

Abstract
The voltage-gated Na+ channel is a critical determinant of the action potential upstroke. Increasing Na+ conductance may speed action potential propagation. Here we propose use of the skeletal muscle Na+ channel SkM1 as a more favorable gene than the cardiac isoform SCN5A to enhance conduction velocity in depolarized cardiac tissue. We used cells which electrically coupled with cardiac myocytes as a delivery platform to introduce the Na+ channels. HEK293 cells were stably transfected with SkM1 or SCN5A. SkM1 had a more depolarized (18mV shift) inactivation curve than SCN5A. We also found that SkM1 recovered faster from inactivation than SCN5A. When coupled with SkM1 expressing cells, cultured myocytes showed an increase in the dV/dtmax of the action potential. Expression of SCN5A had no such effect. In an in vitro cardiac syncytium, coculture of neonatal cardiac myocytes with SkM1 expressing but not SCN5A expressing cells significantly increased the conduction velocity under both normal and depolarized conditions. In an in vitro re-entry model induced by high frequency stimulation, expression of SkM1 also enhanced angular velocity of the induced re-entry. These results suggest that cells carrying a Na+ channel with a more depolarized inactivation curve can improve cardiac excitability and conduction in depolarized tissues.
Keywords: Na+ channel, Gene and cell therapy, Cardiac conduction, Arrhythmia
INTRODUCTION
Reentrant arrhythmias can occur in the setting of slow conduction and unidirectional block 1. Approaches to eliminate this life threatening event include increasing refractoriness 2-3 (with the consequent threat of long Q-T syndrome), converting unidirectional block to bidirectional block by blocking excitatory sodium current 4-6 and more recently the conversion of slow conduction to more rapid conduction 7-9 thereby increasing the wavelength of the propagating wave reducing the potential for reentrant events.
Our group has focused recently on the last of these three approaches, by examining ways to increase the speed of conduction. Conduction velocity is determined by two major variables, the magnitude of the peak inward current and the axial resistance that the myoplasm and gap junctional coupling provide 10. In conditions of post-infarction ischemia the membrane potential is depolarized and gap junctional coupling is reduced 11-12; thus both gap junctional coupling and sodium current represent potential therapeutic targets. Our approach has focused on the inward sodium current, whose magnitude is determined by 1) the number of sodium channels available to be opened, 2) the single channel conductance, 3) the voltage dependence of activation, and 4) the voltage dependence of inactivation 13. When the resting membrane potential is depolarized, a greater fraction of the channels have their inactivation gates closed. This removes a large fraction of the membrane resident sodium channels from participation in the inward current that generates the action potential upstroke, thus slowing conduction. Because of these depolarizing effects on inactivation, our approach has been to work with sodium channels whose voltage dependence of inactivation is more depolarized. In our studies to date 7-8, we have used the skeletal muscle sodium channel (SkM1 or Nav1.4) which inactivates more positively than the cardiac sodium channel isoform (SCN5A or Nav1.5) allowing a greater fraction of these channels to participate in the action potential (AP) upstroke. In prior studies we have used virally-delivered SkM1 to increase action potential upstroke velocity, speed conduction and decrease the inducibility of VT/VF (ventricular tachycardia/ventricular fibrillation) in the 1 week canine infarct as well as to reduce the incidence of spontaneously occurring VT/VF in the mouse heart following acute ischemia and reperfusion 7.
Given concerns about viral delivery and the demonstrated success of cell delivery using adult human mesenchymal stem cells in fabricating biological pacemakers 14-15, we now have explored the possibility of using cell delivery of SkM1 in in vitro cardiac settings. We use HEK293 cells stably transfected with SkM1 or SCN5A genes to test the hypothesis that cellular delivery of a sodium channel with a more depolarized inactivation curve will positively affect cardiac electrical properties associated with the genesis of arrhythmias. We confirm the differences in the inactivation voltage dependence of the channels and the ability of HEK293-SkM1 coupled to canine cardiac myocytes to increase the action potential upstroke velocity (dV/dtmax). We also examine the effects of both the sodium channel genes on the conduction velocity in a culture of neonatal myocytes and their abilities to alter angular velocity. These studies set the stage for future examining the use of canine mesenchymal stem cells transfected with the SkM1 gene to alter conduction velocity and the incidence of arrhythmias in the post-infarction canine heart.
MATERIALS AND METHODS
Cell Culture and Transfection
Human embryonic kidney (HEK) 293 cells (ATCC, Manassas, VA) were maintained in DMEM (Dulbecco’s Modified Eagle’s Medium, GIBCO Invitrogen, Carlsbad, CA) supplemented with 10% FBS (fetal bovine serum, Sigma-Aldrich, St Louis MO) and 1% penicillin-streptomycin (Sigma) at 37°C in a humidified atmosphere incubator with 5% CO2 and 95% air.
HEK293 cells were transfected with the pIRES2-EGFP-SkM1 or -SCN5A construct DNA using Lipofectamine™ 2000 reagent (from Invitrogen) as directed. EGFP expression was examined on the next day. 48 hours after transfection, 500 μg/ml Geneticin (GIBCO Invitrogen) was applied to select the stably transfected cells. The stable cell lines then underwent fluorescence activated cell sorting (USB Research Flow Cytometry Core Facility) to further select GFP-positive cells. The selected cells with high GFP fluorescence were then maintained in Geneticin (500 μg/ml) containing culture medium at 37°C in a humidified atmosphere incubator with 5% CO2 and 95% air.
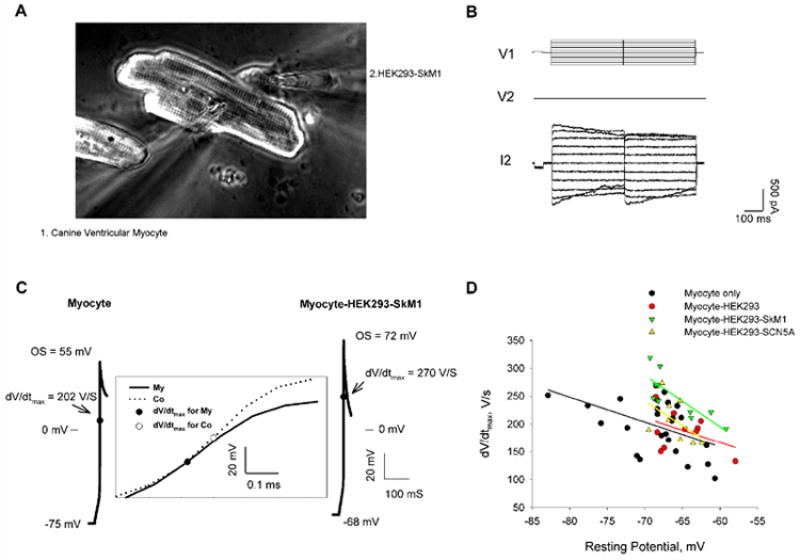
Generation of Cell Pairs between Canine Ventricular Myocytes and HEK293 Cells
Adult mongrel dogs of either sex weighing more than 22 kg were euthanized in accordance with an approved IACUC protocol at Stony Brook University by intravenous injection of sodium pentobarbitone (80mg/kg body weight) and the heart removed. Canine ventricular cells were isolated using a modified Langendorff procedure as previously described 16. Isolated myocytes were stored in Kraft-Brühe (KB) solution at room temperature before coculture with HEK293 cells. The cardiomyocytes in KB solution were plated onto mouse laminin (10 μg/ml, Invitrogen) coated coverslips and incubated in a 37°C, 5% CO2/95% air incubator for 1 hour for attachment and the solution was then replaced by Medium 199 supplemented with 10% FBS, 1% penicillin/streptomycin and 50 μg/ml Gentamicin (Invitrogen). HEK293 cells were typsinized by 0.25% trypsin-EDTA (Gibco Invitrogen) and then collected by centrifugation at 1000 rpm for 4 minutes. The cell pellet was re-suspended in supplemented M199 medium and distributed into the myocyte culture. The cocultures were incubated at 37°C in a humidified atmosphere with 5% CO2. Dual patch clamp experiments were performed 48 to 72 hours after plating. KB solution contains (in mM): KCl, 83; K2HPO4, 30; MgSO4, 5; Na-Pyruvic Acid, 5; β-OH-Butyric Acid, 5; Creatine, 5; Taurine, 20; Glucose, 10; EGTA, 0.5; KOH, 2; and Na2-ATP, 5. The pH was adjusted to 7.2 with KOH.
Electrophysiology
Whole cell patch clamp was used to measure the membrane current in single transfected HEK293 cells. To better control the Na+ current for a more accurate measurement, we used a low Na+ Tyrode’s solution containing (in mM): NaCl 15, TEACl 122.7, KCl 5.4, NaOH 2.3, CaCl2 1.8, MgCl2 1, Glucose 10, and HEPES 10 (pH adjusted to 7.4 with NaOH) to perfuse the cells at room temperature for all Na+ current recordings. Dual patch clamp was used to measure the gap junctional current and action potentials in myocyte-HEK293 cell pairs. Action potentials were stimulated in myocytes with a positive stimulus and recorded by current clamp mode. Cells were superfused with Normal Tyrode’s solution containing (in mM) NaCl 137.7, KCl 5.4, NaOH 2.3, CaCl2 1.8, MgCl2 1, Glucose 10, and HEPES 10 (pH adjusted to 7.4 with NaOH). Electrodes were made from a capillary with a P-87 Flaming/Brown micropipette puller (Sutter Instrument Company, Novato, CA) and filled with (in mM): KCl 50, K-aspartic acid 80, MgCl2 1, EGTA 10, HEPES 10 and Na2-ATP (pH adjusted to 7.2 with KOH). The resistances of the electrodes were between 3 to 4 MΩ. There was a liquid junction potential of ~8 mV between the bath solutions and the electrode solution which was not corrected because exchange between the pipette and cell are never complete 17.
Voltage and current signals were recorded by the amplifiers (Model Axopatch-1B, Axon Instruments Inc.) and digitized through digitizers (Model DIGIDATA 1320A, Axon Instruments) and finally transferred to a personal computer. Data acquisition and analysis were performed by the computer using CLAMPEX 9.2 and CLAMFIT 9.2 software (Axon instruments), respectively.
Generation of the Cardiac Syncytium
Neonatal Sprague-Dawley rats were sacrificed and the ventricular myocytes were isolated by an approved Stony Brook University IACUC protocol as previously described 18. Briefly, the ventricular portion of the hearts was excised and washed free of blood. The tissue was then cut into small pieces and enzymatically digested with trypsin at 4 °C (1mg/ml, USB, Cleveland, OH), then with collagenase at 37°C (1mg/ml, Worthington, Lakewood, NJ) the next morning. Cardiac fibroblasts were removed by ninety minutes of preplating. The isolated ventricular myocytes were then replated at high density (4×105 cells/cm2 for the control myocyte group and 3.5×105 cells/cm2 for the coculture groups) with approximately 50,000 HEK293 cells onto fibronectin-coated polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning, Midland, MI) scaffolds or plastic cover slips in M199 medium (GIBCO Invitrogen) supplemented with 10% fetal bovine serum (GIBCO Invitrogen) for the first 2 days and then reduced to 2%. Cultures were maintained in an incubator at 37°C with 5% CO2 for 4 to 5 days before functional measurements to get confluent monolayer and form gap junctions between each other.
Microscopic Dynamic Functional Measurements and Analysis
All scaffolds were washed and equilibrated at room temperature in normal Tyrode’s solution. The samples were then stained for with di-8 ANEPPS (Molecular Probe, Eugene, OR) for 10 minutes for measuring transmembrane voltage. After washing with Tyrode’s solution for another 10 minutes, the stained samples were transferred into an experimental chamber perfused with Tyrode’s solution at 30 to 32°C. The chamber was placed on an inverted microscope (Nikon TE2000). Propagation measurements were performed as previously described19. Briefly, a Pt line electrode was placed at one end of the scaffold to pace the cells with planar waves at frequencies from 1 Hz to 2 Hz with a 0.5 Hz step and thereafter in 0.2 Hz-increments following a dynamic restitution protocol20, until conduction block. Fluorescence signals were recorded at the other end of the scaffolds (1.1-1.5 cm from the stimulating electrode) with a photomultiplier tube (PMT). Fifteen to twenty transients were collected with IONOPTIX software at each frequency (60 beats pacing per frequency before recording). CV was calculated as described in Fig 5 and 5 transients were taken to average for each frequency. Normal (5.4 mM) K+ and high K+ (10.4 mM) Tyrode’s solutions were used in these experiments.
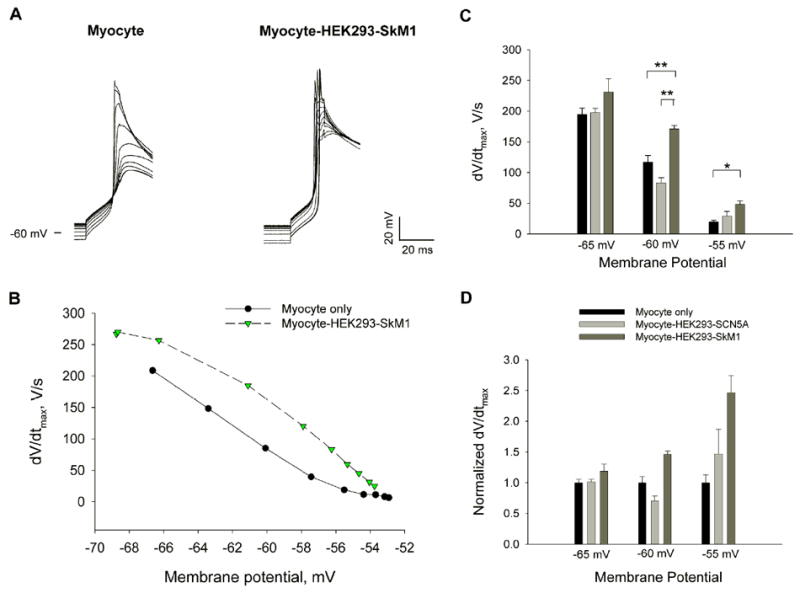
FIGURE 5. Change in dV/dtmax with resting potential.
(A) The change in the upstroke velocity of the AP with perfusion of 10mM K+ solution in a single myocyte and a myocyte coupled with a HEK293-SkM1 cell. (B) dV/dtmax vs. membrane potential during perfusion of 10mM K+ solution in one experiment. (C, D) Comparison of dV/dtmax among three different cultures at -65, -60 and -55 mV (mean±SE, n = 7, 5 and 5 for myocyte, myocyte-HEK293-SCN5A, and myocyte-HEK293-SkM1, respectively; ** P<0.001, * P<=0.002, one way ANOVA test). Data were normalized to the mean value of the single myocyte group at each potential.
Macroscopic Optical Mapping and Analysis
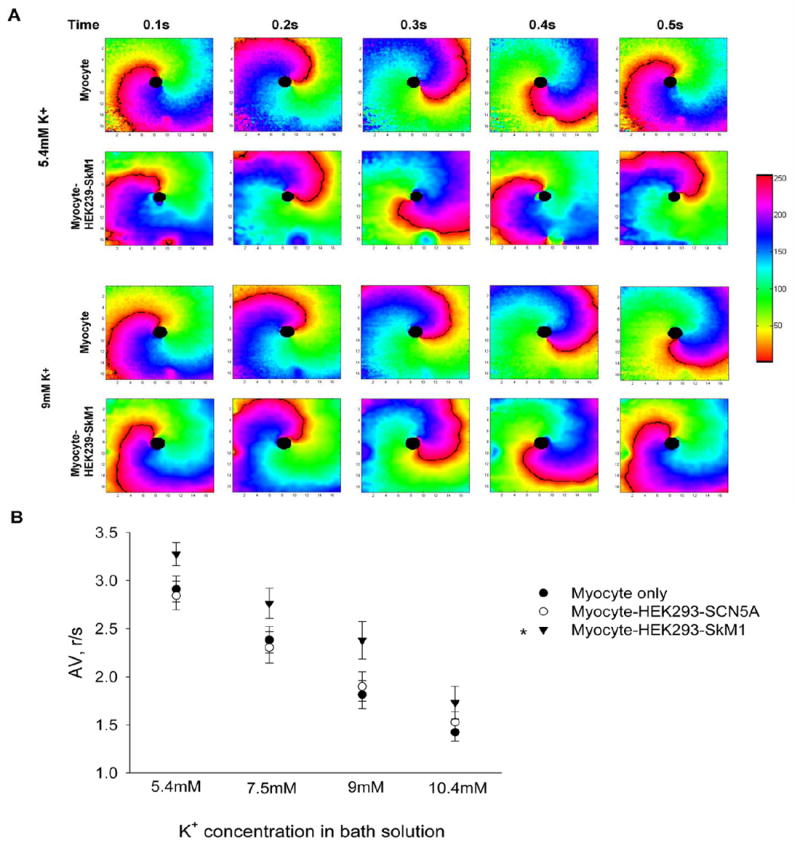
Two-dimensional optical mapping was done with a macroscopic system21, including a CMOS camera (pco, Germany, 200frames per second at1,280×1,024 pixel resolution), an intensifier ((Video Scope International, Dulles, VA), collecting optics (Navitar Platinum lens, 50mm, f/1.0) and filters, excitation light source (Oriel with fiber optics lights guides) and an adjustable imaging stage, over a field of view of 2cm. Samples on coverslips were washed and equilibrated in normal Tyrode’s solution at room temperature. Fluo-4 AM (Invitrogen, Carlsbad, CA) was used to label the cells for tracking Ca2+ waves. After staining, the coverslips were maintained in four different K+ concentrations (normal 5.4 mM, 7.5 mM, 9 mM and high 10.4 mM) for measurements. Propagation movies were acquired using CamWare (pco) data acquisition software. Raw data were binned (2×2) and analyzed in custom-developed Matlab software. Color phase movies were generated using the Hilbert transform 18 after filtering spatially (Bartlett filter, 5-pixel kernel) and temporally (Savitsky-Golay, order 2, width 7). Angular velocity for reentrant propagation was calculated from wavefront tracing across multiple frames.
Statistical Analysis of Data
Data are presented as mean ± standard error. The statistically significant differences between two groups or among three groups were determined by Student’s t-test or one/two way-ANOVA test, respectively. Differences were considered as significant when P≤0.05,
RESULTS
Characterization of the Biophysical Properties of Na+ Channels in SkM1 or SCN5A Expressing Cell Lines
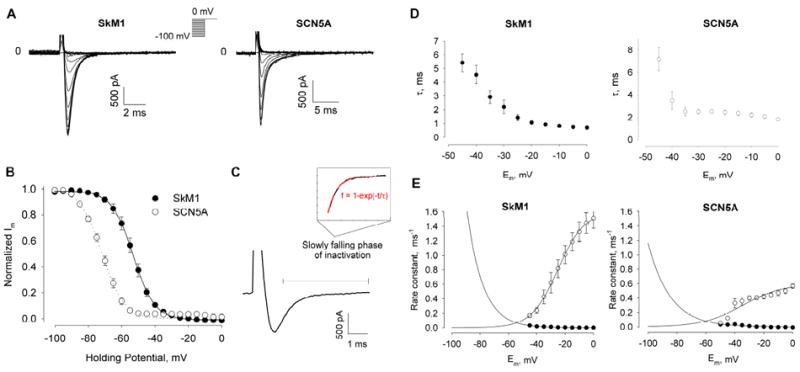
Voltage-gated Na+ channels are responsible for the upstroke and propagation of action potentials in excitable tissues. Although they share similar structures and functions, cardiac and skeletal muscle Na+ channels have different biophysical properties 22-23. The most important difference with respect to our study is their steady state inactivation properties. The cardiac channel has a midpoint of -74 to -80 mV 24-25, compared with a midpoint of around -68mV for the skeletal muscle channel 26. To confirm the more positive position of the skeletal muscle Na+ channel, we investigated the biophysical properties of both channels in our stably transfected cell lines.
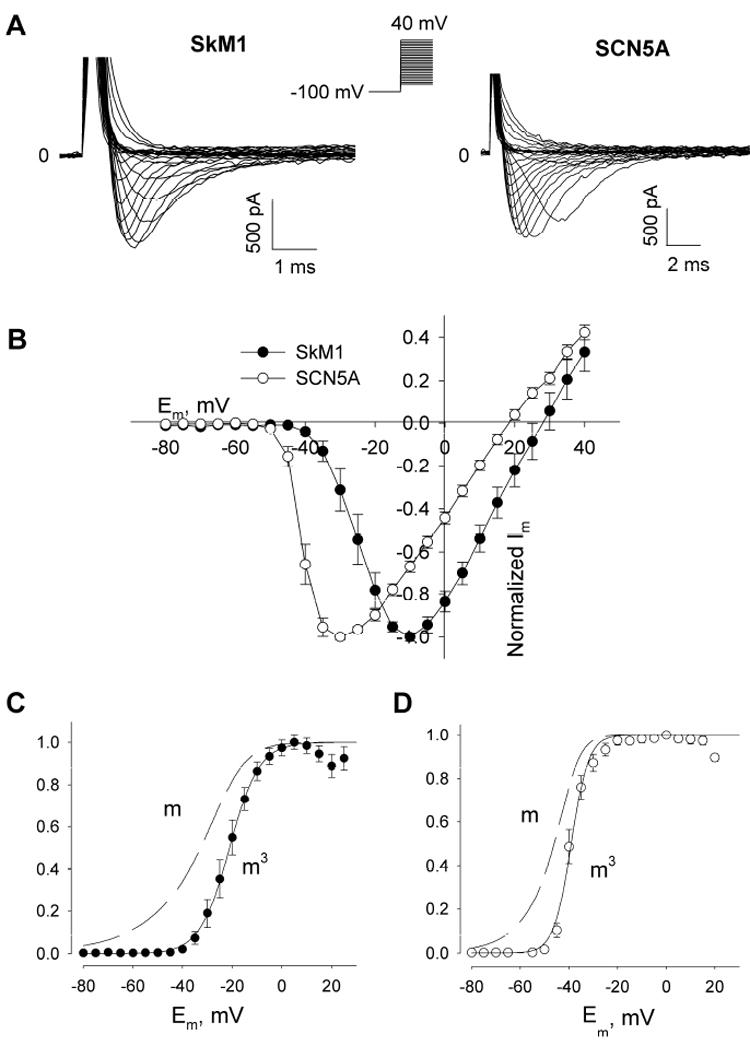
We first measured the voltage-dependent activation of SkM1 and SCN5A current in GFP positive cells (Fig 1A). The capacitances of HEK293-SkM-1 and HEK293-SCN5A cells were 19.72 ± 0.64 pF and 21.45 ± 0.90 pF, respectively. The difference of voltages of maximal currents is about 20 mV between SkM1 and SCN5A (-10 mV for SkM1 and -30 mV for SCN5A). We calculated the equilibrium potential for Na+
ENa was 23.31 mV at 22°C and found comparable reversal potentials for the I/V curves of SkM1 and SCN5A: 28.59±3.04 and 18.50±1.12 respectively (Fig 1B). The peak current density and peak conductance density were 84.53±12.39 pA/pF and 2.19±0.32 nS/pF for SkM1, respectively and 99.64±11.75 pA/pF and 2.05±0.32 nS/pF for SCN5A, respectively. T-tests indicated no significant difference in peak current density or peak conductance density between SkM1 and SCN5A, suggesting comparable expression levels of the two plasmids in HEK293 cells.
FIGURE 1. Activation of SkM1 and SCN5A in HEK293 cells.
(A) Expression of SkM1 or SCN5A in HEK293 cells. Na+ currents were recorded in low Na+ (15 mM) Tyrode’s solution at room temperature. Cells were held at -100 mV and then pulsed to test potentials from -80 mV to +40 mV, with a 5 mV increment. (B) Current-voltage relationship of SkM1 (n=8) and SCN5A (n=10) in HEK293 cells. Data are normalized to the maximum peak current (mean±SE). (C), (D) Study of the m gates of SkM1 and SCN5A. Data of m3 are normalized to the maximal value and fitted to the equation , giving a midpoint of -21.1±0.5 mV and a slope of 5.9±0.4 mV (mean±SE, n = 8) for SkM1 (C), and a midpoint of -39.0±0.4 mV and a slope of 3.6±0.4 mV (mean±SE, n = 10) for SCN5A (D). The m curves were calculated from the curve fit.
To study the activation of these two channels in detail, we calculated the conductance at each test potential as gNa = INa/(Etest-Erev), where Erev is the reversal potential of the I/V curve. Assuming the Hodgkin and Huxley model gNa = m3 hḡNa 27, where ḡNa is a constant and h is assumed as 1 because the cell was held at -100 mV, m3 is then proportional to the conductance. The m gate of the SkM1 channel starts to open around -80 mV and the conductance which is proportional to m3 is half maximal at -20 mV and maximal at +5 mV (Fig 1C); the m gate of SCN5A channel also starts to open at -80 mV, half maximal conductance (proportional to m3) occurs at -40 mV and maximal at -20 mV (Fig 1D).
Next we characterized the steady-state inactivation of SkM1 and SCN5A in HEK293 cells (Fig 2A). Normalized currents were fitted with the Boltzmann equation to obtain a midpoint of -53.1±0.1mV, a slope factor of 6.4 ± 0.1 mV for SkM1 and a midpoint of -71.6 ± 0.5 mV, a slope factor of 6.6±0.4mV for SCN5A (Fig 2B). T-tests indicate a significant difference between the midpoints of both channels (P<=0.005). This relatively positive position of inactivation voltage dependence of SkM1 suggests that it may function better than the cardiac Na+ channel to sustain conduction if cells are depolarized.
FIGURE 2. Inactivation of SkM1 and SCN5A in HEK293 cells.
(A) Representative inactivation of SkM1 and SCN5A currents in HEK293 cells. Cells were held at different holding potentials from -100 mV to 0 mV for 500 msec, with a 5 mV increment, and then pulsed to 0 mV. (B) The inactivation curve (the h∞ curve) of SkM1 (n=12) and SCN5A (n=10). Data are normalized to the maximum peak current and fitted to the Boltzmann equation , where Vh is the midpoint membrane potential and K is the slope factor. (C) Inactivation of a representative Na+ current, starting from 2/3 of the maximal current to full inactivation. (D) The time constant (τ) for each membrane potential of the h gate of channels was obtained by fitting the inactivation phase with an exponential equation f = 1-exp (-t/τ). Data are presented as mean ± SE, n = 8 and 6 for SkM1 and SCN5A, respectively. (E) Calculated α and β values of the h gate of the two channels for each membrane potential. αh values were curve fit by αh = a*exp (-b*Em), where a = 5 ± 2 × 10-4, b = 0.09 ± 0.01 for SkM1; a = 1.5 ± 1.4 × 10-3, b = 0.07 ± 0.02 for SCN5A. βh values were curve fit by , where a = 1.60 ± 0.03, b = 9.37 ± 0.32 and E0 = -25.16 ± 0.45 mV for SkM1; a = 0.60 ± 0.10, b = 14.02 ± 5.62 and E0 = -34.48 ± 6.34 mV for SCN5A.
We also studied the rates of fast inactivation of SkM1 and SCN5A. We took the falling phase (starting from around 2/3 of the maximal current to full inactivation, Fig 2C) of Na+ current measured in the activation experiment, and then fit the data with an exponential function to get a time constant (τ) of inactivation at each membrane potential (Fig 2D). Again employing the Hodgkin & Huxley model of the h gate of Na+ channel 27
we then calculated the α and β values of the h gates of the two channels for each membrane potential (Fig 2E) from the expressions
where h∞ values were obtained from the inactivation curve (Fig 2B). The much higher β values (closing rate constant) of SkM1 illustrate faster kinetics than SCN5A with regard to channel closing.
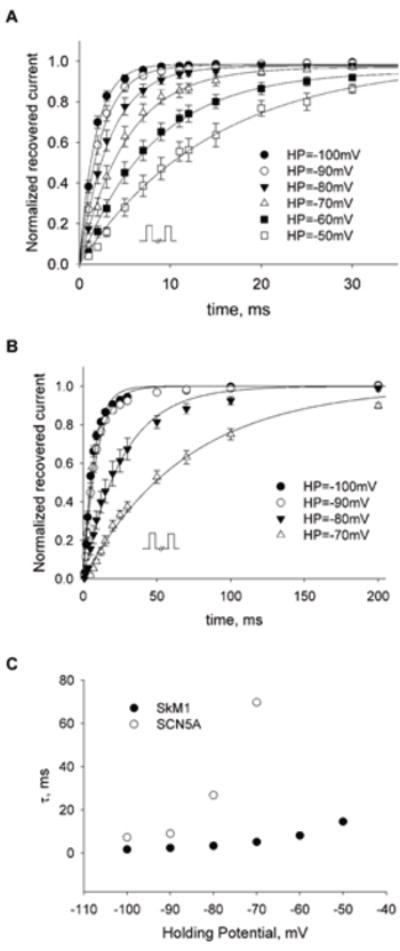
We next measured the recovery of SkM1 and SCN5A from inactivation. We obtained recovery time constants for each channel at a series of holding potentials (Fig 3A&B). These time constants of SkM1 are much smaller than those for the cardiac sodium channel SCN5A at all holding potentials (Fig 3C, Table 1), especially at more depolarized potentials. This suggests a much faster recovery of SkM1 which can potentially contribute to preservation of fast conduction of extra stimuli preventing unidirectional blocks and reentry.
FIGURE 3. Recovery of SkM1 (A) and SCN5A (B) in HEK293 cells.
Cells were held at a holding potential and double-pulsed to 0 mV. Each pulse had a 10 msec duration, with an increasing time interval between the two pulses. Data were normalized to the current value of the first pulse (mean±SE, n = 8 for each holding potential) and curve fit to the equation f = 1- exp (-t/τ). (C) Comparison of recovery time constant t between SkM1 and SCN5A at different holding potentials.
TABLE 1.
Comparison of the recovery time constants between SkM-1 and SCN5A in HEK293 cells
| Holding Potential, mV | τ of SkM-1, mS | τ of SCN5A, mS |
|---|---|---|
| -100 | 1.677 | 7.273 |
| -90 | 2.335 | 9.001 |
| -80 | 3.338 | 26.81 |
| -70 | 5.155 | 69.78 |
| -60 | 8.130 | -- |
| -50 | 14.58 | -- |
Effect of SkM1 on the Maximal Action Potential Rate of Rise (dV/dtmax) in Myocytes Coupled to SkM1 Expressing Cells
To set up a simple cell-to-cell delivery model, myocytes disassociated from canine ventricle were cocultured with HEK293 cells. To examine the electrical coupling between myocytes and HEK cells, we performed double patch clamp to myocyte-HEK cell pairs (Fig 4A&B) in both normal K+ (5.4mM) and high K+ (10.4 mM) Tyrode’s solution at room temperature within 48 to 72 hours after plating. We chose cell pairs with at least 5nS gap junctional conductance (Fig 4B), which hyperpolarized the HEK cells toward the resting potentials of the myocytes.
FIGURE 4. Effect of expression SkM1 on dV/dtmax of the action potential.
- Myocyte only: f = - 4.47× - 109.09, R = 0.4780
- Myocyte-HEK293: f = -4.45× - 100.18, R = 0.4139
- Myocyte-HEK293-SkM1: f = -9.17× - 355.14, R = 0.8106
- Myocyte-HEK293-SCN5A: f = -8.85× - 377.13, R = 0.5001
- Two way ANOVA test suggests significant difference between the SkM1 group and the control groups (P<0.01).
When current clamping the myocytes, we measured the resting potentials and then generated action potentials using a depolarizing stimulus (a 20 msec pulse of 20 to 40 mV) (Fig 4C). Then we compared the dV/dtmax of the APs measured in myocytes only, myocytes coupled with non-transfected HEK293 cells and myocytes coupled with transfected HEK293 cells expressing either SkM1 or SCN5A. We plot the dV/dtmax of the myocytes of the four different groups versus their resting potentials. Compared with the control groups and the Myocyte-HEK293-SCN5A group with similar myocyte resting potentials, the expression of SkM1 was associated with an increased dV/dtmax of the AP of myocytes coupled with HEK293-SkM1 cells (Fig 4D). The resting potentials of all the samples were binned into five different subsets: -80mV, -75mV, -70mV, -65mV, and -60mV (data were included into each subset by most proximal). Then we considered the four culture types as factor A and the five resting potential subsets as factor B. Two-way ANOVA (Holm-Sidak) and post-hoc multiple comparison tests suggested that the myocyte-HEK293-SkM1 coculture but not myocyte-HEK293 or myocyte-HEK293-SCN5A is significantly different from the control (P<0.01). To further study the effect of SkM1 on dV/dtmax at different membrane potentials, we recorded the changes of dV/dtmax with the decrease of the membrane potentials by superfusing the cell pairs with normal K+ and then high K+ solutions (Fig 5A&B). We compared the AP upstrokes of single myocytes and coupled myocytes at around -65 mV, -60 mV and -55 mV and found that SkM1 increased the dV/dtmax significantly at -60 mV and -55 mV (Fig 5C&D). In contrast, expression of SCN5A did not increase the AP upstrokes at these membrane potentials. The absence of an effect of SCN5A likely results from its more negative inactivation curve which may allow SCN5A to increase dV/dtmax only at more hyperpolarized potentials.
Thus, our two cell delivery model suggests that SkM1 currents from the HEK cells traverse the gap junctions and improve the AP upstroke in myocytes and it may function superior to SCN5A especially at depolarized potentials around -60 mV.
Effect of SkM1 on the Conduction Velocity of Action Potential Propagation in a Cell Culture Model
Neonatal rat ventricular myocytes are highly plastic and can reconnect into a tissue-like syncytium having a relatively depolarized resting potential28 after dissociation from the heart. Therefore we cocultured them with the HEK293 cells as an in vitro preparation to study electrical activity of cardiac tissue.
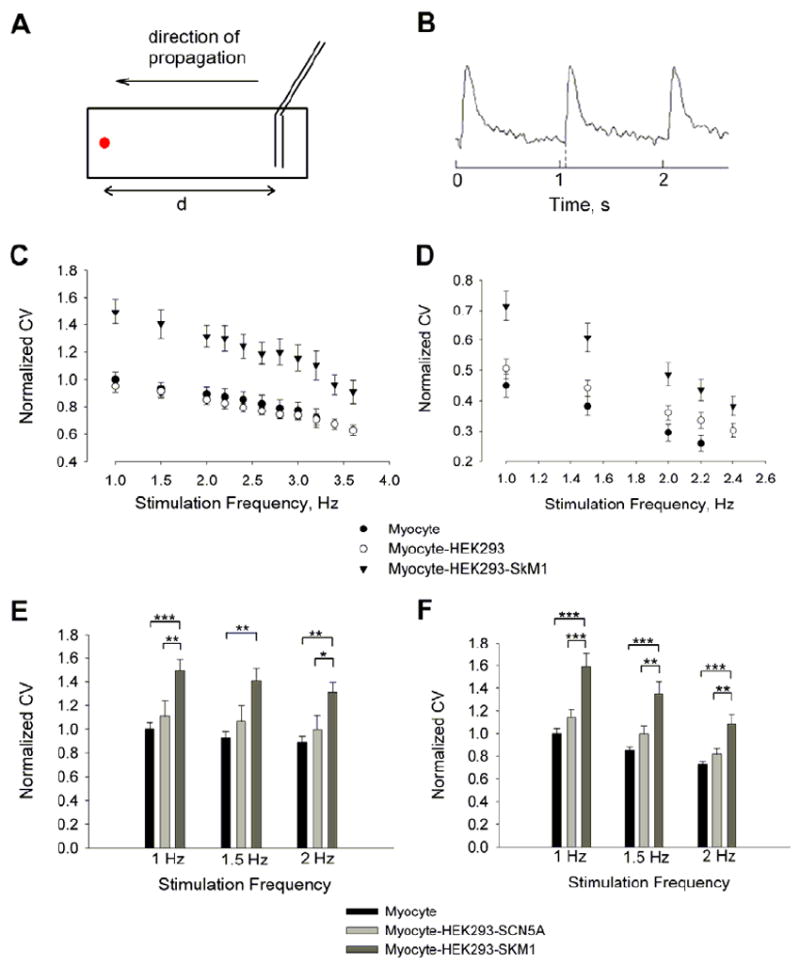
We paced the cultures at several frequencies from one edge of the scaffold with a platinum (Pt) line electrode (Fig6A). Dynamic fluorescence signals of propagated action potentials at the other edges of the scaffolds (1.1-1.5 cm from the electrodes) were recorded by microscopy. Conduction velocity (CV) was calculated as the lag time between the stimulation pulse and acquired action potential divided by the distance (d) between the electrode and the recording site (Fig 6A&B).
FIGURE 6. Effect of expression of SkM1 on conduction velocity (CV) of APs of in vitro cardiac cultures.
(A) Measurement of conduction velocity. A parallel Pt electrode is placed at one edge of the rectangular scaffold, and the recording site is shown as the red circle. d, the distance between the electrode and the recording site. (B) An example of fluorescence signal at the recording site with a 1Hz pacing frequency. The lag time (Δt) was estimated as the distance between the tick (stimulation point) and the dashed line (start of the signal). CV = d / Δt. CV in normal K (5.4 mM) (C) and high K (10.4 mM) (D) Tyrode’s solutions at various stimulation frequencies. The data were normalized to the average CV values of control myocytes in normal Tyrode’s solution at 1Hz for each experiment (mean±SE, n = 10, 12, 11 for myocyte, myocyte-HEK293, and myocyte-HEK293-SkM1, respectively). Comparison of CV between myocyte-HEK293-SkM1 and -SCN5A at 1, 1.5 and 2 Hz in normal Tyrode’s (E) or in high K solution (F), respectively. Data were normalized to the mean CV values of the control myocyte group (mean±SE, n = 10, 11, 10 for myocyte, myocyte-HEK293-SkM1, and myocyte-HEK293-SCN5A, respectively; *** P=<0.001, ** P<0.01, * P<0.025, one way ANOVA test).
At K+ 5.4 and 10.4 mM, CV was significantly higher in myocytes cocultured with HEK293-SkM1 cells compared with myocytes only and myocytes cocultured with non-transfected HEK293 cells at all frequencies (Fig 6C&D). We also compared the effects of SkM1 and SCN5A on CV at frequencies of 1, 1.5 and 2 Hz. The HEK293-SkM1 cells significantly increased the CV at all frequencies. In contrast, the conduction velocities of HEK293-SCN5A cell cocultures were not significantly faster in either normal or high K+ solutions (Fig 6E&F). This lack of effect of SCN5A may derive from the relatively depolarized membrane potential of the neonatal myocytes28.
Thus, our in vitro cardiac syncytium model suggests that introduction of SkM1 can improve conduction velocity better than SCN5A, especially under depolarized conditions.
Cellular Delivery of SkM1 Speed Conduction in an in vitro Arrhythmia (Reentry) Model
We have thus far demonstrated that HEK293 cells expressing SkM1 make gap junctions with cardiac myocytes, increase the maximal rate of rise of the AP and enhance conduction velocity. Next we tested if introducing the SkM1 channel into a cardiac syncytium under an “arrhythmic condition” could also increase conduction velocity and even prevent the occurrence of reentry.
To this end we placed the above described cocultures onto square fibronectin-coated coverslips and punched a hole with a 0.3 cm diameter in the center of the coverslip to generate a damaged zone in the syncytium. Reentry was induced by high-frequency pacing from a point electrode (typically around 3 to 4 Hz). Tyrode’s solutions of four different K+ concentrations (5.4, 7.5, 9 and 10.4 mM) were applied at room temperature. If spiral waves could not be generated in the normal K+ (5.4 mM) condition, a higher K+ solution was used until the conduction was slow enough to induce the reentrant propagation around the hole.
We compared the genesis of re-entry among myocyte only, myocyte-HEK293-SCN5A and myocyte-HEK293-SkM1 cocultures and found that 4 of 7, 3 of 7 and 5 of 7 samples for each of the three groups, respectively, could not generate spiral waves in normal K+ solution. Instead, spiral waves were induced in the 7.5 mM K+ solution for these samples.
Although we did not see a significant difference in the ability to generate spirals in the presence of SkM1, the propagation of the induced spirals was faster in SkM1 cultures as compared to control and SCN5A cultures. Raw data from macroscopic optical mapping were analyzed in custom-developed Matlab software to generate color phase movies (Fig 7A, see Video, Supplemental Digital Content 1 to 4, which show re-entrant propagation induced in a myocyte only sample and a myocyte-HEK293-SkM1 sample in normal K+ (5.4 mM) or high K+ (9 mM) Tyrode’s solution. The color bar shows the fluorescence intensity of the Ca2+ signal. The black line shows the wave front of the propagation). We then calculated the angular velocities (AV; expressed as rotations per second) of each spiral by measuring the time of 3 rotations (360° for each full rotation). We found that AV was significantly increased in SkM1 culture (Fig 7B). This higher AV in SkM1 culture may potentially terminate the propagation of the spirals because when the conduction is too fast there would be insufficient time for the tissue to recover from refractoriness.
FIGURE 7. Effect of expression of SkM1 on angular velocity (AV) of AP propagation.
(A) Phase graphs of re-entrant propagation induced in a myocyte only sample and a myocyte-HEK293-SkM1 sample in 5.4 mM K+ or 9 mM K+ Tyrode’s solution. Phase movies were generated in Metlab software from macroscopic optical mapping data. The color bar shows the fluorescence intensity of the Ca2+ signal. The black line shows the wave front of the propagation. Frames were analyzed every 0.1s to determine the propagation speed. (B) AV of induced re-entry at different K+ concentrations. Data are expressed as mean±SE, n = 7 for each group. (r/s: rotations/sec). Two way ANOVA (Holm-Sidak) test showed a significant difference between Myocyte only and Myocyte-HEK293-SkM1 (P<0.001), and between Myocyte-HEK293-SCN5A and Myocyte-HEK293-SkM1 (P<0.001), but no difference between Myocyte only and Myocyte-HEK293-SCN5A.
DISCUSSION
A Desirable Gene
The voltage gated Na+ channel plays an important role in generation of the upstroke of the AP in that its availability determines the conduction velocity of cardiac tissue. The candidate gene we chose for this antiarrhythmic study was a Na+ channel gene with more favorable characteristics. Compared with existing antiarrhythmic pharmacological therapies which slow or block propagation, we selected this gene to speed conduction, which was effective in the depolarized conditions often associated with an arrhythmic environment.
Our results confirmed the biophysical differences between SCN5A and SkM1 in exogenously expressing cell lines. Among the electrophysiological features of the voltage gated Na+ channel, inactivation is the one most important to cell excitability. It determines the pool of available Na+ channels to fire the AP. Two types of inactivation with different kinetics have been observed in the Na+ channels: fast inactivation within milliseconds and slow inactivation on the order of seconds29. These two separate processes involve different regions of the channel and different mechanisms to inactivate 13. Although both processes may regulate the resting state of Na+ channels and the AP duration, we used the Hodgkin and Huxley model (which describes the fast component of inactivation) to study steady state inactivation because introducing SkM1 into cardiac tissue did not increase the AP duration7 suggesting little if any slow inactivation. In HEK293 cells, we obtained an 18 mV difference between the steady state inactivation curves of SkM1 and SCN5A. At -90 mV, both genes have most of their channels available. At -60 mV, there were about 73% of SkM1 but only 13% of SCN5A available, while at -50 mV, most of SCN5A were inactivated but around 37% of SkM1 channels were still available. Thus the results suggest the fraction of available Na+ channels at depolarized potentials was much higher in cells expressing SkM1 than those expressing SCN5A. In addition, comparing the fast inactivation between SkM1 and SCN5A, we observed slower kinetics for SCN5A. This difference may result from modulation of the C-terminal domain of the SCN5A channel 30. The “faster” fast inactivation of SkM1 may provide another benefit to prevent long QT syndrome3 by preventing late sodium current.
Recovery from fast inactivation also contributes importantly to the availability of Na+ channels for AP propagation. Our results showed SkM1 recovered about 4.3 fold faster at a membrane potential of -100 mV and about 13.5 fold faster at -70 mV than SCN5A. These results demonstrate at a relatively depolarized potential, SCN5A takes an order of magnitude more time to recover than SkM1. This faster recovery of SkM1 may preserve propagation particularly at high frequency stimulation. However, it may have both advantages and disadvantages in avoiding reentrant propagation. On one hand, it may prevent re-entry by protecting from unidirectional block and/or by increasing the wavelength required to accommodate a reentrant wave. On the other hand, it might be difficult to stop existing re-entry because of the faster recovery from inactivation of the tissue. Nevertheless, this fast recovery from inactivation unambiguously speeds conduction in all circumstances.
A Safe Cell Delivery Platform
Because of the ability to insert their own genes into the chromosome of host cells, viruses have been commonly used as vectors to transfer genes into target cells. However, several problems prevent viral gene therapy from becoming an effective and safe treatment. First, immune responses might be stimulated after viral infection. Second, retrovirus vectors may integrate into the genome at any location, which yields a danger for tumorigenesis31. Third, adenoviruses might be safer vectors but have the problem of short-lived expression. In addition, disadvantages of toxicity, inflammation and uncontrolled viral replication or mutation limit the therapeutic use of gene therapy. In contrast, a cell delivery system may overcome some of these problems. We can use the patient’s own stem cells to introduce genes, which should reduce the risks of immunoresponse and toxicity.
Our strategy to deliver the Na+ channel into the cardiac tissue relies on gap junctions which provide the electrical connection between myocytes to conduct the AP in the heart. Three major isoforms of connexin proteins Cx43, Cx45 and Cx40 are prominently expressed in the heart32. We confirmed the expression of Cx43 and/or Cx45 in HEK293 cells and our double patch clamp data showed gap junction formation between the ventricular myocyte and the HEK293 cell. We hypothesized that the Na+ current passing through the membrane of the delivery cell also enters into the myocyte through gap junctions during generation of the AP, so that the AP upstroke can be enhanced. The upstroke of the AP determines the cardiac conduction velocity and its maximal rate of rise (dV/dtmax) is directly determined by the excitatory inward currents. So, measuring the effect of expression of SkM1 on the dV/dtmax was the first step in testing its antiarrhythmic potential. Our results showed that the dV/dtmax was significantly increased in the myocyte coupled with a HEK293-SkM1 cell. To generate a depolarized condition, we used a 10.4 mM K+ solution to decrease the cell resting potentials. We found that the dV/dtmax was also increased in SkM1 cell pairs in the high K+ solution. Analysis of the change of dV/dtmax with membrane potential showed that the increases by SkM1 were most significant at around -60mV and -55mV, with a ratio of 1.5 fold and 2.5 fold respectively. These results are consistent with the inactivation curve of SkM1 we obtained from HEK293 cells. At these voltages, there were big differences in the availability of Na+ channels between SkM1 and SCN5A.
We did not see an effect of SCN5A on the dV/dtmax in myocytes coupled with HEK293-SCN5A. Since the current densities of SkM1 and SCN5A were similar in the two stable cell lines, the absence of an effect of SCN5A is not due to lower expression of SCN5A. Rather, myocytes in primary tissue culture may have relatively lower resting potentials due to less of the inwardly rectifying current (Ik1)33. Most of our cultured myocytes showed membrane potentials between -65 and -68 mV. We assume expression of SCN5A may function effectively at more hyperpolarized potentials which our culture condition did not reach.
After demonstrating that SkM1 increases the AP upstroke, we then tested the effect of SkM1 on conduction velocity. Rat neonatal myocytes were used to generate an in vitro cardiac syncytium. We found that coculturing myocytes with HEK293-SkM1 cells could enhance the conduction velocity of the syncytium at both normal (5.4mM K+) and depolarized (10.4mM K+) conditions. Again, we did not see a significant increase in the conduction velocity in the SCN5A culture under either normal or depolarized conditions. This result may also be due to the relatively positive resting potential of rat neonatal ventricular myocytes. Because many tachyarrhythmias are reentrant, we performed high frequency stimulation in an in vitro model to test if SkM1 could function in an arrhythmic fashion. Although SkM1 did not prevent re-entry, we found that the SkM1 coculture has a faster angular velocity of the induced re-entry than both the control and SCN5A cultures under normal and depolarized conditions. Increased excitability via augmentation or biophysical optimization of the Na+ channels can affect both the inducibility and the stability of reentrant waves. Previous theoretical studies34 have shown that the synergistic decrease of Na+ channel availability, slowed recovery from inactivation and cellular uncoupling, as presumably seen in the impaired myocardium, lead to an increased vulnerable window for reentry induction. Accordingly, our data with SkM1 that corrects 2 out of these 3 effects, revealed a tendency for lower inducibility of reentry in myocyte-HEK293-SkM1 vs. myocyte-HEK293-SCN5A samples (29% vs. 57% in normal K+), though statistical conclusions could not be reached due to insufficient sample number. Reentry stability as a function of Na+ channel availability also has been examined theoretically35, where higher Na+ channel availability lead to destabilization of functional reentrant waves by virtue of increased conduction velocity, and to the reentry breakup and/or termination. In our in vitro model, a small inexcitable obstacle was used rather than a pure functional reentry. Faster angular velocity for the myocyte-HEK293-SkM1 samples translates in possible reentrant wave destabilization, which may or may not result in termination, i.e. wave destabilization can be anti- or pro-arrhythmic. In summary, our proposed approach is likely to reduce the inducibility of reentrant waves, and to facilitate the de-stabilization of reentrant waves that may lead to their easier termination, though further experiments are needed to confirm this possible anti-arrhythmic action.
CONCLUSION
Here we demonstrate the feasibility of cell therapy use to augment cardiac conduction. Our results corroborate the differences in biophysical properties between SkM1 and SCN5A, when the two genes are expressed exogenously in selected cell lines. Consequently, in our two cell delivery model, expression of SkM1, but not SCN5A enhanced the dV/dtmax of the AP upstroke in myocytes which were electrically coupled with HEK293 cells transfected with either of these Na+ channel genes. This increase was particularly significant at a depolarized membrane potential. At a syncytial level, we demonstrated that introducing SkM1 into neonatal cardiac myocyte speeds conduction velocity in both normal and depolarized conditions. In contrast, expression of SCN5A had no significant effect. To mimic an arrhythmic environment, we also generated an in vitro re-entry model with a void center. The spiral propagation induced in SkM1 cultures showed faster angular velocity than control and SCN5A cultures.
Supplementary Material
Supplemental Digital Content 1: myocyte in normal K. wmv
Supplemental Digital Content 2: myocyte-HEKSkM in normal K. wmv
Supplemental Digital Content 3: myocyte in high K. wmv
Supplemental Digital Content 4: myocyte-HEKSkM in high K. wmv
Acknowledgments
Sources of support: Supported by grant HL094410 from the NIH/NHLBI and a grant from NYSTEM
Footnotes
Supplemental Digital Content: Phase movies of re-entrant propagation induced in a myocyte only sample and a myocyte-HEK293-SkM1 sample in normal K+ (5.4 mM) or high K+ (9 mM) Tyrode’s solution. Movies were generated in Metlab software from macroscopic optical mapping data. The color bar shows the fluorescence intensity of the Ca2+ signal. The black line shows the wave front of the propagation.
References
- 1.Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. II. The role of nonuniform recovery of excitability in the occurrence of unidirectional block, as studied with multiple microelectrodes. Circ Res. 1976 Aug;39(2):168–77. doi: 10.1161/01.res.39.2.168. [DOI] [PubMed] [Google Scholar]
- 2.Darpo B, Edvardsson N. Effect of almokalant, a selective potassium channel blocker, on the termination and inducibility of paroxysmal supraventricular tachycardias: a study in patients with Wolff-Parkinson-White syndrome and atrioventricular nodal reentrant tachycardia. Almokalant PSVT Study Group. J Cardiovasc Pharmacol. 1995 Aug;26(2):198–206. doi: 10.1097/00005344-199508000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Darpo B, Edvardsson N. Effects of almokalant, a class III antiarrhythmic agent, on supraventricular, reentrant tachycardias. Almokalant Paroxysmal Supraventricular Tachycardia Study Group. Cardiovasc Drugs Ther. 1997 Jul;11(3):499–508. doi: 10.1023/a:1007761825414. [DOI] [PubMed] [Google Scholar]
- 4.Savelieva I, Camm J. Anti-arrhythmic drug therapy for atrial fibrillation: current anti-arrhythmic drugs, investigational agents, and innovative approaches. Europace. 2008 Jun;10(6):647–65. doi: 10.1093/europace/eun130. [DOI] [PubMed] [Google Scholar]
- 5.Antzelevitch C, Fish JM. Therapy for the Brugada syndrome. Handb Exp Pharmacol. 2006;171:305–30. doi: 10.1007/3-540-29715-4_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weirich J, Wenzel W. Current classification of anti-arrhythmia agents. Z Kardiol. 2000;89(Suppl 3):62–7. [PubMed] [Google Scholar]
- 7.Lau DH, Clausen C, Sosunov EA, Shlapakova IN, Anyukhovsky EP, Danilo P, Jr, et al. Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation. 2009 Jan 6;119(1):19–27. doi: 10.1161/CIRCULATIONAHA.108.809301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Protas L, Dun W, Jia Z, Lu J, Bucchi A, Kumari S, et al. Expression of skeletal but not cardiac Na+ channel isoform preserves normal conduction in a depolarized cardiac syncytium. Cardiovasc Res. 2009 Feb 15;81(3):528–35. doi: 10.1093/cvr/cvn290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takanari H, Honjo H, Takemoto Y, Suzuki T, Kato S, Harada M, et al. Bepridil facilitates early termination of spiral-wave reentry in two-dimensional cardiac muscle through an increase of intercellular electrical coupling. J Pharmacol Sci. 2011;115(1):15–26. doi: 10.1254/jphs.10233fp. [DOI] [PubMed] [Google Scholar]
- 10.Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res. 2004 May 1;62(2):309–22. doi: 10.1016/j.cardiores.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 11.Spear JF, Michelson EL, Moore EN. Cellular electrophysiologic characteristics of chronically infarcted myocardium in dogs susceptible to sustained ventricular tachyarrhythmias. J Am Coll Cardiol. 1983 Apr;1(4):1099–110. doi: 10.1016/s0735-1097(83)80112-0. [DOI] [PubMed] [Google Scholar]
- 12.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997 Feb 18;95(4):988–96. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 13.Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol. 2003 Jun;13(3):284–90. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 14.Potapova I, Plotnikov A, Lu Z, Danilo P, Jr, Valiunas V, Qu J, et al. Human mesenchymal stem cells as a gene delivery system to create cardiac pacemakers. Circ Res. 2004 Apr 16;94(7):952–9. doi: 10.1161/01.RES.0000123827.60210.72. [DOI] [PubMed] [Google Scholar]
- 15.Valiunas V, Doronin S, Valiuniene L, Potapova I, Zuckerman J, Walcott B, et al. Human mesenchymal stem cells make cardiac connexins and form functional gap junctions. J Physiol. 2004 Mar 16;555(Pt 3):617–26. doi: 10.1113/jphysiol.2003.058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zygmunt AC. Intracellular calcium activates a chloride current in canine ventricular myocytes. Am J Physiol. 1994 Nov;267(5 Pt 2):H1984–95. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- 17.Mathias RT, Cohen IS, Oliva C. Limitations of the whole cell patch clamp technique in the control of intracellular concentrations. Biophys J. 1990 Sep;58(3):759–70. doi: 10.1016/S0006-3495(90)82418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bien H, Yin L, Entcheva E. Calcium instabilities in mammalian cardiomyocyte networks. Biophys J. 2006 Apr 1;90(7):2628–40. doi: 10.1529/biophysj.105.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung CY, Bien H, Entcheva E. The role of cardiac tissue alignment in modulating electrical function. J Cardiovasc Electrophysiol. 2007 Dec;18(12):1323–9. doi: 10.1111/j.1540-8167.2007.00959.x. [DOI] [PubMed] [Google Scholar]
- 20.Tolkacheva EG, Schaeffer DG, Gauthier DJ, Krassowska W. Condition for alternans and stability of the 1:1 response pattern in a “memory” model of paced cardiac dynamics. Phys Rev E Stat Nonlin Soft Matter Phys. 2003 Mar;67(3 Pt 1):031904. doi: 10.1103/PhysRevE.67.031904. [DOI] [PubMed] [Google Scholar]
- 21.Entcheva E, Bien H. Macroscopic optical mapping of excitation in cardiac cell networks with ultra-high spatiotemporal resolution. Prog Biophys Mol Biol. 2006 Oct;92(2):232–57. doi: 10.1016/j.pbiomolbio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Nuss HB, Tomaselli GF, Marban E. Cardiac sodium channels (hH1) are intrinsically more sensitive to block by lidocaine than are skeletal muscle (mu 1) channels. J Gen Physiol. 1995 Dec;106(6):1193–209. doi: 10.1085/jgp.106.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–94. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 24.Baba S, Dun W, Cabo C, Boyden PA. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation. 2005 Oct 18;112(16):2386–96. doi: 10.1161/CIRCULATIONAHA.105.534784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baba S, Dun W, Hirose M, Boyden PA. Sodium current function in adult and aged canine atrial cells. Am J Physiol Heart Circ Physiol. 2006 Aug;291(2):H756–61. doi: 10.1152/ajpheart.00063.2006. [DOI] [PubMed] [Google Scholar]
- 26.Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. J Gen Physiol. 1996 May;107(5):559–76. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952 Aug;117(4):500–44. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinson A. The Heart cell in culture. Boca Raton, Fla: CRC Press; 1987. [Google Scholar]
- 29.Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996 Dec;71(6):3098–109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deschenes I, Trottier E, Chahine M. Implication of the C-terminal region of the alpha-subunit of voltage-gated sodium channels in fast inactivation. J Membr Biol. 2001 Sep 15;183(2):103–14. doi: 10.1007/s00232-001-0058-5. [DOI] [PubMed] [Google Scholar]
- 31.Woods NB, Bottero V, Schmidt M, von Kalle C, Verma IM. Gene therapy: therapeutic gene causing lymphoma. Nature. 2006 Apr 27;440(7088):1123. doi: 10.1038/4401123a. [DOI] [PubMed] [Google Scholar]
- 32.Lo CW. Role of gap junctions in cardiac conduction and development: insights from the connexin knockout mice. Circ Res. 2000 Sep 1;87(5):346–8. doi: 10.1161/01.res.87.5.346. [DOI] [PubMed] [Google Scholar]
- 33.Schackow TE, Decker RS, Ten Eick RE. Electrophysiology of adult cat cardiac ventricular myocytes: changes during primary culture. Am J Physiol. 1995 Apr;268(4 Pt 1):C1002–17. doi: 10.1152/ajpcell.1995.268.4.C1002. [DOI] [PubMed] [Google Scholar]
- 34.Qu Z, Karagueuzian HS, Garfinkel A, Weiss JN. Effects of Na(+) channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol. 2004;286(4):H1310–H21. doi: 10.1152/ajpheart.00561.2003. [DOI] [PubMed] [Google Scholar]
- 35.Qu Z, Xie F, Garfinkel A, Weiss JN. Origins of spiral wave meander and breakup in a two-dimensional cardiac tissue model. Ann Biomed Eng. 2000;28(7):755–71. doi: 10.1114/1.1289474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 1: myocyte in normal K. wmv
Supplemental Digital Content 2: myocyte-HEKSkM in normal K. wmv
Supplemental Digital Content 3: myocyte in high K. wmv
Supplemental Digital Content 4: myocyte-HEKSkM in high K. wmv