOS promoter.
FIGURE 2.
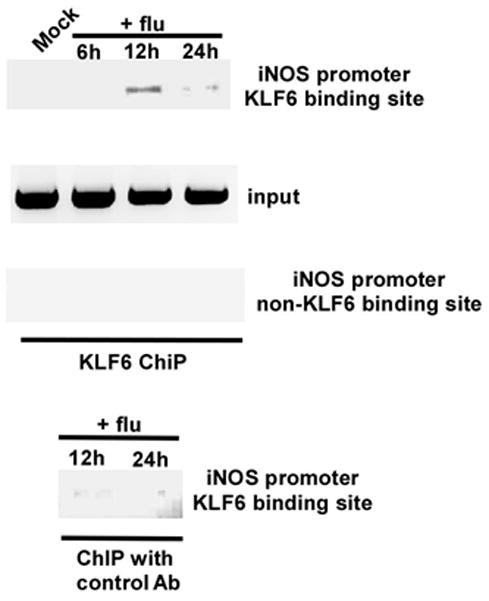
Binding of KLF6 to human iNOS promoter during flu infection. Chromatin immuno-precipitation (ChIP) assay on A549 cells that were either mock infected or infected with flu for 6h, 12h and 24h. Both anti-KLF6 antibody and isotype matched control antibody (control Ab) was used. An upstream region in the human iNOS gene devoid of the KLF6 responsive region was probed as a negative control. The ChIP data is representative of three independent experiments. Input = input DNA for each experiment.
KLF6 regulates apoptosis during infection
Our studies have uncovered an important function of KLF6 during flu infection, since it is required for iNOS expression and subsequent NO production from infected cells. Previous studies have shown that NO is a contributing factor for exaggerated lung disease (due to lung tissue damage) during flu infection (12, 13, 14, 15). Since apoptosis could induce tissue damage, we next investigated whether KLF6 gene is required for apoptosis during infection. Control and KLF6 silenced cells were infected with flu (0.5 MOI) for 48h. After infection, the cells were used to determine apoptosis by annexin V staining. Annexin V staining (which signifies early apoptosis) revealed significant reduction in apoptosis of infected KLF6 silenced cells compared to control cells (Fig. 3). These results demonstrated that KLF6 could directly regulate important cellular activity (i.e. programmed cell death or apoptosis) during flu infection.
FIGURE 3.
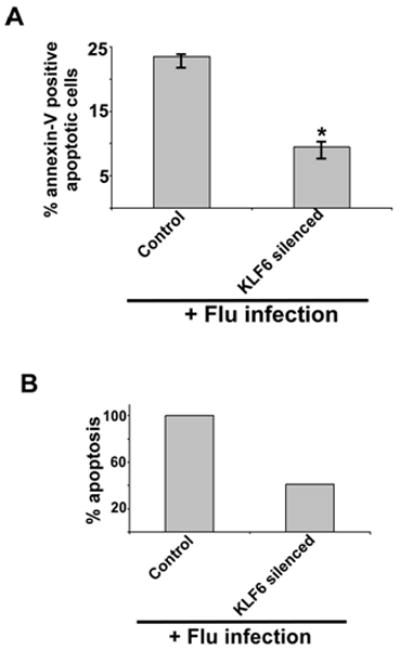
KLF6 expression is required for optimal apoptosis following flu infection. (A) Percentage of apoptotic (annexin V positive cells) cells in flu infected (48h post-infection) control (expressing scrambled shRNA) and KLF6 silenced (expressing KLF6-specific shRNA) stable A549 cells. Annexin V staining quantified by FACS represents mean ± SEM from five independent experiments performed in triplicate, *p < 0.05 by two-tailed t test. (B) The values presented in Fig. 3A were utilized to calculate % apoptosis in flu infected control and KLF6 silenced cells. Apoptosis in flu infected control cells are denoted as 100% apoptosis.
iNOS is required for apoptosis during infection of epithelial cells and macrophages
Based on our results showing requirement of KLF6 for optimal apoptosis and trans-activation of iNOS gene by KLF6, we speculated that iNOS (by generating and producing NO) may contribute to apoptosis during flu infection. So far no studies have evaluated the role of NO (and iNOS) in apoptosis during flu infection. The role of iNOS was examined by inhibiting iNOS enzymatic activity with specific iNOS inhibitor N6-(1-iminoethyl)-L-lysine dihydrochloride (L-NIL) (30, 31, 32, 33). A549 cells were treated with either vehicle (water) or L-NIL (200 μM) during flu (0.5 MOI) infection. A similar concentration of L-NIL (100 μM-900 μM) was used previously to inhibit iNOS activity in lung epithelial cells (including A549 cells) (30, 31, 32, 33). We (data not shown) and others (30, 31, 32, 33) did not observe any cytotoxicity in lung epithelial cells treated with L-NIL (100 μM - 200 μM) for 48h. At 48h post-infection, apoptosis status of infected cells was examined by annexin V staining (annexin V positive and PI negative cells representing apoptotic cells). Inhibition of iNOS activity markedly diminished apoptosis of flu infected cells (Fig. 4A and 4B). Almost 80% reduction in apoptosis was observed in L-NIL treated cells compared to control cells. These results for the first time uncovered the role of NO in inducing apoptosis during flu infection. NO specifically contributes to apoptosis during flu infection, since iNOS inhibition did not significantly alter the necrotic status (annexin V positive and PI positive cells) of infected cells (Fig. 4C).
FIGURE 4.
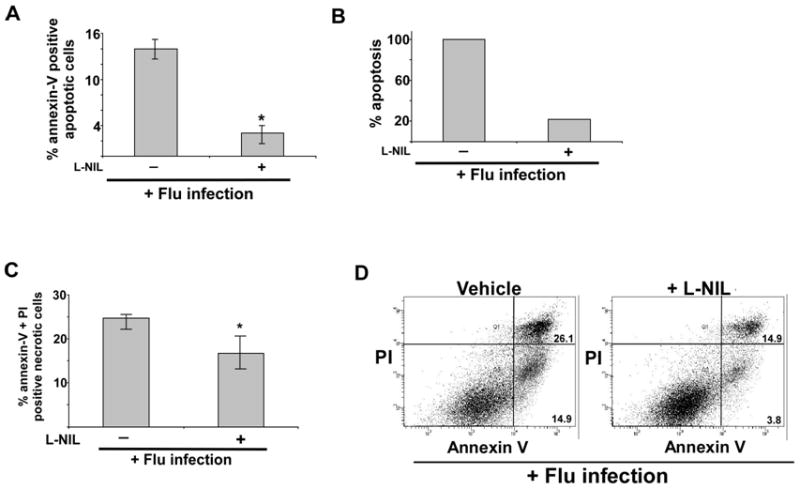
iNOS activity is essential for apoptosis during flu infection of epithelial cells. (A) Percentage of annexin V positive cells (apoptotic cells) were detected by FACS analysis of flu infected (48h post-infection) A549 cells incubated with water (vehicle) or iNOS inhibitor L-NIL. Annexin V staining quantified by FACS represents mean ± SEM from five independent experiments performed in triplicate, *p < 0.05 by two-tailed t test. (B) The values presented in Fig. 4A were utilized to calculate % apoptosis in flu infected cells (+/− L-NIL). Apoptosis in flu infected vehicle treated cells are denoted as 100% apoptosis. (C) Percentage of annexin V and PI positive cells (necrotic cells) were detected by FACS analysis of flu infected (48h post-infection) A549 cells incubated with water (vehicle) or iNOS inhibitor L-NIL. Annexin V + PI staining quantified by FACS represents mean ± SEM from five independent experiments performed in triplicate, *p = 0.12 by two-tailed t test. (D) A representative FACS showing percentage of apoptotic (lower right quadrant – annexin V positive and PI negative cells) and necrotic (upper right – quadrant annexin V and PI positive cells) cells following flu infection of water or L-NIL treated cells.
The role of iNOS in regulating apoptosis was further confirmed by utilizing primary bone marrow derived macrophages (BMDMs) isolated from iNOS knock-out (KO) mice. Wild type (WT) and iNOS KO BMDMs were infected with flu (0.5 MOI) for 48h. After infection, the apoptotic status was evaluated by annexin V staining. As shown in Fig. 5A and 5B, significant decrease (36% reduction in apoptosis) in apoptosis was observed in flu infected iNOS KO BMDMs compared to WT BMDMs. These results once again confirmed that iNOS/NO plays a crucial role in apoptosis induction during flu infection. In contrast, necrotic status of cells remained unchanged in flu infected WT vs. iNOS KO BMDMs (Fig. 5C).
FIGURE 5.
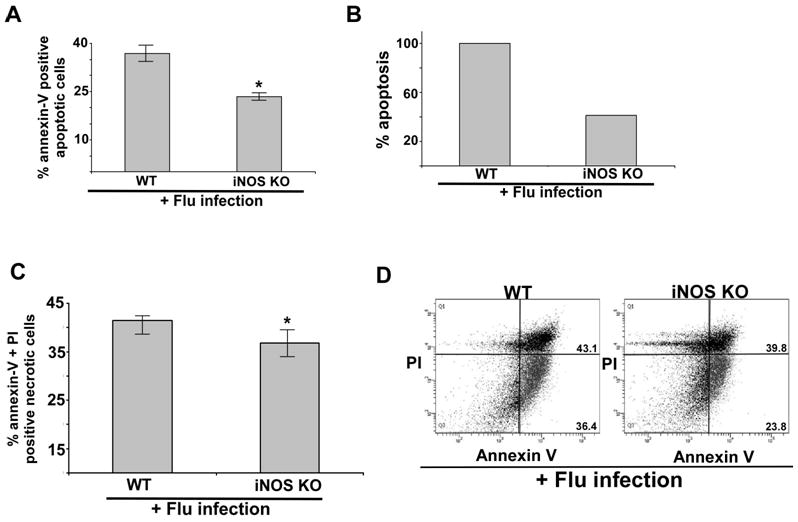
iNOS expression is required for apoptosis during flu infection of primary macrophages. (A) Percentage of annexin V positive cells (apoptotic cells) were detected by FACS analysis of flu infected (48h post-infection) wild-type (WT) and iNOS knock-out (KO) primary bone marrow derived macrophages (BMDMs). Annexin V staining quantified by FACS represents mean ± SEM from four independent experiments performed in triplicate, *p < 0.05 by two-tailed t test. (B) The values presented in Fig. 5A were utilized to calculate % apoptosis in flu infected WT and iNOS KO BMDMs. Apoptosis in flu infected WT BMDMs are denoted as 100% apoptosis. (C) Percentage of annexin V and PI positive cells (necrotic cells) were detected by FACS analysis of flu infected (48h post-infection) WT and iNOS KO) BMDMs. Annexin V + PI staining quantified by FACS represents mean ± SEM from four independent experiments performed in triplicate, *p = 0.09 by two-tailed t test. (D) A representative FACS showing percentage of apoptotic (lower right quadrant – annexin V positive and PI negative cells) and necrotic (upper right quadrant – annexin V and PI positive cells) cells following flu infection WT and iNOS BMDMs.
Binding of KLF6 to iNOS promoter during infection of primary macrophages
Experiments performed with WT and iNOS KO primary BMDMs revealed a role of iNOS in apoptosis during flu infection (Fig. 5). Since KLF6 regulates iNOS expression, we next investigated whether similar to lung epithelial cells, KLF6 also associates with iNOS promoter during flu infection of primary mouse BMDMs. Previous studies have mapped the KLF6 binding site in human iNOS gene to the proximal region (at position −164 to −168) of iNOS gene (29). The consensus CACCC sequence proximal to the human iNOS gene was critical for KLF6 binding to human iNOS gene. We identified similar proximal consensus CACCC sequence (at position −91 to −95) in the mouse iNOS gene.
BMDMs were either mock infected or infected with flu for 6h, 12h and 24h. Chromatin from the BMDMs was immuno-precipitated using either an isotype matched control IgG (negative control) or anti-KLF6 antibody. Primer corresponding to KLF6 binding site on the mouse iNOS promoter was used to analyze KLF6-DNA binding complex by PCR. A primer specific for non-KLF6 binding region on mouse iNOS promoter (an upstream region in the mouse iNOS gene) was used as a negative control. Although KLF6 binding to the mouse iNOS gene was not observed in mock infected BMDMs, flu infection resulted in recruitment of KLF6 to the mouse iNOS promoter (Fig. 6). Interestingly, KLF6 association was detected during early infection (i.e. 6h-12 post-infection) and the association of KLF6 with iNOS promoter was lost at 24h post-infection (Fig. 6). The time-frame of KLF6 interaction with mouse iNOS promoter corresponded with NO production from flu infected BMDMs, since optimal NO production from flu infected BMDMs was observed at 6h-12h post-infection (data not shown). The specificity of KLF6 binding to mouse iNOS promoter is borne out by the observation that amplification with non-specific primers (i.e. primers that do not correspond to KLF6 binding site on mouse iNOS promoter) did not yield any amplified product. Moreover, immuno-precipitation with control IgG did not generate any amplified product (data not shown). These results demonstrated that KLF6 interacts with mouse iNOS promoter during flu infection of primary macrophages.
FIGURE 6.
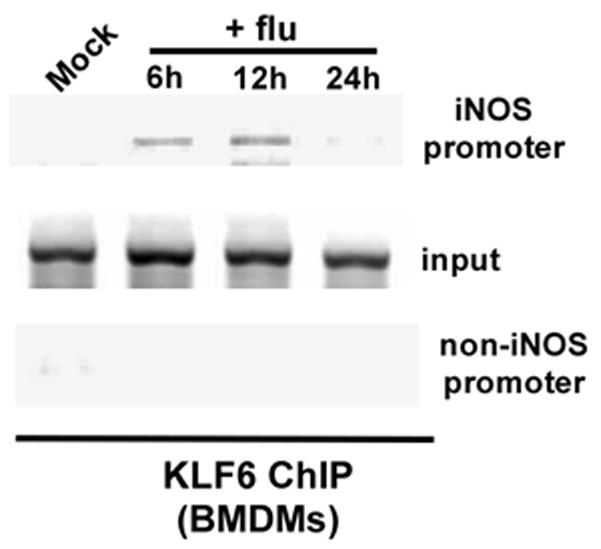
Association of KLF6 to mouse iNOS promoter during flu infection. Chromatin immuno-precipitation (ChIP) assay on primary bone marrow derived macrophages (BMDMs) that were either mock infected or infected with flu for 6h, 12h and 24h. An upstream region in the mouse iNOS gene devoid of the KLF6 responsive region was probed as a negative control. The ChIP data is representative of three independent experiments. Input = input DNA for each experiment.
iNOS expression is essential for optimal apoptosis in flu infected respiratory tract
The in vivo physiological role of iNOS was evaluated by infecting iNOS KO mice with flu. Previous studies have shown that compared to WT mice, iNOS KO mice exhibit reduced lung disease severity following flu infection (13, 14, 15). Interestingly, the reduced lung disease was not due to diminished virus replication (infection), since flu burden in the lungs of WT and iNOS KO mice were similar. In consistent with previous reports, our study revealed that flu replication (as assessed by flu hemagglutinin or HA expression in the lungs) in the airway is similar in WT and iNOS KO mice (Supplemental Fig. 1). Since apoptosis significantly contributes to disease severity, reduced apoptosis in flu infected iNOS KO mice may be one of the contributing factors responsible for diminished airway damage in iNOS KO animals. In order to assess this possibility, WT and iNOS KO mice were inoculated with flu (5 × 104 pfu/mouse) via intra-tracheal route. At 5d post-infection the lungs were collected and in situ TUNEL assay with lung sections was conducted to evaluate the apoptotic status of the respiratory tract. Diminished apoptosis in the lungs of iNOS KO animals were observed compared to WT mice (Fig. 7A). Scanning of the lung sections for TUNEL positive cells (representing apoptotic cells) revealed significantly reduced apoptosis in iNOS KO mice (Fig. 7B). A representative TUNEL stained lung section from flu infected WT and iNOS KO mice is shown in Supplemental Fig. 2. It is interesting to note that consistent with previous report (13) we also observed markedly reduced inflammation (as deduced by cellularity of the airway) in the lung of infected iNOS KO animal (Supplemental Fig. 2). The TUNEL results were further confirmed by performing Western blot analysis with anti-caspase-3 antibody that detects both the full length (35 kDa) and cleaved product (19 kDa) of caspase-3. Cleavage of pro-caspase-3 serves as a hallmark of apoptosis. Lung homogenate prepared from flu infected WT and iNOS KO mice were subjected to Western blot analysis with caspase-3 antibody. While cleaved caspase-3 was abundant in the lungs of WT mice, reduction in cleaved caspase-3 product was noticed in iNOS KO lungs (Fig. 7C and 7D). These results showed that iNOS is a critical regulator of lung apoptosis during flu infection.
FIGURE 7.
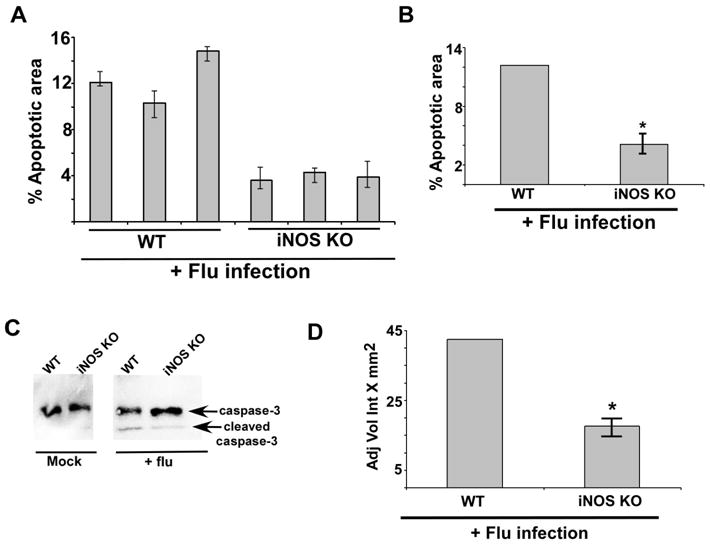
iNOS expression is required for optimal apoptosis in the lung of flu infected mice. (A) For each experimental group lung sections were prepared from three flu infected WT mice and three flu infected iNOS KO mice. The lung sections were used for TUNEL staining. Image J software was used to calculate TUNEL-positive areas (representing apoptosis) in the lung sections as detailed in the methods section. The data is presented as percent apoptotic area. In the figure, each bar represents each infected animal and the values represent the mean ± standard deviation for the percent apoptotic area calculated from nine areas of the lung sections. (B) The values were compiled to calculate the percent apoptotic area in flu infected WT mice vs. iNOS KO mice. The experiment was repeated twice in duplicate. *p = 0.0289 by Student’s t test. (C) Lung homogenate prepared from flu infected WT and iNOS KO mice were subjected to Western blot analysis with anti-caspase-3 antibody. The un-cleaved full-length caspase-3 (35 kDa) and the cleaved caspase-3 product (19 kDa) are indicated by an arrow. (D) For each experimental group lung homogenate was prepared from flu infected WT (four mice) and iNOS KO (four mice) mice. Lung homogenate from each mouse [total=8 mice (WT=4 mice and iNOS KO=4 mice)] were subjected to Western blot analysis with anti-caspase-3 antibody to detect the cleaved caspase-3 product (19 kDa) as shown in Fig. 7C. The 19 kDa caspase-3 band from each infected mouse (total = 8 mice) was quantified by using the Syngene Genetools software and the band density was plotted to show difference in the levels of cleaved caspase-3 product in flu infected WT vs. iNOS KO mice. The band density is represented as adjusted volume intensity X mm2 (Adj Vol Int X mm2). The band density represents mean ± standard from two independent experiments performed in duplicate. *p < 0.05 by two-tailed t test
KLF6 binds to the iNOS promoter during flu infection of mice
Our in vitro study showed that KLF6 is essential for iNOS gene expression by virtue of its binding to the iNOS promoter during flu infection of epithelial cells and macrophages (Figs. 2 and 6). The in vivo physiological relevance of this observation was further confirmed by examining interaction of KLF6 with iNOS promoter in the lungs of mice infected with flu. Tissue ChIP assay was performed by using lung harvested from flu infected mice. Mice were infected with flu via intra-tracheal route. At 5d post-infection, lungs were collected. Lungs isolated from mock infected and flu infected mice were processed for ChIP assay.
Chromatin from the tissue was immuno-precipitated using either isotype matched control IgG (negative control) or anti-KLF6 antibody. Primer corresponding to KLF6 binding site on mouse iNOS promoter was used to analyze KLF6-DNA binding complex by PCR. A primer specific for non-KLF6 binding region on mouse iNOS promoter (an upstream region in the mouse iNOS gene) was used as a negative control. KLF6 binding to mouse iNOS gene was not observed in mock infected animals (Fig. 8). However, flu infection resulted in binding of KLF6 to the mouse iNOS promoter (Fig. 8). The specificity of KLF6 binding to mouse iNOS promoter is evident by the observation that amplification with non-specific primers (i.e. primers that do not correspond to KLF6 binding site on mouse iNOS promoter) did not yield any amplified product. Moreover, immuno-precipitation with control IgG did not generate any amplified product (data not shown). These results demonstrated association of KLF6 with mouse iNOS promoter during flu infection of the respiratory tract.
FIGURE 8.
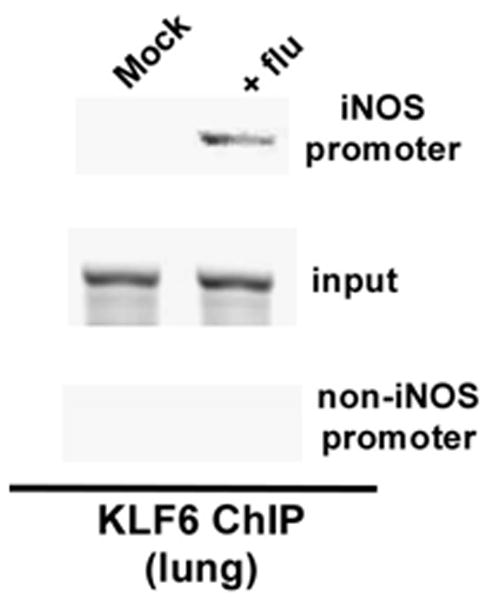
Binding of KLF6 to mouse iNOS promoter during flu infection of mouse respiratory tract. Tissue chromatin immuno-precipitation (ChIP) assay was performed by using lung tissue isolated from mock infected and flu infected (5d post-infection) mice. An upstream region in the mouse iNOS gene devoid of the KLF6 responsive region was probed as a negative control. The ChIP data is representative of three independent experiments. Input = input DNA for each experiment.
Discussion
iNOS gene expression is tightly regulated, since NO mediated tissue damage could lead to enhanced disease phenotype (4–11). Due to the ability of NO to induce apoptosis and enhance inflammation (10, 11, 16), NO produced during virus infection significantly contributes to development of disease state. These events culminate in exacerbated disease condition due to tissue damage. Respiratory viruses like influenza A virus induces iNOS expression to generate NO, which contributes to enhanced disease patho-physiology associated with increased damage to the lung tissue (12, 13, 14, 15). Although iNOS plays a critical role in determining the severity of disease state following flu infection, it is not known whether – a) iNOS/NO is required for apoptosis during flu infection?; b) “non-inflammatory” (apart from NF-κB, which is an “inflammatory” transcription factor) transcription factor(s) are required for iNOS gene expression during flu infection? Our studies have uncovered two mechanisms associated with NO production during flu infection – a) we have demonstrated that NO produced during infection plays an essential role in inducing apoptosis during infection, and b) we have also identified a “non-inflammatory” transcription factor (i.e. KLF6) as a trans-activator of iNOS gene during infection. Based on our studies we propose a model (Fig. 9), whereby flu infection triggers binding of KLF6 to iNOS promoter leading to expression of iNOS gene/protein. NO generated by iNOS is a key contributing factor for induction of apoptotic pathway during infection.
FIGURE 9.
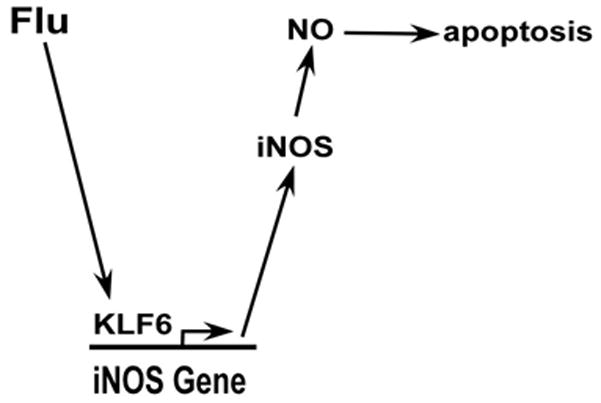
A schematic diagram depicting the role of KLF6 and iNOS during flu infection. Flu infection stimulates binding of KLF6 to the iNOS promoter; resulting in trans-activation of iNOS gene. The enzymatic activity of iNOS generates nitric oxide (NO), which is released from the cells during infection. NO contributes to the infection process by inducing apoptosis.
Apoptosis significantly contributes to pathogenesis and lung-injury/disease pathology associated with flu infection (34–39). Apoptosis of lung epithelial cells during infection with respiratory viruses like respiratory syncytial virus (RSV) results in their sloughing and these cells aggregate along with fibrin, mucin to form plugs associated with airway obstruction, a condition prevalent among RSV infected infants and children (40, 41). Apart from airway obstruction, high degree of apoptosis of both lung epithelial cells and immune cells (e.g. macrophages) during respiratory virus infection results in delayed clearance of dead cells from the airway lumen, which culminates in induction of necrosis and subsequent inflammation of the airway (42). A similar mechanism involving exaggerated apoptosis in the airway may contribute to development of severe lung damage (including progression to disease state like pneumonia) during infection with highly virulent strains of flu (35–39).
Various mechanisms are involved in the induction of apoptosis following flu infection. Apoptosis during flu infection is mediated by Siva-1 expression, extracellular calcium influx, Bax activation, and production of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (43–47). NO induced apoptosis represents one of the key factors contributing to enhanced inflammation and tissue damage (10, 16). Moreover, NO production during infection with respiratory viruses like flu results in exaggerated disease pathology attributed to increased lung inflammation and enhanced tissue damage (13, 14, 15). Although flu infection results in NO production (12), contribution of NO in apoptosis during infection has not been evaluated. Our study showed that NO produced during flu infection plays a critical role in inducing apoptosis. The role of NO in contributing to apoptosis in the airway was also validated in vivo by using flu infected WT and iNOS KO mice. In addition, we have identified KLF6 as an essential transcription factor required for iNOS gene expression during flu infection.
The 16 kB iNOS gene is regulated by numerous transcription factors such as NF-κB, STAT-1a, AP-1, Oct-1 (octamer factor), T-cell factor 4, STAT-3, NF-AT, NF-IL6 (4). Among these transcription factors, “inflammatory” transcription factors (e.g. AP-1, NF-κB, NF-AT and STAT-1a) play an important role during infection and inflammatory response. NO is produced during flu infection primarily due to iNOS induction (12–15). There has been no report that neuronal NOS (nNOS) is involved in NO generation during infection. Thus, inhibition of nNOS activity in A549 cells did not block NO generation following flu infection (Supplemental Fig. 3). The role of NF-κB in iNOS induction during flu infection has been suggested (12). It was also shown that iNOS gene is regulated by IRF-1 during flu infection (12). However, direct interaction/binding of NF-κB and IRF-1 to iNOS promoter during flu infection has not been reported. Moreover, apart from NF-κB, no other transcription factors have been implicated in trans-activation of iNOS gene during flu infection. In the current study we have uncovered the role of a “non-inflammatory” transcription factor (i.e. KLF6) in controlling iNOS gene expression during flu infection. Our studies have also illuminated the role of KLF family of transcription factors as important regulator of gene expression associated with infection. NO production was dependent on infection with viable flu, since significant reduction in NO levels was observed following infection with UV irradiated flu (UV-flu) (Supplemental Fig. 4A). Interestingly, replication incompetent flu (UV-flu) did not completely abrogate NO production, suggesting that viral components from input virus could trigger NO production, however replication is required to sustain this process. The efficiency of UV inactivation was evident from lack of flu HA expression in cells infected with UV-flu (Supplemental Fig. 4B).
The family of Kruppel-like factor (KLF) transcription factors controls a wide spectrum of biological and physiological processes including cell growth, cell proliferation and differentiation (17). The mammalian KLF family consists of seventeen DNA binding proteins that possess three zinc-fingered motifs. KLF transcription factors regulate function of various organ systems including hematological, digestive, cardiovascular, and respiratory systems and thus has been implicated in development/progression of diseases like cancer, metabolic disorders, cardiovascular and inflammatory diseases (17). Among the KLF family, KLF6 is known to regulate gene expression in various tissues by acting as either a trans-activator or repressor of gene expression. KLF6 regulated genes include, E-cadherin (48), TGF-β and TGF-β receptors (49), collagen (50), urokinase plasminogen activator (51). In addition, KLF6 has been identified as a tumor suppressor gene associated mainly with prostate cancer (52). The role of KLF6 in renal ischemia-reperfusion injury (53) and hepatic fibrosis (49, 50, 54) has been previously reported. iNOS expression is also regulated by KLF6 during diverse stress-related patho-physiological condition like heat shock, serum starvation, and hypoxia (29). Although human iNOS gene contains ten consensus KLF6 binding motif “CAAAC”, the proximal 0.63 kb region of iNOS gene is required for KLF6 binding to iNOS gene for trans-activation (29). Another member of KLF family, KLF4 also regulates iNOS and eNOS expression during inflammation of the neuron and endothelium (55, 56). Although KLF factors have been implicated in regulating normal cellular/tissue homeostasis, its role during infection was not known. In the present study we have identified KLF6 as a regulator of iNOS gene expression during flu infection.
Although the mechanism underlying iNOS gene trans-activation by KLF6 is not known, we surprisingly observed that poly-IC (a toll-like receptor 3 or TLR3 ligand) mediated NO production is dependent on KLF6 expression since, KLF6 silencing drastically inhibited NO production following poly-IC treatment (Supplemental Fig. 4C). Several studies have reported that activated TLR3 (and poly-IC treatment) triggers iNOS mediated NO release (57–60), and our result suggested that KLF6 is required for TLR3 mediated iNOS induction. TLR3 is expressed in lung epithelial cells (and TLR3 is activated by poly-IC in these cells) (61, 62) and TLR3 activation during flu infection is not only required for pro-inflammatory response in lung epithelial cells, but TLR3 plays an important role in regulating flu pathogenesis in mice (61, 63, 64, 65). Thus, it is plausible that activation of TLR3 pathway (resulting in activation of NF-κB and MAPK signaling) during flu infection results in KLF6 binding to iNOS promoter leading to iNOS gene expression. During this event, iNOS gene-specific trans-activating property of KLF6 could be achieved by several mechanism(s) – a) “signal specific” phosphorylation of several KLFs (e.g. KLF4) regulates their trans-activating function. For example, phosphorylation of KLF4 by MAPK results in recruitment of KLF4 to the TGF-beta receptor promoter and TGF-beta receptor gene expression (66). Since TLR3 activates MAPK pathway, it is possible that KLF6 phosphorylation promotes KLF6 binding to the iNOS promoter; b) Interaction of KLFs with proteins like Sp1 (67), p53 (68) and NF-κB p65 (69) subunit (following NF-κB activation) dictate their transcriptional activity. A similar scenario may exist during flu infection, whereby upon NF-κB activation (via TLR3), KLF6 may interact with NF-κB subunits or yet to be identified cellular proteins. These interactions may play a pivotal role in determining iNOS-gene specific trans-activation function of KLF6; c) Apart from phosphorylation, the trans-activating property of KLFs are also known to be regulated by additional post-translational modifications like SUMOylation (70, 71). KLF6 may undergo similar modification following flu infection. In the future, we will dissect the exact mechanism regulating iNOS-gene specific trans-activating function of KLF6 during flu infection and characterize the upstream signaling pathway (and mediators) involved in this process.
Our studies have demonstrated that KLF6 binds to iNOS promoter during flu infection to trans-activate iNOS gene expression. The importance of KLF6 during this process is evident from association of KLF6 with the iNOS promoter during flu infection of human lung epithelial cells, primary mouse macrophages and mouse lung. KLF6 is also expressed in the respiratory tract (72) (Fig. 8) and KLF4 [both KLF4 and KLF6 belong to group-2 of KLF factors (29)] expression is potentially associated with fibrosis and enhanced airway inflammation (73). Therefore, we envision that KLF6 may also contribute to exaggerated lung disease during flu infection, since KLF6 regulates expression of iNOS; which is involved in apoptosis/tissue damage. Further studies are required to investigate the functions of KLF6 in the airway tract by using conditionally knocked-out transgenic mouse once they are available since KLF6 knockout mice are embryonic lethally.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant AI083387 (to S.B.), AI071118 (to G.T.C.) and a grant from Center for Innovation in Prevention and Treatment of Airway Diseases (to S.B.). V.M. and J.A.S. was supported by NIH/NIDCR grant # DE14318 for the COSTAR program.
We thank Dr. Serpil C. Erzurum, M.D. (Department of Pathobiology, Cleveland Clinic, Cleveland, OH) for providing the iNOS-luciferase reporter plasmid. We thank Drs. Ibtissam Echchgadda, Ph.D. and Bandana Chatterjee, Ph.D. (Department of Molecular Medicine, University of Texas Health Science Center or UTHSCSA, San Antonio, TX) for their excellent suggestions. We thank Dr. Linda Roman (Department of Biochemistry, UTHSCSA) for assisting us with the in vitro neuronal nitric oxide synthase assay. We would like to thank Dr. Benjamin J. Daniel, Ph.D. and Karla M. Gorena from the UTHSCSA FACS facility for assistance. UTHSCSA FACS Core Facility was supported by the Cancer Center Program grant NIH P30 CA54174.
Abbreviations
- Flu
influenza A virus
- iNOS
inducible nitric oxide synthase
- NO
nitric oxide
- KLF6
Kruppel-like factor 6
- BMDMs
bone marrow-derived macrophages
- WT
wild type
- KO
knock-out
- ChIP
Chromatin immuno-precipitation
- L-NIL
N6-(1-iminoethyl)-L-lysine dihydrochloride
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end-labeling
- PI
propidium iodide
References
- 1.Kunisaki KM, Janoff EN. Influenza in immunosuppressed populations: a review of infection frequency, morbidity, mortality, and vaccine responses. Lancet Infect Dis. 2009;9:493–504. doi: 10.1016/S1473-3099(09)70175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tscherne DM, García-Sastre A. Virulence determinants of pandemic influenza viruses. J Clin Invest. 2011;4:6–13. doi: 10.1172/JCI44947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruuskanen O, Lahti E, Jennings LC, Murdoch DR. Viral pneumonia. Lancet. 2011;377:1264–1275. doi: 10.1016/S0140-6736(10)61459-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. 2010;15:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Vareille M, Kieninger E, Edwards MR, Regamey N. The airway epithelium: soldier in the fight against respiratory viruses. Clin Microbiol Rev. 2011;24:210–29. doi: 10.1128/CMR.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Reilly P, Hickman-Davis JM, McArdle P, Young KR, Matalon S. The role of nitric oxide in lung innate immunity: modulation by surfactant protein-A. Mol Cell Biochem. 2002;234–235(1–2):39–48. [PubMed] [Google Scholar]
- 7.Richardson AR, Libby SJ, Fang FC. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science. 2008;319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 8.Zheng S, De BP, Choudhary S, Comhair SA, Goggans T, Slee R, Williams BR, Pilewski J, Haque SJ, Erzurum SC. Impaired innate host defense causes susceptibility to respiratory virus infections in cystic fibrosis. Immunity. 2003;18:619–630. doi: 10.1016/s1074-7613(03)00114-6. [DOI] [PubMed] [Google Scholar]
- 9.Zheng S, Xu W, Bose S, Banerjee AK, Haque SJ, Erzurum SC. Impaired nitric oxide synthase-2 signaling pathway in cystic fibrosis airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2004;287:L374–81. doi: 10.1152/ajplung.00039.2004. [DOI] [PubMed] [Google Scholar]
- 10.Bove PF, van der Vliet A. Nitric oxide and reactive nitrogen species in airway epithelial signaling and inflammation. Free Radic Biol Med. 2006;41:515–527. doi: 10.1016/j.freeradbiomed.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Akaike T, Maeda H. Nitric oxide and virus infection. Immunology. 2000;101:300–308. doi: 10.1046/j.1365-2567.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uetani K, Der SD, Zamanian-Daryoush M, de La Motte C, Lieberman BY, Williams BR, Erzurum SC. Central role of double-stranded RNA-activated protein kinase in microbial induction of nitric oxide synthase. J Immunol. 2000;165:988–996. doi: 10.4049/jimmunol.165.2.988. [DOI] [PubMed] [Google Scholar]
- 13.Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD. Rapid interferon gamma-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J Exp Med. 1998;188:1541–1546. doi: 10.1084/jem.188.8.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, Dietzschold B, Maeda H. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci U S A. 1996;93:2448–2453. doi: 10.1073/pnas.93.6.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burggraaf S, Bingham J, Payne J, Kimpton WG, Lowenthal JW, Bean AG. Increased inducible nitric oxide synthase expression in organs is associated with a higher severity of H5N1 influenza virus infection. PLoS One. 2011;6:e14561. doi: 10.1371/journal.pone.0014561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kharitonov SA, Barnes PJ. Nitric Oxide, Nitrotyrosine, and Nitric Oxide Modulators in Asthma and Chronic Obstructive Pulmonary Disease. Curr Allergy Asthma Rep. 2003;3:121–129. doi: 10.1007/s11882-003-0024-7. [DOI] [PubMed] [Google Scholar]
- 17.McConnell BB, V, Yang W. Mammalian Krüppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oishi Y, Manabe I, Tobe K, Ohsugi M, Kubota T, Fujiu K, Maemura K, Kubota N, Kadowaki T, Nagai R. SUMOylation of Krüppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPAR- Nat Med. 2008;14:656–666. doi: 10.1038/nm1756. [DOI] [PubMed] [Google Scholar]
- 19.Sabbah A, Chang T, Harnack R, Frohlich V, Dube PH, Tominaga K, Xiang Y, Bose S. Activation of innate immune antiviral response by Nod2. Nature Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ueba O. Respiratory syncytial virus - concentration and purification of the infectious virus. Acta Med Okayama. 1978;32:265–272. [PubMed] [Google Scholar]
- 21.Kota S, Sabbah A, Chang TH, Harnack R, Xiang Y, Meng Y, Bose S. Role of human beta-defensin-2 during tumor necrosis factor-alpha/NF-kB mediated innate anti-viral response against human respiratory syncytial virus. J Biol Chem. 2008;283:22417–22429. doi: 10.1074/jbc.M710415200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Racoosin EL, Swanson JA. Macrophage colony stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J Exp Med. 1989;170:1635–1648. doi: 10.1084/jem.170.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao H, Hwang J, Moscat J, Diaz-Meco MT, Leitges M, Kishore N, Li X, Rahman I. Protein Kinase C Mediates Cigarette Smoke/Aldehyde- and Lipopolysaccharide-induced Lung Inflammation and Histone Modifications. J Biol Chem. 2010;285:5405–5416. doi: 10.1074/jbc.M109.041418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolodziejski PJ, Koo JS, Eissa NT. Regulation of inducible nitric oxide synthase by rapid cellular turnover and cotranslational down-regulation by dimerization inhibitors. Proc Natl Acad Sci U S A. 2004;101:18141–18146. doi: 10.1073/pnas.0406711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bose S, Kar N, Maitra R, Didonato J, Banerjee AK. Temporal activation of NF-κB regulates an interferon independent innate anti-viral response against cytoplasmic RNA viruses. Proc Natl Acad Sci USA. 2003;100:10890–10895. doi: 10.1073/pnas.1832775100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Echchgadda I, Chang T, Sabbah A, Bakri I, Ikeno Y, Hubbard G, Chatterjee B, Bose S. Oncolytic targeting of androgen-sensitive prostate tumor by the respiratory syncytial virus: A Consequence to impaired type-I interferon-dependent antiviral response. BMC Cancer. 2011;11:43. doi: 10.1186/1471-2407-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Echchgadda I, Kota S, DeLa Cruz I, Sabbah A, Chatterjee B, Bose S. Anti-cancer oncolytic activity of respiratory syncytial virus. Cancer Gene Ther. 2009;16:23–935. doi: 10.1038/cgt.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banihashemi B, Vlad R, Debeljevic B, Giles A, Kolios MC, Czarnota GJ. Ultrasound imaging of apoptosis in tumor response: novel preclinical monitoring of photodynamic therapy effects. Cancer Res. 2008;68:8590–6. doi: 10.1158/0008-5472.CAN-08-0006. [DOI] [PubMed] [Google Scholar]
- 29.Warke VG, Nambiar MP, Krishnan S, Tenbrock K, Geller DA, Koritschoner NP, Atkins JL, Farber DL, Tsokos GC. Transcriptional activation of the human inducible nitric-oxide synthase promoter by Kruppel-like factor 6. J Biol Chem. 2003;278:14812–14819. doi: 10.1074/jbc.M300787200. [DOI] [PubMed] [Google Scholar]
- 30.Land SC, Rae C. iNOS initiates and sustains metabolic arrest in hypoxic lung adenocarcinoma cells: mechanism of cell survival in solid tumor core. Am J Physiol Cell Physiol. 2005;289:C918–33. doi: 10.1152/ajpcell.00476.2004. [DOI] [PubMed] [Google Scholar]
- 31.Badn W, Visse E, Darabi A, Smith KE, Salford LG, Siesjö P. Postimmunization with IFN-gamma-secreting glioma cells combined with the inducible nitric oxide synthase inhibitor mercaptoethylguanidine prolongs survival of rats with intracerebral tumors. J Immunol. 2007;179:4231–4238. doi: 10.4049/jimmunol.179.6.4231. [DOI] [PubMed] [Google Scholar]
- 32.Rae C, Cherry J, Land F, Land S. Endotoxin-induced nitric oxide production rescues airway growth and maturation in atrophic fetal rat lung explants. Biochem Biophys Res Commun. 2006;349:416–425. doi: 10.1016/j.bbrc.2006.08.067. [DOI] [PubMed] [Google Scholar]
- 33.Darling K, Evans T. Effects of nitric oxide on Pseudomonas aeruginosa infection of epithelial cells from a human respiratory cell line derived from a patient with cystic fibrosis. Infect Immun. 2003;71:2341–2349. doi: 10.1128/IAI.71.5.2341-2349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC., Sr Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195:1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinshaw VS, Olsen CW, Dybdahl-Sissoko N, Evans D. Apoptosis: a mechanism of cell killing by influenza A and B viruses. J Virol. 1994;68:3667–3673. doi: 10.1128/jvi.68.6.3667-3673.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, Yang Y, Zhou X, Liu X, Song H, He Y, Huang P. Highly pathogenic avian influenza A virus H5N1 NS1 protein induces caspase-dependent apoptosis in human alveolar basal epithelial cells. Virol J. 2010;7:51. doi: 10.1186/1743-422X-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu X, Masic A, Li Y, Shin Y, Liu Q, Zhou Y. The PI3K/Akt pathway inhibits influenza A virus-induced Bax-mediated apoptosis by negatively regulating the JNK pathway via ASK1. J Gen Virol. 2010;91:1439–1449. doi: 10.1099/vir.0.018465-0. [DOI] [PubMed] [Google Scholar]
- 38.Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. 2008;205:3065–3077. doi: 10.1084/jem.20080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brydon EW, Morris SJ, Sweet C. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol Rev. 2005;29:837–850. doi: 10.1016/j.femsre.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 40.Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. Pathological changes in virus infections of the lower respiratory tract in children. J Clin Pathol. 1970;23:7–18. doi: 10.1136/jcp.23.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol. 2007;20:108–119. doi: 10.1038/modpathol.3800725. [DOI] [PubMed] [Google Scholar]
- 42.Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 43.Shiozaki T, Iwai A, Kawaoka Y, Takada A, Kida H, Miyazaki T. Requirement for Siva-1 for replication of influenza A virus through apoptosis induction. J Gen Virol. 2011;92(Pt 2):315–25. doi: 10.1099/vir.0.028316-0. [DOI] [PubMed] [Google Scholar]
- 44.Ueda M, Daidoji T, Du A, Yang CS, Ibrahim MS, Ikuta K, Nakaya T. Highly pathogenic H5N1 avian influenza virus induces extracellular Ca2+ influx, leading to apoptosis in avian cells. J Virol. 2010;84:3068–3078. doi: 10.1128/JVI.01923-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLean JE, Datan E, Matassov D, Zakeri ZF. Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J Virol. 2009;83:8233–8246. doi: 10.1128/JVI.02672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brincks EL, Kucaba TA, Legge KL, Griffith TS. Influenza-induced expression of functional tumor necrosis factor-related apoptosis-inducing ligand on human peripheral blood mononuclear cells. Hum Immunol. 2008;69:634–646. doi: 10.1016/j.humimm.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wurzer WJ, Ehrhardt C, Pleschka S, Berberich-Siebelt F, Wolff T, Walczak H, Planz O, Ludwig S. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem. 2004;279:30931–30937. doi: 10.1074/jbc.M403258200. [DOI] [PubMed] [Google Scholar]
- 48.DiFeo A, Narla G, Camacho-Vanegas O, Nishio H, Rose SL, Buller RE, Friedman SL, Walsh MJ, Martignetti JA. E-cadherin is a novel transcriptional target of the KLF6 tumor suppressor. Oncogene. 2006;25:6026–6031. doi: 10.1038/sj.onc.1209611. [DOI] [PubMed] [Google Scholar]
- 49.Kim Y, Ratziu V, Choi SG, Lalazar A, Theiss G, Dang Q, Kim SJ, Friedman SL. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem. 1998;273:33750–33758. doi: 10.1074/jbc.273.50.33750. [DOI] [PubMed] [Google Scholar]
- 50.Ratziu V, Lalazar A, Wong L, Dang Q, Collins C, Shaulian E, Jensen S, Friedman SL. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis. Proc Natl Acad Sci USA. 1998;95:9500–9505. doi: 10.1073/pnas.95.16.9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kojima S, Hayashi S, Shimokado K, Suzuki Y, Shimada J, Crippa MP, Friedman SL. Transcriptional activation of urokinase by the Kruppellike factor Zf9/COPEB activates latent TGF-beta1 in vascular endothelial cells. Blood. 2000;95:1309–1316. [PubMed] [Google Scholar]
- 52.Reeves HL, Narla G, Ogunbiyi O, Haq AI, Katz A, Benzeno S, Hod E, Harpaz N, Goldberg S, Tal-Kremer S, Eng FJ, Arthur MJ, Martignetti JA, Friedman SL. Kruppel-like factor 6 (KLF6) is a tumor-suppressor gene frequently inactivated in colorectal cancer. Gastroenterology. 2004;126:1090–1103. doi: 10.1053/j.gastro.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Tarabishi R, Zahedi K, Mishra J, Ma Q, Kelly C, Tehrani K, Devarajan P. Induction of Zf9 in the kidney following early ischemia/reperfusion. Kidney Int. 2005;68:1511–1519. doi: 10.1111/j.1523-1755.2005.00563.x. [DOI] [PubMed] [Google Scholar]
- 54.Starkel P, Sempoux C, Leclercq I, Herin M, Deby C, Desager JP, Horsmans Y. Oxidative stress, KLF6 and transforming growth factor-beta up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J Hepatol. 2003;39:538–546. doi: 10.1016/s0168-8278(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 55.Kaushik DK, Gupta M, Das S, Basu AJ. Krüppel-like factor 4, a novel transcription factor regulates microglial activation and subsequent neuroinflammation. Neuroinflammation. 2010;7:68. doi: 10.1186/1742-2094-7-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;4:13769–13779. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 57.Flandin J, Chano F, Descoteaux A. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-gamma-primed macrophages. Eur J Immunol. 2006;36:411–420. doi: 10.1002/eji.200535079. [DOI] [PubMed] [Google Scholar]
- 58.Pindado J, Balsinde J, Balboa M. TLR3-dependent induction of nitric oxide synthase in RAW 264.7 macrophage-like cells via a cytosolic phospholipase A2/cyclooxygenase-2 pathway. J Immunol. 2007;179:4821–4828. doi: 10.4049/jimmunol.179.7.4821. [DOI] [PubMed] [Google Scholar]
- 59.Tumurkhuu G, Koide N, Dagvadorj J, Noman A, Khuda I, Naiki Y, Komatsu T, Yoshida T, Yokochi T. B1 cells produce nitric oxide in response to a series of toll-like receptor ligands. Cell Immunol. 2010;261:122–127. doi: 10.1016/j.cellimm.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Lewis R, Kolesnik T, Kuang Z, D’Cruz A, Blewitt M, Masters S, Low A, Willson T, Norton R, Nicholson S. TLR regulation of SPSB1 controls inducible nitric oxide synthase induction. J Immunol. 2011;187:3798–3805. doi: 10.4049/jimmunol.1002993. [DOI] [PubMed] [Google Scholar]
- 61.Ciencewicki J, Brighton L, Wu W, Madden M, Jaspers I. Diesel exhaust enhances virus- and poly(I:C)-induced Toll-like receptor 3 expression and signaling in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1154–L1163. doi: 10.1152/ajplung.00318.2005. [DOI] [PubMed] [Google Scholar]
- 62.Hou Y, Zhou Y, Zheng X, Wang H, Fu Y, Fang Z, He S. Modulation of expression and function of Toll-like receptor 3 in A549 and H292 cells by histamine. Mol Immunol. 2006;43:1982–1992. doi: 10.1016/j.molimm.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 63.Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M, Si-Tahar M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 64.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007;178:3368–3372. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]
- 66.Li H, Han M, Bernier M, Zheng B, Sun S, Su M, Zhang R, Fu J, Wen J. Krüppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J Biol Chem. 2010;285:17846–17856. doi: 10.1074/jbc.M109.076992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Botella L, Sánchez-Elsner T, Sanz-Rodriguez F, Kojima S, Shimada J, Guerrero-Esteo M, Cooreman M, Ratziu V, Langa C, Vary C, Ramirez J, Friedman S, Bernabéu C. Transcriptional activation of endoglin and transforming growth factor-beta signaling components by cooperative interaction between Sp1 and KLF6: their potential role in the response to vascular injury. Blood. 2002;100:4001–4010. doi: 10.1182/blood.V100.12.4001. [DOI] [PubMed] [Google Scholar]
- 68.Rubinstein M, Idelman G, Plymate S, Narla G, Friedman S, Werner H. Transcriptional activation of the insulin-like growth factor I receptor gene by the Kruppel-like factor 6 (KLF6) tumor suppressor protein: potential interactions between KLF6 and p53. Endocrinology. 2004;145:3769–3777. doi: 10.1210/en.2004-0173. [DOI] [PubMed] [Google Scholar]
- 69.Feinberg M, Cao Z, Wara A, Lebedeva M, Senbanerjee S, Jain M. Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem. 2005;280:38247–38258. doi: 10.1074/jbc.M509378200. [DOI] [PubMed] [Google Scholar]
- 70.Du J, Bialkowska A, McConnell B, Yang V. SUMOylation regulates nuclear localization of Kruppel-like factor 5. J Biol Chem. 2008;283:31991–32002. doi: 10.1074/jbc.M803612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei H, Wang X, Gan B, Urvalek A, Melkoumian Z, Guan J, Zhao J. Sumoylation delimits KLF8 transcriptional activity associated with the cell cycle regulation. J Biol Chem. 2006;281:16664–16671. doi: 10.1074/jbc.M513135200. [DOI] [PubMed] [Google Scholar]
- 72.Laub F, Aldabe R, Ramirez F, Friedman S. Embryonic expression of Krüppel-like factor 6 in neural and non-neural tissues. Mech Dev. 2001;106:167–170. doi: 10.1016/s0925-4773(01)00419-1. [DOI] [PubMed] [Google Scholar]
- 73.Kuhn AR, Schlauch K, Lao R, Halayko AJ, Gerthoffer WT, Singer CA. MicroRNA expression in human airway smooth muscle cells: role of miR-25 in regulation of airway smooth muscle phenotype. Am J Respir Cell Mol Biol. 2010;42:506–513. doi: 10.1165/rcmb.2009-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.