

Abstract
The ability to induce antibody responses to pathogens while maintaining the quiescence of autoreactive cells is an important aspect of immune tolerance. During activation of Toll-like receptor-4 (TLR4), dendritic cells (DCs) and macrophages (MFs) repress autoantibody production through their secretion of IL-6 and soluble CD40L (sCD40L). These soluble mediators selectively repress B cells chronically exposed to antigen, but not naïve cells, suggesting a means to maintain tolerance during TLR4 stimulation, yet allow immunity. In this study, we identify TNFα as a third repressive factor, which together with IL-6 and CD40L, account for nearly all the repression conferred by DCs and MFs. Like IL-6 and sCD40L, TNFα did not alter B cell proliferation or survival. Rather, it reduced the number of antibody secreting cells. To address whether the soluble mediators secreted by DCs and MFs functioned in vivo, we generated mice lacking IL-6, CD40L and TNFα. Compared to wildtype mice, these mice showed prolonged anti-nuclear antibody responses following TLR4 stimulation. Further, adoptive transfer of autoreactive B cells into chimeric IL-6-/- × CD40L-/- × TNFα-/- mice showed that pre-plasma cells secreted autoantibodies independent of germinal center formation or extrafollicular foci. These data indicate that in the absence of genetic predisposition to autoimmunity, loss of endogenous IL-6, CD40L, and TNFα promotes autoantibody secretion during TLR4 stimulation.
Introduction
Toll-like receptor-4 (TLR4) is a germline encoded innate immune receptor that recognizes lipopolysaccharide (LPS) and promotes the secretion of inflammatory mediators and antibodies (1). TLR4 plays a role in the immune response to tissue injury (7, 8, 12-16) and multiple self-proteins released from necrotic cells or damaged tissues have been found to activate and/or bind TLR4 (reviewed in 2, 3). These include heat shock protein gp96 (4), HMGB1 (reviewed 5, 6), dysfunctional mitochondria (7), and endogenous lipoproteins (8-11). The findings that host-derived endogenous ligands, or DAMPs, bind TLR4 suggests the possibility that autoimmunity could arise from an inability to regulate TLR4-expressing autoreactive B cells or myeloid cells. In support of this, gene deletion and overexpression studies identify TLR2, TLR4, and TLR7 as contributing to autoantibody titers, renal disease, and the heightened cytokine production found in lupus-like autoimmune disease in mice (4, 17). TLR4 is also required for autoimmunity and organ injury in experimental lupus induced by pristine (18). Further, repeat injections of low-dose LPS into lupus-prone mice accelerate the development of disease, increases autoantibody levels, and worsens renal impairment (19-21). This indicates that innate immune responses involving TLR4 contribute to pathogenic autoantibody production in lupus-prone mice.
B lymphocytes express most TLRs and respond to their ligands by proliferating, expressing co-stimulatory molecules, and secreting immunoglobulin (Ig). We identified that autoreactive B cells are regulated during TLR4 activation by myeloid dendritic cells (DCs) and macrophages (MF) (22, 23). Regulation of autoantibody secretion is conferred in part by IL-6 and soluble CD40L (sCD40L) secreted from TLR4-activated MFs and myeloid DCs. A unique feature of DC/MF-mediated tolerance is that B cells chronically exposed to self-antigen are affected by IL-6 and sCD40L, while antigenically naïve B cells are unaffected. The data show that chronic B cell antigen receptor (BCR) signaling, as a consequence of self-antigen exposure, integrates with acute IL-6 receptor or CD40 signals to repress TLR4-induced Ig secretion in B autoreactive, but not in antigenically naïve B cells (23). DC/MF defects are apparent in lupus-prone MRL/lpr mice where reduced secretion of IL-6 and sCD40L occurs coincident with a diminished ability to repress TLR4-induced Ig secretion (22, 23). Collectively the data indicate that DCs and MFs are key regulatory cells that maintain B cell tolerance during TLR4-induced innate immune responses, and that defects in this tolerance mechanism might contribute to the early autoantibody production associated with disease.
In this study, we describe TNFα as a third factor produced by LPS-activated DCs and MFs that represses autoantibody production, and we assess whether DCs and MFs regulate autoreactive B cells in vivo. We find that compared to DCs and MFs from C57BL/6 (B6) mice, those from lupus-prone MRL/lpr mice produce less TNFα suggesting that diminished TNFα may contribute to a break in tolerance. In vivo we show that IL-6, CD40L, and TNFα regulate B cell tolerance during innate immune responses as mice deficient for these three factors (IL-6-/- × CD40L-/- × TNFα-/-; 3XKO) exhibited prolonged anti-nuclear and anti-nucleosome antibody responses. Adoptive transfer studies show that Sm-specific pre-plasma cells that were adoptively transferred into bone marrow chimeras lacking IL-6, CD40L, and TNFα secreted anti-Sm during an ongoing TLR4 response. The findings that loss of IL-6, sCD40L and TNFα leads to a breach in tolerance in the absence of genetic predisposition establishes that the regulation of autoreactive B cells in vivo relies on DCs and MFs during TLR4-induced innate immune responses.
Materials and Methods
Mice
Ars/A1, 125Tg, 2-12H, and 2-12H/Vκ8 mice have been previously described (24-28). C57BL/6J, MRL/MpJ-Faslpr/J (MRL/lpr), IL-6-/-, CD40L-/- and TNFα-/- mice were purchased from the Jackson Laboratory. IL-6-/-, CD40L-/- and TNFα-/- mice were crossed to generate the IL-6-/- × CD40L-/- × TNFα-/- (3XKO) mice. B6.SJL-Ly5.2/Cr mice (C57BL/6 mice expressing the Ly5.1 antigen from SJL/J mice) were purchased from the National Cancer Institute. The Institutional Animal Care and Use Committee at the University of North Carolina approved all animal experiments.
Bone Marrow Transplant
Six- to eight-week-old B6.SJL-Ly5.2/Cr mice were lethally irradiated (900 rads). Twenty-four hours later 5-8×106 bone marrow cells from C57BL/6 or 3XKO donor mice were transferred. These chimeras are referred to as B6 chimeras and 3XKO chimeras, respectively. After 8 weeks, the expression of Ly5.1 or Ly5.2 was assessed as a means to distinguish donor and recipient cells following reconstitution.
Bone marrow-derived DC (BMDC) and MF (BMMF) Cultures
BMDCs (95% pure) and BMMFs (98% pure) were generated as previously described (22). Conditioned medium (CM) was made from 1×104 BMDCs and BMMFs (0.2 ml) cultured 4 days with or without Sigma LPS (30 μg/ml) or InvivoGen LPS (10 ng/ml).
Reagents and Antibodies
LPS was purchased from Sigma Aldrich (Escherichia coli 055:B5) and Invitrogen (Escherichia coli 0111:B4). R848 was purchased from Enzo Life Sciences, CpG-B (1826) and non-CpG (2138) oligodeoxynucleotides (ODN) from Coley Pharmaceutical Groups. Recombinant IL-6 and antibodies to IL-6, CD40L, B220, Thy1.2, CD11b, CD11c, CD21, CD23, CD138, CD19 CD90.2, streptavidin PE-Cy5.5 and hamster IgG3 were purchased from BD Biosciences. TEPC 183, and rabbit IgG were purchased from Sigma-Aldrich, mouse GM-CSF, IL-4, and M-CSF from PeproTech, CFSE, and streptavidin Alexa 488 from Invitrogen, and recombinant TNFα and recombinant soluble CD40L from R&D Systems. Monoclonal antibodies 54.1 (3-83 idiotype), 187.1 (anti-κ), HB100 (anti-IgMa), B7.6 (anti-IgM), RS3.1 (IgMa), and PL2-6 (anti-nucleosome) were purified from hybridoma culture supernatants. Rabbit polyclonal anti-TNFα was obtained from Vic Johnson (CDC/NIOSH/HELD, West Virginia) and affinity purified using Protein A.
B Cell Purification and Cell Sorting
Splenic B cells were negatively selected using the StemSep B cell enrichment kit (StemCell Technologies). B cell purity ranged from 85-97%. B cell populations from 2-12H mice (92% IgMa (27) were sorted on a MoFlo high-speed sorter (DakoCytomation) as previously described (29). Briefly, CD19+ cells were gated for CD21, CD23, and CD138. Cells were divided into populations by staining patterns: follicular (FO; CD19hi CD21med, CD23hi, CD138lo), marginal zone (MZ; CD19hi CD21hi, CD23lo/med, CD138lo) and pre-plasma cells (prePC; CD19hi, CD138med). Populations were >90% pure on re-analysis.
B Cell Culture
Purified B cells (1×105 per well in a 96-well plate) were cultured with 30 μg/ml LPS for 4 days. Recombinant IL-6, rsCD40L, rTNFα, CHO-TNFα, BMDC or BMMF conditioned media (CM) (25% of final volume) were added to B cell cultures on day 0. The IL-6 in CM was neutralized with either anti-IL-6 antibody or a control rat IgG1 antibody (54.1). Soluble CD40L in CM was neutralized with either anti-CD40L or control hamster IgG3 antibody. TNFα in CM was neutralized with either anti-TNFα or control rabbit IgG.
ELISA
IgMa/κ (encoded by 2-12H/Vk8, Ars/A1 and 125Tg) was captured with anti-κ (clone 187.1). Total IgM (B6 mice) was captured with anti-IgM (clone 33-60), nucleosome-specific Ig with histones (4 μg/well; Immunovision) and dsDNA (1 μg/well; Sigma), and TNFα with anti-TNFα (clone TN3-19, eBioscience). Captured antibodies were detected with biotinylated anti-IgMa (clone HB100), anti-IgM (clone B7.6), anti-mouse Ig (IgG, IgM, and IgA), or polyclonal anti-TNFα (eBioscience), respectively. TEPC 183 (IgMa/κ), PL2-6 (anti-nucleosome) and rTNFα served as standards. All assays were visualized using streptavidin-alkaline phosphate (Southern Biotech) and 4-nitrophenyl phosphate disodium salt hexahydrate (Sigma). Data were plotted as percent of control calculated relative to cultures of LPS-stimulated B cells.
Antinuclear Antibody (ANA) Assay
Hep-2 substrate slides (Antibodies Inc.) were used to detect serum (1:50 dilution) autoantibodies. Nuclear and cytoplasmic staining was identified using anti-mouse IgG-Alexa 488 or IgM-Alexa 647 (Invitrogen). Based on the guidelines for fluorescent antibody reagents established by the Center for Disease Control, the fluorescence of the IgM and IgG staining was quantitated using a scale of 0–4, and the values from multiple mice compiled.
Immunohistochemistry
Chimeric mice were pretreated with LPS (25 μg) on days -10 and -3. On day 0, 15 × 106 negatively selected B cells (2-12H) were tail vein injected. On days 4 and 7, spleens were flash frozen in Optimal Cutting Temperature Compound (OCT; TissueTek), sectioned to 8μm, and fixed in acetone/methanol (1:2) for 5 minutes. Rehydrated sections were blocked for 1 hour in Superblock (Pierce) diluted 1:2 in PBS containing 10 μg/ml 2.4G2 (anti-FcγRII/III), stained for 1 hour, and mounted with DAPI Prolong Gold (Invitrogen) or FluroSave (Calbiochem). Images were viewed using digital deconvolution microscopy and processed using Slidebook software (Intelligent Imaging Innovations). The numbers of IgMa+ and CD138+ cells in each field were quantified. These cells were present throughout the red pulp and areas of the follicles. The percent of transferred cells that became ASCs was determined by calculating the percent of IgMa+ cells that were also CD138+. To assess splenic architecture (Figure 5A) of 3XKO, 3XKO chimera, and B6 mice, we counted at least 8 (and as many as 44) follicles from each group and enumerated the follicles that contained distinct PALs. The data are expressed as percent of total follicles counted.
Figure 5. Innate immune responses induce the activation of pre-plasma cells in mice lacking IL-6, CD40L and TNFα (3XKO).
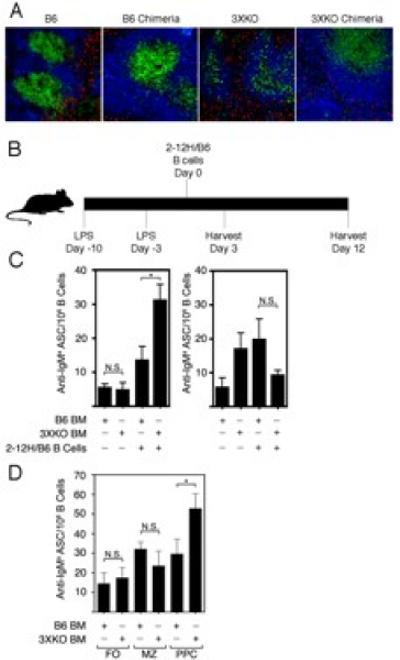
(A) Spleen sections of non-chimeric B6, chimeric B6, non-chimeric 3XKO and chimeric 3XKO stained with B220 (blue), Thy1.2 (green), CD11b and CD11c (both red). Images were captured at 10X magnification. (B) Chimeric mice (B6 chimeras and 3XKO chimeras) were injected with LPS (1.25 mg/kg at day -10 and -3). On day 0, 1.5×106 purified Sm-specific B cells (2-12H/B6) were adoptively transferred. (C) IgMa ASCs were quantitated by ELISPOT three (left) and twelve (right) days post B cell (2-12H) transfer. (D) Sorted FO, MZ, and prePC subsets (2-12H) were transferred and IgMa ASCs quantitated by ELISPOT 3 days post transfer. Data are representative of at least three individual experiments totaling 3-6 mice for each experimental condition. Error bars depict SEM. (** p≤0.01, * p≤0.05, NS; not significant, p>0.05).
In vivo stimulation
To induce an innate immune response, an injection of 1.25 mg/kg, 0.9 mg/kg, or 0.625 mg/kg (25μg, 18 μg, 12.5 μg/injection/mouse) of LPS were injected i.p. on days 0, 7, and 14 (Figure 4), or on days -10 and -3 (Figure 5).
Figure 4. Innate immune responses induce autoantibodies in mice lacking IL-6, CD40L, and TNFα (3XKO).
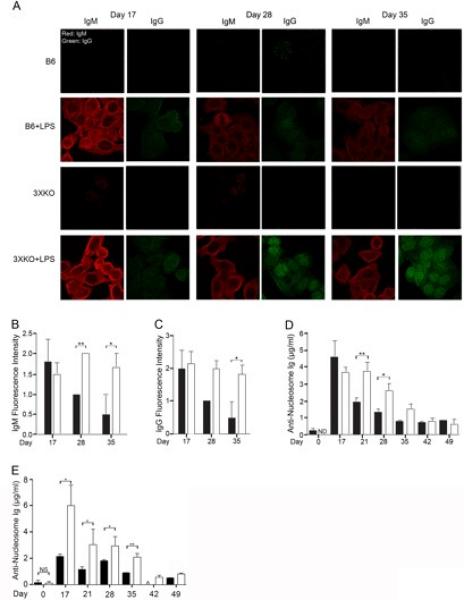
(A) Representative IgM (red) and IgG (green) Hep-2 staining from B6 (top 2 rows) and 3XKO (bottom 2 rows), with (rows 2 and 4) and without (rows 1 and 3) LPS (1.25mg/kg days 0, 7, and 14). Images were captured at 60X magnification. (B) Compilation of fluorescence intensity of IgM autoantibodies from LPS-treated B6 (black) and 3XKO (white) mice. n=10 (C) Compilation of fluorescence intensity of IgG autoantibodies from LPS-treated B6 (black) and 3XKO (white) mice. n=10 (D) Total anti-nucleosome (IgG, IgM, IgA) levels from LPS-treated (1.25 mg/kg) B6 (black) and 3XKO (white) mice were quantitated on the indicated days. n= 7. (E) Total anti-nucleosome (IgG, IgM, IgA) levels from LPS-treated (0.9 mg/kg) B6 (black) and 3XKO (white) mice were quantitated on the indicated days. n= 5. Error bars depict SEM. **p≤0.01, * p≤0.05, NS; not significant, p>0.05).
ELISPOT
Following adoptive transfer, 5×106 splenocytes were plated onto anti-kappa (187.1) or anti-IgMa (RS3.1) coated plates. Bound antibody was detected using biotin-labeled anti-IgM (B7.6) and streptavidin-HRP (BD Biosciences), and visualized using 3-amino-9-ethylcarbazole (Sigma-Aldrich) at 24 hours. Spots were enumerated using the Immunospot Analyzer and Cellular Technologies software. To assess the effect of TNFα on ASC formation, 4 × 103 cultured B cells were serially diluted and cultured on anti-kappa coated ELISPOT plates. Eight hours later, plates were washed and bound antibody was visualized as above.
CFSE-based Proliferation Assay
B cell proliferation was assessed by the dilution of 5-carboxyfluorescein diacetate succinimidyl ester (CFSE) loaded cells as previously described (23).
Statistical analysis
Antibody or cytokine secretion between treated and untreated cultures was analyzed using a one-sampled t test. Comparison of antibody production between experimental groups was analyzed using the Wilcoxon rank-sum test. Statistical analysis was preformed with GraphPad Prism (v4.0c, 2005).
Results
DCs and MFs regulate Ig secretion from TLR4-activated autoreactive B cells
Self-antigens and pathogen-associated antigens bind TLR4 thus requiring that tolerance mechanisms regulate autoreactive B cells to protect from autoimmunity (6, 30). As shown in Figure 1A, and previously reported (22, 23), LPS stimulated Sm-specific B cells from 2-12H/Vκ8 mice are repressed by IL-6 and sCD40L, the secreted products of LPS-activated dendritic cells (DCs) and macrophages (MFs). In this model, conditioned medium (CM) from bone marrow-derived DCs (BMDCs) and MFs (BMMFs), recombinant IL-6 (rIL-6) and recombinant soluble CD40L (rsCD40L) repressed 50% to 75% of Ig secretion (Figure 1A). Similarly, the secreted products from DCs and MFs repressed 40 to 60% of Ig secretion from 2-12H B cells (Figure 1B), but not from B6 B cells, which are predominately antigenically naive (Figure 1C). These findings show that during TLR4 activation, DCs and MFs secrete soluble factors that repress TLR4-induced Ig secretion from B cells chronically exposed to self-antigen, but not antigenically naive cells. In this in vitro culture model, one possible caveat was that the dose of LPS used for maximal B cell Ig secretion (30 μg/ml) was in excess of that required to activate DCs and MFs (6, 31, 32) and could induce non-physiological levels of IL-6 and sCD40L. To address whether the repressive effect was the result of the dose of LPS, or a unique property of the LPS, we used a second source of LPS and a lower dose (10 ng/ml). This dose induces cytokine secretion by MFs and approximates the endotoxin concentration when cells are treated with live E. coli (one bacterium per MF; (31). We found that MF CM prepared using InvivoGen LPS (10 ng/ml) was as efficient at repressing Ig secretion as the 30 μg/ml dose of Sigma LPS; neither dose repressed TLR4-induced Ig from B6 B cells (Figure 1D). This indicates that repression of LPS-induced Ig secretion by DCs and MFs is not dependent on the source or dose of the LPS.
Figure 1. TLR4, but not TLR7 or TLR9 stimulated DCs and MFs regulate Sm-specific B cells.
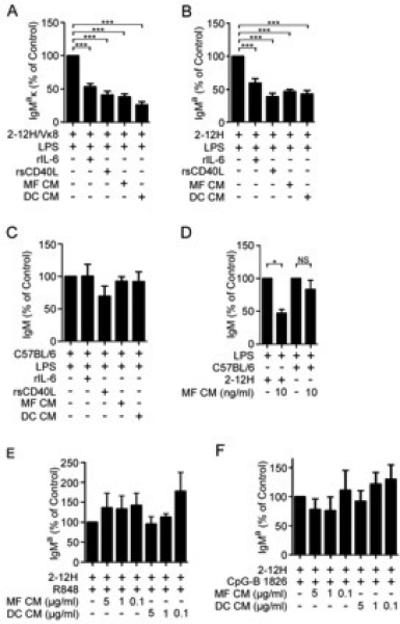
(A) Purified 2-12H/Vκ8 (1×105), (B) 2-12H, or (C) B6 B cells were stimulated with Sigma LPS (30 μg/ml) and cultured with conditioned medium (CM) from LPS-activated BMMF or BMDC, recombinant IL-6, or sCD40L. (D) 2-12H or B6 B cells stimulated with 1 μg/ml InvivoGen LPS and cocultured with MF CM prepared from low dose LPS stimulation (InvivoGen; 10 ng/ml). (E) 2-12H B cells (1×105) stimulated with R848 (1 μg/ml), or (F) CpG-B (1 μg/ml) cocultured with BMDC or BMMF CM prepared using varying doses of R848 or CpG-B. Secreted antibody was measured by ELISA from day 4 culture supernatant. LPS-stimulated purified B cells (100%) secreted an average of 7.7 μg/ml IgMa/κ (2-12H/Vκ8), 38.5 μg/ml IgMa (2-12H), or 23.3 μg/ml IgM (B6). R848-stimulated cells (100%) secreted 8.5 μg/ml (2-12H), and CpG-stimulated cells (100%) secreted 5.5 μg/ml (2-12H). There was no detectable Ig from cells stimulated with the non-CpG ODN. Data represent at least three independent experiments. Error bars depict SEM. (*** p≤0.001, ** p≤0.01, * p≤0.05, NS; not significant, p>0.05).
The mechanisms that maintain autoreactive B cells in an unresponsive state during TLR activation have not been well studied. In ssDNA-specific B cells, activation of TLR9 is regulated by the exclusion of TLR and BCR-containing vesicles from the late endosome (33). In HEL-specific B cells, chronic binding of high affinity antigen is sufficient to repress TLR4 and TLR9 (34, 35). Mechanisms that regulate autoantibody secretion through TLR7 have not been described. Although TLR7 and TLR9 are localized within late endosomes while TLR4 is surface expressed, they are similar in that they are coupled to MyD88. To address whether IL-6 and sCD40L repressed autoreactive B cells stimulated through TLR7 and TLR9, we prepared CM from MFs and DCs stimulated with R848, CpG ODN, or control CpG, then assessed whether the day 4 CM repressed TLR7- or TLR9-stimulated B cells. Supernatants from R848 stimulated MFs or DCs failed to repress R848 stimulated 2-12H B cells (Figure 1E). Similarly, CpG stimulation of MFs or DCs failed to induce a soluble factor that repressed CpG-treated 2-12H B cells (Figure 1F). This indicates that Ig secretion from autoreactive Sm-specific B cells stimulated through TLR7 and TLR9 is not regulated by the same mechanism as TLR4.
DCs and MFs repress autoantibody secretion through TNFα
Regulation of TLR4-induced Ig secretion occurs by the secretion of IL-6 from LPS-activated DCs, and IL-6 and sCD40L from MFs. To address whether IL-6 and sCD40L were the only repressive factors involved, we assessed the ability of DC CM from IL-6 deficient mice to repress LPS-induced Ig secretion, since the predominant repressive factor secreted by DCs is IL-6. However, we found that CM from IL-6-deficient DCs still repressed 45% of Ig secretion, compared to 60% repression by B6 DC CM (Figure 2A). This suggests that DCs secrete another factor(s) that represses autoantibody production during innate immune responses.
Figure 2. TNFα represses LPS-induced Ig secretion by Sm-specific B cells.
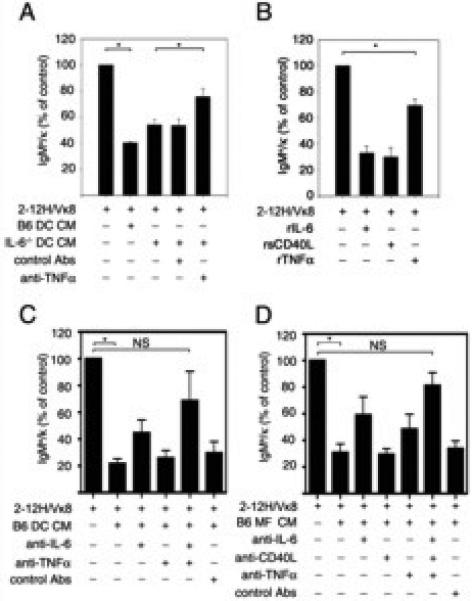
Purified Sm-specific (2-12H/Vκ8) B cells (1×105) were stimulated with LPS (30 μg/ml) and cultured with (A) conditioned medium (CM) from LPS-activated B6 or IL-6-/- BMDCs and anti-TNFα antibody (324 μg), or an isotype matched control antibody, (B) recombinant IL-6 (rIL-6; 20 ng/ml), recombinant sCD40L (rsCD40L; 100 ng/ml) or recombinant TNFα (rTNFα; 50 ng/ml), (C) CM from LPS-activated B6 DCs and anti-IL-6 (50 μg/ml), anti-TNFα (324 μg), or isotype matched control antibody (D) CM from LPS-activated B6 MF and anti-IL-6 (50 μg/ml), anti-CD40L (10 μg/ml), anti-TNFα (324 μg), or isotype matched control antibody. Secreted IgMa/κ was measured by ELISA from day 4 culture supernatant. LPS-stimulated purified B cells (100%) secreted an average of 3-15 μg/ml IgMa/κ. Data represent at least three independent experiments. Error bars depict SEM. (* p≤0.05, NS; not significant, p>0.05).
To identify other factor(s) that regulated autoantibody secretion, we neutralized DC CM from IL-6-/- mice (IL-6-/- DC CM) with a panel of antibodies to cytokines and chemokines. We found that neutralizing antibodies to TNFα restored Ig secretion from LPS-stimulated 2-12H/Vκ8 B cells to 78% of control, while addition of an unrelated antibody did not have a significant effect (Figure 2A). This suggests that in addition to IL-6, DCs secrete TNFα to regulate TLR4-induced Ig secretion.
To corroborate the finding that TNFα regulates autoreactive B cells, we assessed whether recombinant TNFα (rTNFα) repressed LPS-induced Ig secretion. We found that 50 ng/ml of rTNFα repressed 30% of Ig secretion by 2-12H/Vκ8 B cells (Figure 2B). Although rTNFα was repressive, it was considerably less effective compared to rIL-6 or rsCD40L (Figure 2B; (22, 23). To ensure that the low level of repression was not due to limited bioactivity of the rTNFα, we expressed murine TNFα trimers in CHO cells. Addition of increasing amounts of highly bioactive TNFα to the B cell cultures did not further diminish the level of Ig secretion (data not shown). These data indicate that during LPS stimulation, TNFα modestly regulates Ig secretion by autoreactive B cells.
To determine whether IL-6, sCD40L and TNFα were the only repressive factors that regulated autoreactive B cells, we neutralized CM from B6 DCs (Figure 2C) and MFs (Figure 2D) with antibodies to IL-6, CD40L and TNFα. Neutralization of IL-6 or TNFα from DC CM had a partial effect while neutralizing both factors restored Ig secretion to levels that were not significantly different from LPS-stimulated B cells (74%; Figure 2C). Similarly, neutralizing MF CM with antibodies to IL-6, CD40L, or TNFα partially restored Ig secretion while neutralizing with antibodies to all three factors restored Ig secretion to 81% of control, a level not statistically different from LPS-stimulated B cells (Figure 2D). Collectively, the data show that TNFα represses autoantibody secretion; however TNFα is not as potent as IL-6 and sCD40L in regulating Ig secretion.
TNFα represses autoreactive B cells, but not antigenically naïve cells
Our previous data showed that IL-6 and sCD40L repressed B cells that were chronically exposed to self-antigen including Sm-, Ars- and HEL-specific B cells (HEL-Ig × sHEL). However, it did not significantly diminish the amount of Ig secreted by B cells from B6 or HEL-Ig mice (22, 23). We compared the ability of rTNFα to selectively repress LPS-induced Ig secretion from B6 B cells compared to several transgenic lines specific for self-antigens. B cells from Ars/A1 immunoglobulin transgenic mice bind Ars and ssDNA and are repressed by IL-6 and sCD40L (22-24). TNFα repressed 26% of Ig secretion by Ars-specific B cells, 17% of insulin-specific (125Tg) Ig secretion, and 30% of Sm-specific (2-12H/Vκ8) Ig secretion (Figure 3A right panel). However, TNFα did not repress Ig secretion by antigenically naïve B cells from B6 mice (Figure 3A left panel). These data show that TNFα modestly represses Ig secretion from B cells of multiple autoreactive specificities suggesting that chronic exposure to self-antigen integrated with co-ligation of TNF receptor(s) stimulation attenuates TLR-induced Ig secretion.
Figure 3. TNFα represses autoantibody secretion by reducing the number of ASCs.
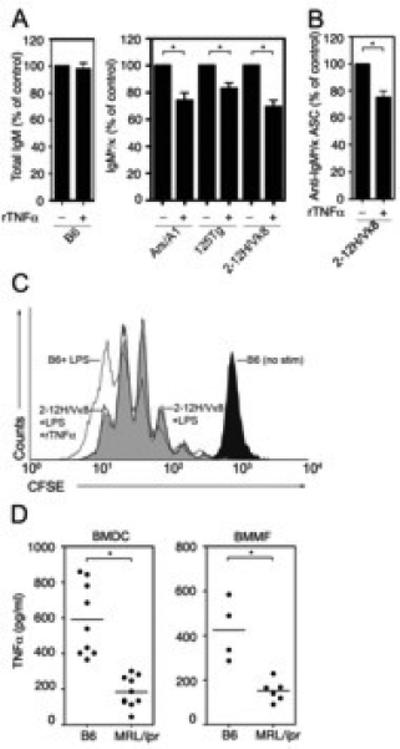
(A) Purified B cells from the indicated mice were stimulated with LPS (30 μg/ml) and cultured with rTNFα (50 ng/ml). Total IgM (left) or IgMa/κ (right) was measured by ELISA from day four culture supernatant. LPS-stimulated B cells (100%) secreted 23-51μg/ml total IgM or 1-6 μg/ml IgMa/κ. ((B) Sm-specific (2-12H/Vκ8) B cells were stimulated with LPS (30 μg/ml) in the presence or absence of rTNFα (50 ng/ml) for 3 days. The frequency of ASCs was determined by ELISPOT. (C) CFSE loaded B cells from 2-12H/Vκ8 mice were incubated in the presence (black line) or absence (gray shade) of rTNFα (50 ng/ml). The proliferation indices were 5.6 and 5.5, respectively. The CFSE dilution of LPS stimulated is shown as a reference (D) BMDCs (left) or BMMF (right) from B6 or MRL/lpr were stimulated with LPS (30 μg/ml). TNFα was quantitated by ELISA from day four culture supernatants. Data represent at least three independent experiments. Error bars depict SEM. (* p≤0.05).
TNFα represses Ig secretion by diminishing the number of antibody secreting cells
We previously found that sCD40L and IL-6 did not affect the proliferation of autoreactive B cells; rather, they regulated autoantibody production by diminishing the number of B cells that became antibody secreting cells (ASCs; (23). To define whether TNFα functioned by a similar mechanism, we used ELISPOT to compare ASC formation in the presence or absence of TNFα. We found that TNFα treatment of LPS-stimulated B cells (2-12H/Vκ8) diminished the number of ASCs by 25% (Figure 3B). This modest decrease is consistent with the effect of rTNFα on Ig secretion (Figure 2B and 3A right panel).
TNFα might also diminish the rate of B cell proliferation as a means of affecting Ig secretion. To assess this, we compared the rate of proliferation of B cells from 2-12H/Vκ8 mice that were untreated or treated with TNFα. The dilution of CFSE was similar between untreated B cells from B6 and 2-12H/Vκ8 mice (proliferative index (PI)= 7.6 vs 5.5). Addition of rTNFα to B cells from 2-12H/Vκ8 mice did not alter the rate of proliferation, as the profiles of CSFE dilution were indistinguishable (Figure 3C; PI = 5.5 -TNFα; 5.6 +TNFα). This indicates that the ability of TNFα to diminish Ig secretion does not reflect diminished proliferation of autoreactive cells.
DCs and MFs from lupus-prone mice are defective in secreting TNFα
Defects in the ability of DCs and/or MFs to secrete repressive factors might underlie the break in tolerance associated with autoimmunity. Our studies of IL-6 and sCD40L showed that DCs and MFs from lupus-prone mice are defective in repressing Ig secretion, coincident with the reduced secretion of these repressive factors (22, 23, 36). To determine whether DCs and MFs from lupus-prone mice were defective in secreting TNFα, we quantitated TNFα by ELISA. LPS-stimulated BMDCs from lupus-prone MRL/lpr mice secreted significantly less TNFα than LPS-stimulated B6 BMDCs (Figure 3D left). Macrophages from MRL/lpr mice also secreted less TNFα compared to B6 (Figure 3D right). Taken together, the data show that DCs and MFs from MRL/lpr mice are defective in secreting IL-6, sCD40L and TNFα, soluble mediators that regulate autoreactive B cells during TLR4 stimulation (23, 36).
In the absence of IL-6, CD40L and TNFα, LPS stimulation induces ASC formation
In non-autoimmune mice, innate stimulation induces autoantibody production, however these responses begin to resolve within four weeks (19-21). To define whether IL-6, sCD40L and TNFα were important in maintaining B cell tolerance in vivo, we generated mice deficient in the repressive factors (IL-6-/- × CD40L-/- × TNFα-/-; 3XKO). 3XKO mice aged for one year in a pathogen free environment did not spontaneously develop autoimmunity as evidenced by the lack of inflammation/mononuclear cell inflammation in the kidneys, and the absence of serum anti-nucleosome antibody (data not shown). To assess the role of the repressive factors during TLR4 stimulation in vivo, we LPS (1.25 mg/kg) injected 3XKO and B6 mice and compared the course of autoantibody production. After three intraperitoneal injections (days 0, 7, and 14), we monitored the formation of serum autoantibodies by Hep-2 staining and anti-nucleosome antibody by ELISA. We hypothesized that if IL-6, sCD40L, and TNFα were necessary to repress autoreactive B cells in vivo, then the duration of the LPS-induced autoantibody response would be prolonged, although both strains of mice were expected to eventually resolve the LPS challenge since neither was genetically predisposed to disease. Three days after the last LPS injection (day 17), we found that the IgM and IgG staining of Hep-2 cells from both B6 and 3XKO mice was apparent indicating that the absence of the IL-6, CD40L and TNFα did not ablate autoantibody secretion (Figure 4A row 2 compared to 4). In B6 mice, the robust IgM response waned over time while the response in 3XKO was prolonged (Figure 4A row 2 compared to 4; IgM stain; scoring in Figure 4B). The IgG response became evident in both B6 and 3XKO on day 17 (Figure 4C). In 3XKO mice the IgG response was sustained over time and exhibited a nuclear staining pattern around day 28 (Figure 4A row 2 compared to 4; IgG stain). In B6 mice, the response was diminished by day 28 and a nuclear staining pattern was never evident. Continued monitoring of 3XKO mice showed that the IgG response remained high at day 42, but was attenuated by day 60 (data not shown). In the absence of LPS, the 3XKO mice did not show elevated serum antinuclear antibody levels (Figure 4A rows 1 and 3). Mice lacking IL-6, CD40L, and TNFα also exhibited a prolonged serum anti-nucleosome response (Figure 4D). B6 and 3XKO mice also mounted comparable anti-nucleosome responses at day 17 (3 days after LPS priming). However, at day 21 (1 week after LPS priming), the B6 mice reduced the anti-nucleosome response by >50% while the antibody levels in the 3XKO mice remained unchanged. Consistent with the presence of IgM and IgG in the Hep2 staining, we found that the anti-nucleosome antibody response contained both IgM and IgG, although IgM predominated (data not shown). This suggests that the nuclear IgG response in the Hep-2 staining might be due to the low levels of anti-nucleosome IgG, while the IgM staining (day 17), is a specificity other than anti-nucleosome, such as anti-mitochondrial or anti-smooth muscle antibody, which stain the cytoplasm. The diminished ability of 3XKO mice to resolve the autoantibody response remained evident for three weeks. At week five (day 35) the anti-nucleosome response in 3XKO mice was reduced to levels similar to those in B6 mice. The eventual restoration of tolerance is consistent with the fact that the 3XKO mice lack genetic modifiers that predispose to autoimmune disease. To ensure this was not due to the dose of LPS, we injected two other cohorts of mice with lower doses of LPS (0.9 mg/kg and 0.625 mg/kg). We found that 0.9 mg/kg of LPS induced a robust anti-nucleosome antibody response in the 3XKO mice that failed to resolve until day 35 (Figure 4E); however, this dose was suboptimal for the B6 mice. This suggests that the threshold for LPS-induced Ig secretion is lower in the absence of the repressive cytokines. The low dose of LPS (0.625 mg/kg) failed to induce an anti-nucleosome antibody response above the prebleed in the B6 mice (data not shown). Taken together, the data indicate that loss of IL-6, CD40L and TNFα does not result in spontaneous autoimmunity, however it leads to prolonged autoantibody production in response to LPS. This is consistent with an in vivo role for IL-6, sCD40L, and/or TNFα in resolving autoantibody secretion following TLR4 stimulation.
Sm-specific B cells become activated when transferred into chimeric 3XKO mice
Previous studies showed that mice lacking CD40L and TNFα have immune defects including the inability to form germinal centers, abnormal class switch recombination, and poorly defined splenic B and T cell regions (37-40). These deficiencies could alter the course of antibody production in the 3XKO mice due to splenic abnormalities not related to tolerance. To address this issue, we generated chimeras by transplanting bone marrow from B6 or 3XKO mice (Ly5.1) into lethally irradiated B6.SJL-Ly5.2/Cr mice to generate B6 and 3XKO chimeras. As shown in Figure 5A, the organization of the splenic B cell follicles and T cell regions of the 3XKO chimera was comparable to the chimeric B6 mice and B6 control mice while the non-chimeric 3XKO mice mostly showed poorly defined B and T regions. We found that about 25% of the architecture within the nonchimeric 3XKO spleen showed organized B and T regions, while the red pulp did not display any apparent abnormalities (see M&M for quantitation method). This might provide an explanation for the IgG ANA response in 3XKO mice despite lacking TNFα (Figure 4A row 4).
To determine whether autoreactive B cells from mice lacking the repressive factors would break tolerance during TLR4 stimulation, we transferred Sm-specific B cells (2-12H) into chimeric B6 and 3XKO mice. Chimeric mice were given two LPS injections (1.25 mg/kg) seven days apart, followed by intravenous injection of purified B cells from 2-12H/B6 mice (IgMa allotype) (Figure 5B). Three days post-transfer we enumerated Sm-specific (anti-IgMa) ASC (Figure 5C) by ELISPOT. The data show that transferred 2-12H B cells were modestly activated in the LPS-primed B6 mice compared to mice that did not have cells transferred indicating that sufficient LPS remained in the system for B cell activation (Figure 5C). Compared to B6 chimeras where IL-6, CD40L and TNFα remain intact, the transfer of 2-12H B cells into 3XKO chimeric mice showed a 2.5-fold increase in the formation of ASCs by day 3 post-transfer (Figure 5C left panel). Twelve days after B cell transfer into 3XKO, donor IgMa+ ASCs were no longer evident (Figure 5C right panel). Similar results were found using anti-Sm ELISPOTS. Additionally, there were no anti-Sm (or IgMa+) ASCs present in the bone marrow of the chimeric mice at any time points (data not shown), indicating that plasma cells had not migrated to the bone marrow. Mice that were not injected with LPS did not show any antibody responses indicating that the response seen in treated mice was the result of LPS-mediated B cell activation. These data indicate that the absence of IL-6, sCD40L, and TNFα in hematopoietic cells, allows autoreactive B cells to secrete Ig during a TLR4 response demonstrating that these factors play a role in B cell tolerance in vivo.
In the absence of IL-6, CD40L and TNFα, pre-plasma cells become activated
The marginal zone (MZ) and pre-plasma cell (prePC) subsets have been described as early responders during innate responses (41-43). To define the splenic B cell subsets responsible for the day 3 response, we adoptively transferred sorted follicular (FO; CD19hi CD21med, CD23hi, CD138lo), marginal zone (MZ; CD19hi CD21hi, CD23lo/med, CD138lo) and pre-plasma cells (prePC; CD19hi, CD138med) from 2-12H mice into LPS-primed B6 and 3XKO chimeric mice. Three days post-transfer, the numbers of IgMa-specific ASCs were enumerated by ELISPOT. As shown in Figure 5D, the numbers of ASCs that formed from transferred MZ and FO were equal between B6 and 3XKO chimeras. However, the numbers of transferred pre-PCs that became ASCs in the 3XKO chimeras was increased approximately 2-fold compared to transfer into B6 chimeras. This indicates that the pre-PCs from 2-12H mice exhibit enhanced Ig secretion in the absence of IL-6, CD40L and TNFα.
The production of Ig occurs in the absence of extrafollicular foci or germinal centers
The differentiation of B cells into antibody secreting cells can occur in extrafollicular areas or germinal centers (44-48). In addition, prePCs can become activated and secrete antibody in the absence of forming a defined structure (29). To identify the location of the Ig secreting, Sm-specific B cells in the 3XKO chimeras, we stained spleen sections four, and seven days following transfer of 2-12H B cells (IgMa+). As shown in Figure 6A, IgMa+ cells were evident in the spleens of B6 chimeras (left panels) and 3XKO chimeras (right panel) four days after B cell (2-12H) transfer, but absent in mice not receiving cells. Single IgMa+ cells were distributed in the follicles (boxed areas 4, and 7 but lacking in box 1) and extrafollicular space (boxed areas 3, 6, and 9), suggesting a lack of proliferation or the formation of foci. The transferred cells however, were excluded from the T cell areas. Similar results were found on day seven (data not shown). Note however, that the infiltration of DC/MFs into the follicular areas in the 3XKO chimera (compared to B6 chimera) was not a reproducible finding. To quantitate the numbers of ASCs, we stained serial sections of spleens with antibodies to IgMa and CD138 then compared the numbers of IgMahi cells that were CD138+ between B6 and 3XKO chimeras. A representative image of the dual stained cells that were enumerated from throughout the red pulp and follicles is shown in Figure 6B. We found that in the 3XKO chimeras, 28% of IgMa+ cells co-stained with CD138. In comparison, 15% of IgMa+ cells costained with CD138 in the B6 chimera (Figure 6C). This 1.8-fold change (Figure 6C) is similar to the numbers of ASC enumerated by ELISPOT, and suggests that the IgMa+/CD138+ cells are responsible for Ig secretion. This supports the data in Figure 5 showing that activation of transferred Sm-specific cells occurs in the absence of IL-6, CD40L, and TNFα, but not in wildtype mice. Further, the timing of Ig secretion and the finding that secretion occurs in the absence of organized foci or germinal centers suggests that the transferred cells rapidly become antibody secreting cells without proliferating. This is consistent with previous reports of the activation of the prePCs in autoimmune mice (29).
Figure 6. Secretion of Ig by prePCs occurs in the follicles and extrafollicular regions.
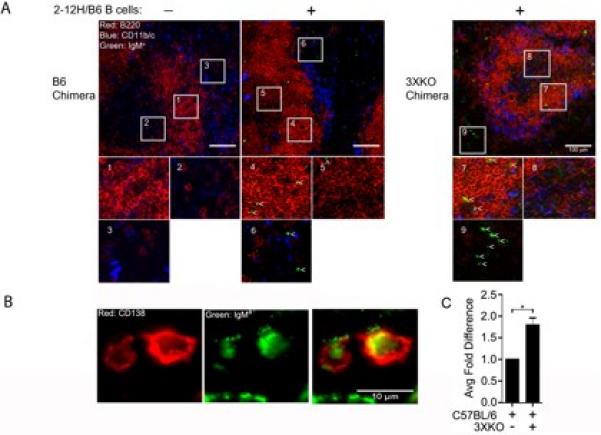
Chimeric B6 and 3XKO mice were injected with LPS (1.25 mg/kg on day -10 and -3). On day 0, PBS or 15 × 106 B cells from 2-12H/B6 mice were adoptively transferred. (A) On day 4, spleens were harvested, sectioned and stained with anti-IgMa (green), anti-B220 (red), and anti-CD11b/CD11c (blue). (Left panels) B6 chimera with and without 2-12H B cells, (Right panel) 3XKO chimera with 2-12H B cells. White boxes within the image demark areas of interest that are magnified below each image. In the magnified images, white carets identify examples of transferred (IgMa +) B cells. (B) Magnified images depicting IgMa+/CD138+ cells. Left panel: CD138+ (red), middle panel: IgMa+ (green), and right panel: merged image. (C) Compilation of 128 IgMa+ cells from B6 and 121 from 3XKO mice receiving 2-12H B cells. Three mice were analyzed 4-7 days after transfer of splenic 2-12H B cells. The percentage of IgMa+ cells that costained with CD138+ cells did not change between d4 and d7. Error bars depict SEM. (* p≤0.05).
Discussion
The genetic and environmental components that underlie SLE are complex, with many genes likely contributing indirectly to the breakdown of tolerance. Several mechanisms have been identified that regulate autoreactive B cells activated through the BCR; however, less is known about how B cells are regulated during TLR activation. Our studies examining the tolerance mechanisms that regulate TLR4 have shown that the secreted products of activated DCs and MFs regulate B cells chronically exposed to self-antigen. These products, IL-6 and sCD40L, repress TLR4-induced Ig by diminishing the number of antibody secreting cells (22, 23, 36). In this study, we identify TNFα as a third factor secreted by DCs and MFs that represses autoreactive B cells. We show that loss of DC/MF-mediated tolerance promotes autoantibody responses from activated pre-plasma cells in mice lacking IL-6, CD40L, and TNFα. B cell activation occurs in the absence of genetic predisposition, does not involve the formation of germinal centers or extrafollicular foci, and is not sustained. This suggests that loss of DC/MF-mediated tolerance could be an early event in the onset of autoimmunity.
The finding that multiple factors secreted by DCs and MFs repress autoreactive B cells suggests there is a high level of redundancy in maintaining tolerance during TLR4 stimulation. Neutralization studies showed that loss of any single repressive factor did not restore Ig secretion (Figure 2C and D), or alter the expression levels of the remaining factors (unpublished observation). However, neutralizing DC or MF conditioned medium with antibodies to IL-6, TNFα, and CD40L restored Ig secretion (Figure 2C and D), (23) and DCs and MFs from the 3XKO mice repressed approximately 30% of Ig secretion by autoreactive B cells (unpublished observations). These data indicate that IL-6 and sCD40L are the primary soluble factors regulating autoreactive B cells, but that other factors such as TNFα play a redundant, but less effective, role. The high level of redundancy in the factors makes it difficult to assess their in vivo role using conditional knockouts. The identity of the factors conferring additional repressive activity is not under investigation.
The role of TNFα in autoimmunity is seemingly contradictory. Some studies indicate that TNFα plays an inhibitory role (49-52), while other studies show it promotes the inflammatory symptoms in murine and human SLE (53-55). These opposing functions of TNFα are much like those described for the other repressive factors. For example, IL-6 promotes terminal differentiation of naïve B cells but represses autoreactive B cells (22, 56). However, serum IL-6 levels are elevated in SLE and contribute to the inflammation associated with disease (57). Similarly, CD40L determines whether activated B cells differentiate into memory or plasma cells, however soluble CD40L also represses autoreactive B cells (23, 58, 59). The pleiotropic roles for IL-6, CD40L and TNFα may indicate that different locations and/or concentrations of the repressive factors determine tolerance versus inflammation. For example, IL-6 produced by kidney tubular epithelial cells might contribute to inflammation but the serum levels seen in disease, albeit elevated compared to healthy controls, are below that required to regulate autoreactive B cells (53, 60).
Our data show that the loss of IL-6, CD40L, and TNFα leads to the activation of autoreactive B cells. We found that antibody secretion originated from autoreactive prePC transferred into 3XKO bone marrow chimeras (Figure 5C, 5D), despite the fact that these mice are not genetically predisposed to autoimmunity. Moreover, the endogenous Ig responses in 3XKO mice were prolonged compared to those seen in B6 mice (Figure 4A and 4D). This raises the possibility that defects in DC/MF-mediated tolerance lead to early autoantibody production perhaps playing a role in initiating the cascade of autoimmune events leading to disease. Other evidence that supports this idea come from our published work showing that myeloid cells from lupus-prone mice are defective in secreting IL-6, sCD40L, and TNFα (23, 36); Figure 3D), and from the work of others showing that gene ablation of TNFα promotes the onset of lupus-like disease in non-autoimmune-prone NZW mice (49), treatment of young lupus-prone NZB/W F1 mice with recombinant treatment (rTNFα) diminishes disease (50), anti-TNFα therapy in rheumatoid arthritis evokes autoimmunity (52), and human hyper IgM syndrome patients express elevated levels of antinuclear antibodies and have decreased levels of CD40L consistent with a role in B cell tolerance (23, 61).
Pre-plasma cells (or plasmablasts) represent an early stage of plasma cell development characterized by expression of CD138 on B cells that retain expression of B220, CD19 and surface Ig (B220low CD19+, surface Iglow CD138int/high). These cells are typically short-lived and in non-autoimmune 2-12H mice, prePCs are enriched in autoreactive specificities with Sm-specific prePCs represent approximately 15-20% of B6 B cells (29). They fail to upregulate Blimp-1 and do not spontaneously become ASCs. During TLR4 stimulation, these cells are regulated by IL-6 and sCD40L (Figure 1B). In autoimmune-prone hosts, prePCs become activated but do not proliferate or form germinal centers (GCs); rather, they spontaneously secrete antibody (29). Similarly, in mice lacking IL-6, CD40L, and TNFα, prePCs become short-lived antibody secreting cells without forming foci in the extracellular space or entering germinal centers (Figures 5 and 6). The short-lived response likely reflects the lack of self-renewal in the adoptive transfer model (29) because analysis of endogenous cells (Figure 4) showed Ig levels remained elevated for 2-3 weeks. It is also possible that the lack of expansion of the transferred cells might reflect poor survival of the CD138+ Sm B cells due to the lack of a secondary event conferred by genetic predisposition. Regardless, the findings confirm the critical role of IL-6, sCD40L, and/or TNFα in regulating pre-PCs, and indicate that antigen encounter in the absence of tolerance mechanisms can lead to antibody secretion during TLR4 responses. It remains unclear why transferred FO and MZ B cells were not activated in the 3XKO mice (Figure 5D) since these subsets are regulated by IL-6, and/or sCD40L (23). One possibility is that despite in vitro susceptibility to IL-6 and sCD40L, in vivo FO and MZ cells might be allowed to differentiate to the prePC stage before reaching another tolerance checkpoint. Alternatively, the transferred MZ and FO cells might not get adequate signals to further differentiate (62).
The production of autoantibodies to nuclear antigens is strongly dependent on TLR engagement with the resulting autoantibodies capable of forming immune-complexes that promote inflammation, glomerular deposition and kidney damage (63-66). Our studies of B cell tolerance during TLR4 stimulation show that IL-6, sCD40L and TNFα maintain autoreactive B cells in an unresponsive state by regulating the number of antibody secreting cells (Figure 3B; 22, 23). This mechanism of tolerance is selective in that IL-6, sCD40L, and TNFα only repress B cells chronically exposed to self antigen, but not antigenically naïve cells (Figure 3A, (22, 23). This occurs through receptor crosstalk wherein phospho-ERK is restricted from the nucleus through a mechanism that integrates the signals produced from chronic BCR ligation with acute signaling via IL-6 receptor or CD40 leading to the attenuation of Blimp-1 and XBP-1 expression and TLR4-induced Ig secretion (Lee, Rutan, and Vilen, manuscript submitted). In vivo, we show that the regulation of autoreactive B cells also requires DC/MF-mediated tolerance as mice deficient in IL-6, sCD40L and TNFα show prolonged autoantibody responses, and adoptively transferred autoreactive prePCs become activated and secrete autoantibody (Figures 4 and 5). It remains unclear whether this mechanism impacts non-autoreactive B cells chronically encountering foreign antigen on follicular dendritic cells (FDCs) during GC responses. The requirement for IL-6, CD40L, and TNFα in maintaining B cell unresponsiveness in the absence of other genetic modifiers suggests that this tolerance mechanism might be disrupted in lupus-prone mice. Indeed, we find that DCs and MFs from lupus-prone mice secrete significantly less IL-6, sCD40L and TNFα (Figure 3D; 23, 36), and that B cells from lupus-prone mice fail to be repressed by these soluble factors coincident with the failure to exclude phospho-ERK from the nucleus upon IL-6 receptor or CD40 engagement (Lee, Rutan, and Vilen, manuscript submitted). This raises the possibility that defects in DC/MF-mediated tolerance lead to early autoantibody production perhaps playing a role in initiating the cascade of autoimmune events leading to disease. The events that underlie the diminished secretion of IL-6, CD40L, or TNFα by lupus-prone DC/MFs, and those that attenuate the susceptibility of autoreactive B cells from lupus-prone mice remain unclear, but studies are underway that address the role of immune complexes in this process.
Acknowledgments
We thank Dr. Vic Johnson for anti-TNFα, Dr. Ramiro Diz for his help with microscopy, Dr. Steve Clarke for critically reading the manuscript, and members of the Vilen and Clarke labs for helpful discussions.
Abbreviations
- TLR4
Toll-like receptor-4
- LPS
lipopolysaccharide
- DAMP
damage-associated molecular pattern
- Ig
immunoglobulin
- SLE
systemic lupus erythematosus
- MF
macrophage
- DC
dendritic cell
- BCR
B cell receptor
- IL-6
interleukin-6
- sCD40L
soluble CD40-ligand
- TNFα
tumor necrosis factor alpha
- ASC
antibody secreting cell
- BMDC
bone marrow derived dendritic cell
- BMMF
bone marrow derived macrophage
- B6
C57BL/6
- 3XKO
IL-6-/- × CD40L-/- × TNFα-/- mice
- CM
conditioned medium
- PI
proliferative index
- MZ
marginal zone B cell
- FO
follicular B cell
- PC
plasma cell
Footnotes
This work was supported by National Institutes of Health (NIAID) grant R01 AI070984. M.R.G. was supported by F31 AI71292, S.R.L by NIAMS postdoctoral training grant AR07416, N.J.W and S.Z.J by predoctoral training grants AI07273 and ES007126. A.B.W. and S.Z.J. were supported by a Howard Hughes Medical Institute (HHMI) grant as part of the Med into Grad Scholar program at UNC.
Conflict-of-interest disclosure: The authors declare no conflicting financial interests.
References
- 1.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 4.Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, Li Z. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol. 2006;177:6880–6888. doi: 10.4049/jimmunol.177.10.6880. [DOI] [PubMed] [Google Scholar]
- 5.Rakoff-Nahoum S, Medzhitov R. Role of toll-like receptors in tissue repair and tumorigenesis. Biochemistry. 2008;73:555–561. doi: 10.1134/s0006297908050088. [DOI] [PubMed] [Google Scholar]
- 6.Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 2010;87:989–999. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- 7.Nishina PM, Wang J, Toyofuku W, Kuypers FA, Ishida BY, Paigen B. Atherosclerosis and plasma and liver lipids in nine inbred strains of mice. Lipids. 1993;28:599–605. doi: 10.1007/BF02536053. [DOI] [PubMed] [Google Scholar]
- 8.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas LM, Salter RD. Activation of macrophages by P2X7-induced microvesicles from myeloid cells is mediated by phospholipids and is partially dependent on TLR4. J Immunol. 2010;185:3740–3749. doi: 10.4049/jimmunol.1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorne E, Dupont H, Abraham E. Toll-like receptors 2 and 4: initiators of non-septic inflammation in critical care medicine? Intensive Care Medicine. 2010;36:1826–1835. doi: 10.1007/s00134-010-1983-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol. 2008;83:1174–1180. doi: 10.1189/jlb.0407203. [DOI] [PubMed] [Google Scholar]
- 12.Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J Immunol. 2004;172:20–24. doi: 10.4049/jimmunol.172.1.20. [DOI] [PubMed] [Google Scholar]
- 13.Oyama J, Blais C, Jr., Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 14.Wright SD, Burton C, Hernandez M, Hassing H, Montenegro J, Mundt S, Patel S, Card DJ, Hermanowski-Vosatka A, Bergstrom JD, Sparrow CP, Detmers PA, Chao YS. Infectious agents are not necessary for murine atherogenesis. J Exp Med. 2000;191:1437–1442. doi: 10.1084/jem.191.8.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll-like receptor 4 polymorphisms and atherogenesis. New Engl J Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 16.Ameziane N, Beillat T, Verpillat P, Chollet-Martin S, Aumont MC, Seknadji P, Lamotte M, Lebret D, Ollivier V, de Prost D. Association of the Toll-like receptor 4 gene Asp299Gly polymorphism with acute coronary events. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:e61–64. doi: 10.1161/01.ATV.0000101191.92392.1D. [DOI] [PubMed] [Google Scholar]
- 17.Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, Tron F, Gilbert D, Musette P. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol. 2009;183:6207–6216. doi: 10.4049/jimmunol.0803219. [DOI] [PubMed] [Google Scholar]
- 18.Summers SA, Hoi A, Steinmetz OM, O'Sullivan KM, Ooi JD, Odobasic D, Akira S, Kitching AR, Holdsworth SR. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun. 2010;35:291–298. doi: 10.1016/j.jaut.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Hang L, Slack JH, Amundson C, Izui S, Theofilopoulos AN, Dixon FJ. Induction of murine autoimmune disease by chronic polyclonal B cell activation. J Exp Med. 1983;157:874–883. doi: 10.1084/jem.157.3.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fournie GJ, Lambert PH, Meischer PA. Release of DNA in circulating blood and induction of anti-DNA antibodies after injection of bacterial lipopolysaccharides. J Exp Med. 1974;140:1189–1206. doi: 10.1084/jem.140.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izui S, Lambert PH, Fournie GJ, Turler H, Miescher PA. Features of systemic lupus erythematosus in mice injected with bacterial lipopolysaccharides: identificantion of circulating DNA and renal localization of DNA-anti-DNA complexes. J Exp Med. 1977;145:1115–1130. doi: 10.1084/jem.145.5.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kilmon MA, Rutan JA, Clarke SH, Vilen BJ. Low-affinity, Smith antigen-specific B cells are tolerized by dendritic cells and macrophages. J Immunol. 2005;175:37–41. doi: 10.4049/jimmunol.175.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kilmon MA, Wagner NJ, Garland AL, Lin L, Aviszus K, Wysocki LJ, Vilen BJ. Macrophages prevent the differentiation of autoreactive B cells by secreting CD40 ligand and interleukin-6. Blood. 2007;110:1595–1602. doi: 10.1182/blood-2006-12-061648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benschop RJ, Aviszus K, Zhang X, Manser T, Cambier JC, Wysocki LJ. Activation and anergy in bone marrow B cells of a novel immunoglobulin transgenic mouse that is both hapten specific and autoreactive. Immunity. 2001;14:33–43. doi: 10.1016/s1074-7613(01)00087-5. [DOI] [PubMed] [Google Scholar]
- 25.Diz R, McCray SK, Clarke SH. B cell receptor affinity and B cell subset identity integrate to define the effectiveness, affinity threshold, and mechanism of anergy. J Immunol. 2008;181:3834–3840. doi: 10.4049/jimmunol.181.6.3834. [DOI] [PubMed] [Google Scholar]
- 26.Borrero M, Clarke SH. Low-affinity anti-Smith antigen B cells are regulated by anergy as opposed to developmental arrest or differentiation to B-1. J Immunol. 2002;168:13–21. doi: 10.4049/jimmunol.168.1.13. [DOI] [PubMed] [Google Scholar]
- 27.Santulli-Marotto S, Retter MW, Gee R, Mamula MJ, Clarke SH. Autoreactive B cell regulation: peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity. 1998;8:209–219. doi: 10.1016/s1074-7613(00)80473-2. [DOI] [PubMed] [Google Scholar]
- 28.Acevedo-Suarez CA, Hulbert C, Woodward EJ, Thomas JW. Uncoupling of anergy from developmental arrest in anti-insulin B cells supports the development of autoimmune diabetes. J Immunol. 2005;174:827–833. doi: 10.4049/jimmunol.174.2.827. [DOI] [PubMed] [Google Scholar]
- 29.Culton DA, O'Conner BP, Conway KL, Diz R, Rutan J, Vilen BJ, Clarke SH. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol. 2006;176:790–802. doi: 10.4049/jimmunol.176.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richez C, Blanco P, Rifkin I, Moreau JF, Schaeverbeke T. Role for toll-like receptors in autoimmune disease: the example of systemic lupus erythematosus. Joint, bone, spine. 2011;78:124–130. doi: 10.1016/j.jbspin.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Bjorkbacka H, Fitzgerald KA, Huet F, Li X, Gregory JA, Lee MA, Ordija CM, Dowley NE, Golenbock DT, Freeman MW. The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiolog Genom. 2004;19:319–330. doi: 10.1152/physiolgenomics.00128.2004. [DOI] [PubMed] [Google Scholar]
- 32.Hirohashi N, Morrison DC. Low-dose lipopolysaccharide (LPS) pretreatment of mouse macrophages modulates LPS-dependent interleukin-6 production in vitro. Infection and immunity. 1996;64:1011–1015. doi: 10.1128/iai.64.3.1011-1015.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Neill SK, Veselits ML, Zhang M, Labno C, Cao Y, Finnegan A, Uccellini M, Alegre ML, Cambier JC, Clark MR. Endocytic sequestration of the B cell antigen receptor and toll-like receptor 9 in anergic cells. Proc Natl Acad Sci USA. 2009;106:6262–6267. doi: 10.1073/pnas.0812922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rui L, Healy JI, Blasioli J, Goodnow CC. ERK signaling is a molecular switch integrating opposing inputs from B cell receptor and T cell cytokines to control TLR4-driven plasma cell differentiation. J Immunol. 2006;177:5337–5346. doi: 10.4049/jimmunol.177.8.5337. [DOI] [PubMed] [Google Scholar]
- 35.Rui L, Vinuesa CG, Blasioli J, Goodnow CC. Resistance to CpG DNA-induced autoimmunity through tolerogenic B cell antigen receptor ERK signaling. Nature Immunol. 2003;4:594–600. doi: 10.1038/ni924. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert MR, Carnathan DG, Cogswell PC, Lin L, Baldwin AS, Jr., Vilen BJ. Dendritic cells from lupus-prone mice are defective in repressing immunoglobulin secretion. J Immunol. 2007;178:4803–4810. doi: 10.4049/jimmunol.178.8.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renshaw BR, Fanslow WC, 3rd, Armitage RJ, Campbell KA, Liggitt D, Wright B, Davison BL, Maliszewski CR. Humoral immune responses in CD40 ligand-deficient mice. J Exp Med. 1994;180:1889–1900. doi: 10.1084/jem.180.5.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korner H, Cook M, Riminton DS, Lemckert FA, Hoek RM, Ledermann B, Kontgen F, Fazekas de St Groth B, Sedgwick JD. Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur J Immunol. 1997;27:2600–2609. doi: 10.1002/eji.1830271020. [DOI] [PubMed] [Google Scholar]
- 39.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cook MC, Korner H, Riminton DS, Lemckert FA, Hasbold J, Amesbury M, Hodgkin PD, Cyster JG, Sedgwick JD, Basten A. Generation of splenic follicular structure and B cell movement in tumor necrosis factor-deficient mice. J Exp Med. 1998;188:1503–1510. doi: 10.1084/jem.188.8.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 42.Herlands RA, William J, Hershberg U, Shlomchik MJ. Anti-chromatin antibodies drive in vivo antigen-specific activation and somatic hypermutation of rheumatoid factor B cells at extrafollicular sites. Eur J Immunol. 2007;37:3339–3351. doi: 10.1002/eji.200737752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peeva E, Michael D, Cleary J, Rice J, Chen X, Diamond B. Prolactin modulates the naive B cell repertoire. J Clin Invest. 2003;111:275–283. doi: 10.1172/JCI16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cozine CL, Wolniak KL, Waldschmidt TJ. The primary germinal center response in mice. Curr Opin Immunol. 2005;17:298–302. doi: 10.1016/j.coi.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Elgueta R, de Vries VC, Noelle RJ. The immortality of humoral immunity. Immunol Rev. 2010;236:139–150. doi: 10.1111/j.1600-065X.2010.00924.x. [DOI] [PubMed] [Google Scholar]
- 47.Garcâia De Vinuesa C, Gulbranson-Judge A, Khan M, O'Leary P, Cascalho M, Wabl M, Klaus GG, Owen MJ, MacLennan IC. Dendritic cells associated with plasmablast survival. Eur J Immunol. 1999;29:3712–3721. doi: 10.1002/(SICI)1521-4141(199911)29:11<3712::AID-IMMU3712>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 48.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 49.Kontoyiannis D, Kollias G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur J Immunol. 2000;30:2038–2047. doi: 10.1002/1521-4141(200007)30:7<2038::AID-IMMU2038>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 50.Jacob CO, McDevitt HO. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature. 1988;331:356–358. doi: 10.1038/331356a0. [DOI] [PubMed] [Google Scholar]
- 51.Ramos-Casals M, Brito-Zeron P, Munoz S, Soria N, Galiana D, Bertolaccini L, Cuadrado MJ, Khamashta MA. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine. 2007;86:2–251. doi: 10.1097/MD.0b013e3181441a68. [DOI] [PubMed] [Google Scholar]
- 52.Debandt M, Vittecoq O, Descamps V, Le Loet X, Meyer O. Anti-TNF-alpha-induced systemic lupus syndrome. Clin Rheumatol. 2003;22:56–61. doi: 10.1007/s10067-002-0654-5. [DOI] [PubMed] [Google Scholar]
- 53.Boswell JM, Yui MA, Burt DW, Kelley VE. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J Immunol. 1988;141:3050–3054. [PubMed] [Google Scholar]
- 54.Brennan DC, Yui MA, Wuthrich RP, Kelley VE. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J Immunol. 1989;143:3470–3475. [PubMed] [Google Scholar]
- 55.Maury CP, Teppo AM. Tumor necrosis factor in the serum of patients with systemic lupus erythematosus. Arthritis Rheum. 1989;32:146–150. doi: 10.1002/anr.1780320206. [DOI] [PubMed] [Google Scholar]
- 56.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 57.Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR. Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol. 1991;147:117–123. [PubMed] [Google Scholar]
- 58.Gray D, Dullforce P, Jainandunsing S. Memory B cell development but not germinal center formation is impaired by in vivo blockade of CD40-CD40 ligand interaction. J Exp Med. 1994;180:141–155. doi: 10.1084/jem.180.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silvy A, Lagresle C, Bella C, Defrance T. The differentiation of human memory B cells into specific antibody-secreting cells is CD40 independent. Eur J Immunol. 1996;26:517–524. doi: 10.1002/eji.1830260303. [DOI] [PubMed] [Google Scholar]
- 60.Baer PC, Wegner B, Geiger H. Effects of mycophenolic acid on IL-6 expression of human renal proximal and distal tubular cells in vitro. Nephrol Dial Transplant. 2004;19:47–52. doi: 10.1093/ndt/gfg429. [DOI] [PubMed] [Google Scholar]
- 61.Hervâe M, Isnardi I, Ng YS, Bussel JB, Ochs HD, Cunningham-Rundles C, Meffre E. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med. 2007;204:1583–1593. doi: 10.1084/jem.20062287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fuentes-Panana EM, Bannish G, Monroe JG. Basal B-cell receptor signaling in B lymphocytes: mechanisms of regulation and role in positive selection, differentiation, and peripheral survival. Immunol Rev. 2004;197:26–40. doi: 10.1111/j.0105-2896.2004.0105.x. [DOI] [PubMed] [Google Scholar]
- 63.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Ann Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 64.Hurst J, von Landenberg P. Toll-like receptors and autoimmunity. Autoimmunity Rev. 2008;7:204–208. doi: 10.1016/j.autrev.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 65.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 66.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, Theofilopoulos AN. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci USA. 2009;106:12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]