

Abstract
Objective
Nicotinic acid (NA) treatment has been associated with benefits in atherosclerosis that are usually attributed to effects on plasma lipoproteins. The NA receptor GPR109A is expressed in monocytes and macrophages, suggesting a possible additional role for NA in modulating function of these immune cells. We hypothesize that NA has the potential to act directly on monocytes to alter mediators of inflammation that may contribute to its antiatherogenic effects in vivo.
Methods and Results
In human monocytes activated by Toll-like receptor (TLR)-4 agonist lipopolysaccharide, NA reduced secretion of proinflammatory mediators: TNF-α (by 49.2±4.5%); interleukin-6 (by 56.2±2.8%), and monocyte chemoattractant protein-1 (by 43.2±3.1%) (P<0.01). In TLR2 agonist, heat-killed Listeria monocytogenes-activated human monocytes, NA reduced secretion of TNF-α (by 48.6±7.1%), interleukin-6 (by 60.9±1.6%), and monocyte chemoattractant protein-1 (by 59.3±5.3%) (P<0.01; n=7). Knockdown of GPR109A by siRNA resulted in a loss of this anti-inflammatory effect in THP-1 monocytes. However, inhibition of prostaglandin D2 receptor by MK0524 or COX2 by NS398 did not alter the anti-inflammatory effects of NA observed in activated human monocytes. Preincubation of THP-1 monocytes with NA 0.1 mmol/L reduced phosphorylated IKKβ by 42±2% (P<0.001) IKB-α by 54±14% (P<0.01). Accumulation of nuclear p65 NF-κB in response to lipopolysaccharide treatment was also profoundly inhibited, by 89±1.3% (n=4; P<0.01). NA potently inhibited monocyte adhesion to activated HUVEC, and VCAM, mediated by the integrin, very late antigen 4. Monocyte chemotaxis was also significantly reduced (by 45.7±1.2%; P<0.001).
Conclusion
NA displays a range of effects that are lipoprotein-independent and potentially antiatherogenic. These effects are mediated by GPR109A and are independent of prostaglandin pathways. They suggest a rationale for treatment with NA that is not dependent on levels of plasma cholesterol and possible applications beyond the treatment of dyslipidemia.
Keywords: atherosclerosis, cholesterol-lowering drugs, macrophages, receptors, vascular biology
In patients with prior myocardial infarction, nicotinic acid (NA) reduces long-term mortality1 and may confer additional antiatherogenic benefits when used in conjunction with statins.2,3 These effects are generally attributed to favorable actions on the lipoprotein profile, which include LDL-cholesterol reduction and HDL-cholesterol elevation.
In addition, NA reduces systemic markers of inflammation (eg, high-sensitivity C-reactive protein, monocyte chemoattractant protein-1 [MCP-1], and TNF-α) and increases adiponectin, an adipokine with insulin sensitizing, antiatherogenic, and anti-inflammatory properties.2,4 These observations raise the possibility of additional nonlipoprotein-mediated effects of NA. The receptor for NA, GPR109A, is abundantly expressed in adipocytes, where it suppresses free fatty acid release5 and has G-protein coupled receptor-mediated anti-inflammatory effects.6 It is also expressed in immune cells including monocytes, macrophages, neutrophils, dendritic cells, and skin Langerhans cells, but a clear role for this receptor in these cells has yet to be elicudated.7–9 Monocytes and macrophages are key mediators of inflammation; they play a central role in the genesis and pathology of atherosclerosis and represent an important therapeutic target.10 Given the anti-inflammatory effects of NA in adipocytes, the presence of GPR109A in macrophages suggests a potentially more complex macrophage-mediated role for NA in the modulation of atherosclerosis. A recent study using the LDL-receptor–/– mouse model of atherosclerosis showed significant inhibition of disease progression with dietary NA treatment, which was not accompanied by changes in the lipoprotein profile; this effect was not observed in equivalent mice that were GPR109A-deficient.11 Significantly, the effects of NA were also abrogated by bone marrow transplantation from GPR109A-deficient mice into atherosclerosis-prone recipients, suggesting that the observed effects of NA were mediated by bone marrow-derived cells.11 Unknown, however, is whether these and other anti-inflammatory effects of NA pertain in human monocytes and the cellular mechanisms through which this drug may act. In addition, it is not known whether anti-inflammatory effects may be altered by interactions with drugs used in the treatment of the side effects associated with niacin, such as administration of prostaglandin D2 (PGD2) receptor antagonists to counteract flushing. Here, we test the effects of NA on human monocytes and explore its mechanisms of action on a range functions that are relevant to atherogenesis.
Methods
Human Monocyte Isolation and Cell Treatments
Blood from healthy volunteers was taken according Oxford University local protocols and informed consent was obtained. Monocytes were isolated by density gradient centrifugation using Optiprep™ (Axis-shield, Kimbolton, UK), washed twice using sterile PBS and cultured in X-Vivo 10 media (Fisher Scientific, Loughborough, UK) containing 1% autologous serum.
Cells were pretreated for 18 hours with NA 0.1 mmol/L (Sigma Aldrich, Poole, UK), following which lipopolysaccharide (LPS, 50 ng/mL) (Sigma Aldrich, Poole, UK) was added for a further 6 or 24 hours. TLR receptor agonists heat-killed Listeria monocytogenes (HKLM), TLR2, LPS, TLR4, and Poly(I:C), Poly(I:C) low molecular weight, and TLR 3 from a human TLR agonist kit (Invivogen, San Diego CA) were added at the recommended concentrations. PPARγ antagonist, GW9662 (10 μg/mL) (Sigma Aldrich, Poole, UK) and GW1929 (1 μg/mL) (Tocris, Bioscience, Bristol, UK) and the selective prostaglandin D2 receptor 1 antagonist, MK0524 (100 μmol/L) (Santa Cruz Biotechnology, Santa Cruz CA) or the selective cyclooxygenase-2 inhibitor NS398 (100 μmol/L) (Sigma Aldrich, Poole, UK) were added at the same time as NA treatments. At the end of the incubation times, the surrounding cell culture media was collected and snap frozen then stored at −80°C until analysis.
Measurement of Secreted Cytokines and Chemokines and PGD2
TNF-α, interleukin (IL)-6, and chemokine MCP-1 were measured in cell culture supernatants removed from monocytes by a Luminex™ Multiplex bead-based system using Milliplex™ MAP kits, from the Human Cytokine/Chemokine panel. PGD2 was measured by ELISA kit (Cayman Chemical, Tallin, Estonia) according to the manufacturer’s instructions.
siRNA Knockdown of GPR109A in THP-1 Cells
THP-1 cells in suspension were transfected with GPR109A siRNA Flexitube Gene Solution (Qiagen, UK), mock and green fluorescent protein vectors or Hi GC Stealth Control scrambled siRNA (Invitrogen, Paisley, UK) were used as controls. Cells were transfected with 1 μg of siRNA or 0.5 μg of green fluorescent protein vector using the THP-1 Amaxa® Cell Line Nucleofector kit V (Lonza, Verviers, Belgium) and the Nucleofector II system (Amaxa, Cologne, Germany) according to manufacturer’s protocol. Transfected cells were incubated with phorbol 12-myristate 13-acetate (PMA, 100 ng/mL) for 24 hours, then washed twice with PBS, after which NA and LPS treatments were performed as described above. The percentage of live/dead green fluorescent protein-transfected cells were quantified by FACS using Fixable Live/Dead Violet stain (Invitrogen, Paisley, UK).
THP-1 Cell Culture
Please refer to the Methods section in the online-only Data Supplement.
Assessment of the NF-κB Pathway
Please refer to the Methods section in the online-only Data Supplement.
Monocyte Binding Assays
Flow chamber assays were performed as previously described.12 For details of cell treatments and static adhesion assay protocol, please refer to the Methods section in the online-only Data Supplement.
Monocyte Chemotaxis Assay
For the chemotaxis assays, freshly isolated human monocytes were suspended in chemotaxis buffer, RPMI with HEPES (25 mmol/L), and 0.1% BSA, with or without NA 0.1 mmol/L, and applied to a 96-well Neuroprobe ChemoTx™ membrane (Receptor Technologies, Adderbury, UK), 8-μm pore size at a density of approximately 400,000 cells per well. The lower chamber contained either media taken from human umbilical endothelial cells stimulated with TNF-α (10 ng/mL) for 8 hours or chemotaxis buffer alone. After 4 hours incubation at 37°C in a 5% CO2 cell culture incubator, the cells on the upper layer of the membrane were removed with a cotton swab and the membrane rinsed with PBS. Migrated cells attached to the lower area of the membrane were fixed in paraformaldehyde (4%) then mounted with mounting media containing DAPI. Migration of the cells was quantified by taking 4 images under a fluorescent microscope from each membrane with a minimum of 4 membranes per treatment. Stained nuclei were then counted using image software Image Pro Plus™ (Media Cybernetics, Silver Spring, MD).
Measurement of mRNA by Quantitative RT-PCR
Please refer to the Methods section in the online-only Data Supplement.
Statistical Methods
Values are expressed as mean±SEM for replicates between experiments. Wilcoxon signed-rank nonparametric test was used to determine differences between data sets from secreted cytokines. Data for all other analyses were analyzed using one-way ANOVA with a post-hoc Dunn’s Multiple comparison test, significance was set at *P<0.05.
Results
NA Suppresses TLR2- and TLR4-Induced Release of Inflammatory Mediators
To investigate the effect of NA on TLR2- and TLR4-mediated secretion of proinflammatory mediators, we incubated human monocytes with or without NA 0.1 mmol/L for 18 hours, after which LPS (TLR4 agonist) or HKLM (TLR2 agonist) was added for a further 12 hours. Pilot dose response experiments (100 nmol/L to 0.1 mmol/L) established optimal dosing of NA (Figure I in the online-only Data Supplement) and excluded detrimental effects on cell viability (Figure II in the online-only Data Supplement). Addition of LPS stimulated the release of TNF-α, IL-6, and MCP-1 by between 3000- and 6000-fold. NA reduced secretion of each these proinflammatory mediators: TNF-α (by 49.2±4.5%); IL-6 (by 56.2±2.8%) and MCP-1 (by 43.2±3.1%) (P<0.01; n=7) (Figure 1 A-C). Addition of HKLM stimulated release of TNF-α, IL-6, and MCP-1 by between 1000- and 5000-fold. NA reduced secretion of each these proinflammatory mediators: TNF-α (by 48.6±7.1%), IL-6 (by 60.9±1.6%), and MCP-1 (by 59.3±5.3%) (P<0.01; n=7) (Figure 1D–1F). Quantitative RT-PCR showed that mRNA for each of these inflammatory mediators was similarly reduced in both freshly isolated human monocytes and in cells of the THP-1 monocytic line (Figure III in the online-only Data Supplement). The TLR3 agonist viral double-stranded RNA was also used in this assay with minimal response in TNF-α secretion and no effect of NA (data not shown).
Figure 1.

Nicotinic acid (NA) attenuates Toll-like receptor 4 (TLR4) (lipopolysaccharide [LPS])-mediated cytokine release from human monocytes in vitro (A–C). Monocytes were preincubated with NA (0.1 mmol/L) then stimulated with LPS, 50 ng/ml or heat-killed Listeria monocytogenes (HKLM). Data are shown as mean±SEM (n=7, **P<0.01 vs cells incubated with LPS only) (A–C). NA attenuates TLR2 (HKLM) mediated cytokine release from human monocytes in vitro. (n=4, *P<0.05) (D–F). Phosphorylated proteins of components of the NFκB pathway by Western blotting in THP-1 cell lysates (G and H): Total IkB-α vs phosphorylated IKB-α, total IKKβ vs phosphorylated IKKβ (n=3; **P<0.01; ***P<0.001; LPS 50 ng/ml; NA 0.1 mmol/L vs LPS (50 ng/ml). Transcriptional activity of phosphorylated NFκB using an ELISA, NF-κB present in nuclear extracts binds to the NF-κB (p65) response element. LPS (50 ng/ml)+NA (0.1 mmol/L) vs LPS (50 ng / ml), was increased 4.4-fold in nuclear extracts from LPS-treated monocytes. This effect was profoundly inhibited with NA pretreatment to below that detected in nonstimulated cell extracts. I, Nuclear NF-κB (p65) was detected by immunofluorescence in THP-1 cells treated with LPS for 30 minutes, this was not detectable in under basal conditions or in cells that had been pretreated with NA (0.1 mmol/L). Alexa 488 shows NFκB (p65) DAPI was used as a nuclear stain (J). MCP-1 indicates monocyte chemoattractant protein-1; IL6, interleukin-6.
NA Acts on the NFκB Transcription Pathway
The NF-κB pathway is a key mediator of inflammation and is activated via toll-like receptors (TLRs) resulting in increased cytokine and chemokine production.13 Activation of NF-κB and release of its subunits play a key role in the early development of endothelial dysfunction and atherosclerosis. Moreover, transcription of IL-6, TNFα, and MCP-1 is regulated by the transcription factor NFκB.14,15 In the THP-1 human monocytic cell line, LPS treatment for 6 hours resulted in a 2-fold increase in phosphorylated IKKβ and phosphorylated IKB-α. Preincubation with NA 0.1 mmol/L reduced phosphorylated IKKβ by 42±2% (n=3; P<0.001) and phosphorylated IKB-α by 54±14% (n=3; P<0.01) relative to total (Figure 1G and 1H). Transcription factor activity was determined by ELISA.
NF-κB present in nuclear extracts binds to the NF-κB (p65) response element and was increased 5.4±1-fold (n=4; P<0.01) in nuclear extracts from LPS-treated monocytes. This effect was profoundly inhibited, by 89±1.3% (n=4; P<0.01), with NA pretreatment, to below the level detected in nonstimulated cell extracts (Figure 1I). Accumulation of nuclear p65 NF-κB in THP-1 in response to LPS treatment was shown by immunofluorescence and was abolished by NA pretreatment (Figure 1J).
Effects of NA Mediated by GPR109A
Incubations of non-siRNA transfected and scrambled siRNA transfected THP-1 monocytes and GPR109a siRNA were carried out in parallel. As observed in previous experiments, preincubation with NA attenuated LPS-induced TNF-α, IL-6, and MCP-1 release in both the nontransfected and scrambled siRNA transfected cells, but in the cells, which had GPR109A knockdown by siRNA, this effect was abolished, Figure 2A–2C. Cell viability following transfection was unaffected as demonstrated by FACS live/dead staining and transfection efficiency was established using transfection of green fluorescent protein (Figure IV in the online-only Data Supplement). Confirmation of gene and protein knockdown was confirmed by PCR and Western blot respectively (Figure IV in the online-only Data Supplement).
Figure 2.

Attenuation by nicotinic acid (NA) (0.1 mmol/L) of induced cytokine release from human THP-1 monocytes in vitro, this effect is abolished with silencing of GPR109A (A–C) (n=4, *P<0.05, **P<0.01, ***P<0.001, Student t-test). Pretreatment of freshly isolated human monocytes with NA (0.1 mmol/L) or acipimox for 18 hours results in a reduction in lipopolysaccharide (LPS)-stimulated TNFα release (D) (n=3, *P<0.05). MCP-1 indicates monocyte chemoattractant protein 1; IL6, interleukin-6.
GPR109A Agonist Acipimox Reduces Cytokine and Chemokine Release
Pretreatment of freshly isolated human monocytes with the GPR109A acipimox 0.1 mmol/L for 18 hours resulted in a reduction in LPS-stimulated TNF-α release, which was comparable to that in monocytes pretreated for 18 hours with NA 0.1 mmol/L (Figure 2D) (n=3, *P<0.05).
NA-Induced Reduction in Cytokine and Chemokine Release is Unaffected by the Prostaglandin D2 Receptor 1 Antagonist MK0524 or COX2 Inhibitor NS398
NA at the optimal dose used to inhibit an inflammatory response resulted in an increase in PGD2 release (Figure V in the online-only Data Supplement). To test whether activation of the prostaglandin pathway was involved in the anti-inflammatory effects observed with NA treatment, freshly isolated human monocytes were pretreated with the prostaglandin D2 receptor antagonist MK0524. As with the previous cytokine measurements, addition of LPS stimulated the release of TNF-α, IL-6, and MCP-1 by between 4000- to 12,000-fold. NA pretreatment again reduced secretion of each these proinflammatory mediators: TNF-α (by 91±1.6%); IL-6 (by 76±6.5%) and MCP-1 (by 85±4%) (P<0.01; n=3). The addition with niacin of either the COX-2 inhibitor, NS398, or the prostaglandin D2 receptor antagonist MK0524, had no effect on the inhibition of LPS-stimulated cytokine or chemokine release (Figure 3A–3C). This lack of effect demonstrates the persistence of the anti-inflammatory effect of NA despite inhibition of the eicosanoid pathway in two distinct regions.
Figure 3.

The nicotinic acid (NA)-mediated reduction in lipopolysaccharide (LPS) stimulated TNF-α release does not involve activation of the prostaglandin pathway via prostaglandin D2 (PGD2) receptor activation as shown by incubation with the PGD2 receptor antagonist, MK0524, nor by inhibition of COX-2 using NS398 (A–C). Incubation with the PPARγ antagonist GW9662, along with NA and LPS did not affect the reduction in TNF-α release observed with NA treatment (D). CD36 was increased by 2.7±0.3-fold (P<0.05; n=3) in response to PPARγ agonist GW1929 and the PPARγ antagonist GW9662 reduced CD36 mRNA expression to virtually undetectable levels (E). MCP-1 indicates monocyte chemoattractant protein 1; IL6, interleukin-6.
NA-Induced Reduction in TNFα Release is Independent of PPARγ Activation
Because PPARγ can exert anti-inflammatory effects through antagonism of NFκB,16 and NA is able to increase PPARγ activity,7 we sought to ascertain whether inhibition of PPARγ would affect the NA-induced reduction of TNFα release from human monocytes. Incubation with the PPARγ antagonist GW9662, along with NA and LPS did not affect the reduction in TNFα release observed with NA treatment (Figure 3D). Incubation with the PPARγ agonist GW1929 increased CD36 expression by 2.7±0.3-fold, whereas addition of the antagonist GW9662 negated this effect (Figure 3E). Changes in CD36 gene expression confirmed that PPARγ was inactivated under these experimental conditions.
NA Potently Inhibits Monocyte Adhesion to Activated HUVEC and to VCAM via Very Late Antigen 4 Mediated Mechanisms
To obtain functional assessments of monocyte adhesion, we used a cell adhesion assay under flow conditions. Monocyte-endothelial binding under flow conditions was assessed in a parallel plate flow chamber. Monocytes bound readily to activated HUVEC, whereas NA treatment for 1 hour reduced adhesion of monocytes by 79.2±1.2% compared to non-treated cells (P<0.01; n=3) (Figure 4A and Movies I–III in the online-only Data Supplement).
Figure 4.

Nicotinic acid (NA) reduces adhesion of human monocytes to activated (A) HUVEC and to (B) VCAM-coated plates under flow conditions. Cells were incubated with NA (0.1 mmol/L) for 1 hour then perfused (shear stress, 1 dyne cm−2) over HUVEC that had previously been activated with TNF-α (10 ng / ml) or over VCAM-coated plates. Adhesion to HUVEC or VCAM coated plates was assessed after 5 minutes. Images were taken from 10 fields of view (FOV) and data are shown as mean number of cells in each FOV (n=3, **P<0.01 vs cells incubated without NA). Binding of THP-1 cells under flow conditions; short-term response to phorbol 12-myristate 13-acetate (PMA) treatment for 1 minute then exposure to NA (0.1 mmol/L) for 1 minute (C). Fluorescent immunohistochemistry showing active membrane localization very late antigen 4 (VLA-4) after PMA stimulation for 1 minute was abolished by NA pretreatment for 1 hour (D).
VCAM-coated plates were substituted for activated HUVEC to investigate whether the mechanism of reduced adhesion was mediated by its ligand the integrin, very late antigen 4 (VLA-4) on monocytes. PMA was used to activate VLA-4 and resulted in a 71.9±4.1% (P<0.01) increase in the mean number of monocytes bound to VCAM-coated plates, compared to that of nontreated cells. Pretreatment of monocytes with NA for 1 hour prior to PMA reduced adhesion by 75.1±2.4% (P<0.001; n=3) compared with that of stimulated with PMA alone (Figure 4B). To show that binding was VLA-4 specific, cells were incubated with saturating concentrations of anti-VLA-4 antibody for 1 hour. No binding of cells was observed in any of the fields of view (Figure 4B and Movies IV–VII in the online-only Data Supplement).
Using the same cell preparation, PMA treatment for 1 minute resulted in an increase in monocyte binding compared to that observed in the nontreated cells by 83±10.7% (P<0.01; n=2) in line with that observed in previous experiments. However, after 1 minute of NA treatment, binding was reduced to levels seen in NA-untreated cells undergoing the same procedure (Figure 4C).
Confocal microscopy showed an increase in detectable membrane VLA-4 with PMA treatment. This increase was not seen in cells that had been pretreated with NA prior to PMA treatment (Figure 4D).
NA-Induced Reduction VLA-4-Mediated Adhesion Is Unaffected by the Prostaglandin D2 Receptor 1 Antagonist MK0524 or COX2 Inhibitor NS398
In a static cell adhesion assay, the decrease in adhesion of THP-1 cells to VCAM coated plates observed with NA treatment was also unaffected by MK0524 or NS398 (Figure 5A).
Figure 5.

Reduction of very late antigen 4 (VLA-4) adhesion to VCAM-coated plates is unaffected by inhibition of components of the prostaglandin pathway (A). Fluorescent immunohistochemistry showing active membrane localization VLA-4 is lost with nicotinic acid (NA) treatment (0.1 mmol/L), which is maintained with the addition of the PGD2 receptor antagonist, MK0524, or COX-2 inhibitor, NS398 (B).
The addition of MK0524 had no effect on the reduction in VLA-4 expression,as shown by immunofluorescence (Figure 5B).
NA Potently Inhibits Monocyte Migration Toward Activated HUVEC
Using a modified Boyden chamber assay we investigated whether NA treatment altered the chemotactic activity of human monocytes. Chemotaxis of human monocytes in response to media taken from activated HUVEC was reduced by 45.7%±1.2%; (P<0.001; n=4, Figure 6).
Figure 6.
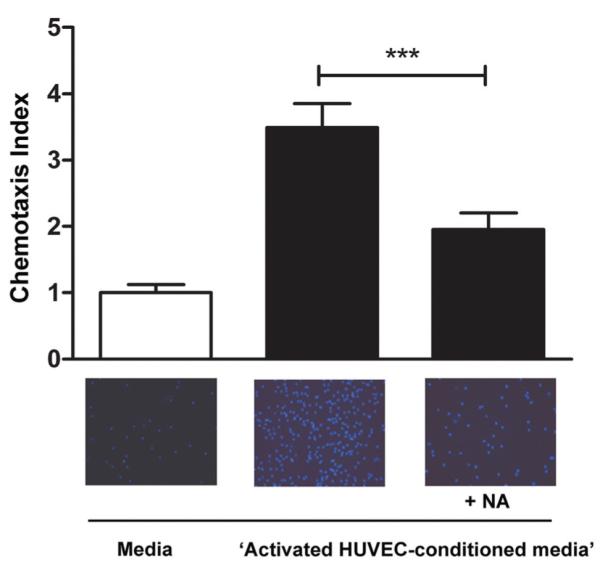
Nicotinic acid (NA) reduces migration of human monocytes toward media taken from activated HUVEC. Cells were suspended in buffer±NA (0.1 mmol/L) and placed on top of a polycarbonate membrane (pore size 8 μm). Data shown are the chemotaxis index; number of cells migrated/number of cells in the control media not exposed to media taken from activated HUVEC (n=4, ***P<0.001 vs cells incubated without the addition of NA, 0.1 mmol/L).
Discussion
Under conditions of inflammation associated with cardiovascular disease, increased secretion of proatherogenic, proinflammatory cytokines, and chemokines contribute significantly to the recruitment of inflammatory T-cells and macrophages into atherosclerotic lesions.17
This study shows, for the first time in human monocytes, substantial anti-inflammatory effects of NA. NA (1) reduced chemokine and cytokine production in response to a range of TLR ligands; (2) suppressed signaling through NF-κB; (3) decreased adhesion to activated endothelial cells, through effects on VLA-4; and (4) reduced chemotaxis toward conditioned media. Each of these monocyte and macrophage functions is of potential relevance in atherogenesis2,18,19 and could potentially contribute to the atheroprotective effects of NA. Obtained in cell culture, the observed effects were independent of the NA-induced changes in lipoproteins that are observed in vivo.
Monocyte recruitment to activated vascular endothelium and chemotaxis within the subendothelial space are key events in atherogenesis. Interventions that reduce monocyte recruitment, through reduced chemokine signaling20,21 or through reduction in endothelial adhesion, eg, mediated by VCAM-1,22 can reduce atherosclerosis in experimental models. Lukasova et al have recently shown that NA reduces chemotaxis of mouse peritoneal macrophages in response to MCP-1,11 but the effects on the earlier event of endothelial binding have not been explored. We undertook a series of functional assays in human monocytes demonstrating marked reduction in adhesion to activated HUVEC that is mediated, predominantly through a reduction in the availability of the monocyte surface integrin VLA-4 (ligand for VCAM-1).23,24 Using immunofluorescence, we found that the rapid reduction in monocyte–VCAM-1 binding after treatment was associated with almost total loss of antibody binding consistent with the allosteric regulation of integrins to modulate ligand affinity.25,26 This mechanism is also consistent with the rapidity of pharmacological action of NA on this process, which occurred within minutes.
Because many genes important in the development of atherosclerosis are under control of NF-κB, we examined activity in this transcriptional regulatory pathway. NA substantially reduced the phosphorylated intermediates IKKβ and IKB-α. Phosphorylated p-65 in the nuclear fraction was also reduced with confirmation of suppressed gene transcription in a functional reporter assay. The current data confirm and complement studies in which NA increased the activity of the transcription regulator PPAR-γ7 and increased the expression of ABCA1 cholesterol transporter in a monocyte cell line.27,28 Ricote et al have shown that PPAR-γ is a negative regulator of macrophage function, in part through antagonism of NF-κB.16 Using the irreversible inhibitor GW9662 to block PPAR-γ activity we have shown that PPAR-γ is not necessary for the anti-inflammatory action of NA on NF-κB.
Cutaneous flushing is a common side-effect of taking niacin and represents a major cause for lack of adherence to treatment. The mechanism is through PGD2 release from immune cells in the skin including the dendritic Langerhans cells and macrophages that act on the PGD2 receptor DP1.8,29,30 An approach to overcome this problem is coadministration of laropiprant (a selective prostaglandin D2 receptor anagonist), which has improved compliance of this drug.31,32 Of concern is the possibility that by inhibiting PGD2 action via its receptor, the beneficial anti-inflammatory effects of niacin might diminish. Indeed PGD2 can inhibit the mobilization of antigen-presenting dendritic cells in response to an inflammatory insult.33 In line with previous findings, niacin treatment in resulted in an increase in PGD2 release from human THP-1 monocytes.34 However, the anti-inflammatory effects with niacin treatment measured by release of proinflammatory mediators and alterations in cell adhesion prevailed despite adding the either PGD2 receptor antagonist, laropiprant, or by inhibiting COX-2. These findings indicate that the anti-inflammatory effects of NA are independent of alterations to prostaglandins and would not be predicted to be susceptible to attenuation by laropiprant, when used clinically.
The observed effects of NA are mediated through its receptor GPR109A. We have been able to demonstrate: (1) siRNA knockdown of GPR 109A abrogates the inhibition of LPS-stimulated cytokine release from human monocytes and (2) treatment with the GPR109A agonist acipimox results in a similar reduction in cytokine production to that observed with NA.
These findings are consistent with a recent study in LDL-receptor–/– knockout mice which reported novel GPR109A receptor mediated antiatherosclerotic effects of niacin administered in the diet, which were not dependent on alterations in lipoproteins.11 Moreover, these beneficial effects were abrogated in LDL-receptor–/– and GPR109A–/– double knockout mice. Through bone marrow transplantation of GPR109A competent cells, mediation of antiatherosclerotic mechanisms was shown to be via this receptor in bone marrow-derived cells.
The dose of NA associated with anti-inflammatory effects in human monocytes in the current study was identical to that which induced G-protein coupled receptor-mediated suppression of cytokines and increased adiponectin expression in human adipocytes.6 Because this dose range also inhibits lipolysis in adipocytes (a recognized pharmacological action of NA in humans)5,35 the effects observed in cell culture have plausible relevance in the clinical setting.
TLRs recognize conserved patterns on bacterial and viral pathogens, but can also have a detrimental effect in atherosclerosis. Macrophages in human atherosclerotic plaque express TLR2 and TLR4,36,37 and TLR2 signaling mediates both plaque inflammation and matrix degradation.38 Conversely TLR3 may confer athero-protective effects.39 Our findings that NA suppresses TNF-α release in response to TLR2 and TLR4 stimulation (but has no effect on TLR-3) accord further with a pharmacological role for this drug in the suppression of the monocyte/macrophage activity relevant to atherogenesis.
Further anti-inflammatory effects of NA have been reported in endothelial cells showing a reduction in TNF-α– induced inflammation and monocyte adhesion.40 In a rabbit model of acute vascular inflammation, niacin added to the diet resulted in a reduction in endothelial expression of adhesion molecules and MCP-1.41 These effects are presumably not GPR109A-mediated because the receptor is not expressed in vascular tissues.42
Collectively, our findings are of potential significance in atherosclerosis where each of these affected processes has a significant role in lesion development. It has long been postulated that NA has beneficial effects in the treatment of atherosclerosis and its complications, but conventional explanations have relied on favorable modification of plasma lipoproteins. The identification of alternative modes of action, eg, directly through GPR109A on monocytes is important because it suggests a potential rationale for treatment with NA that is not dependent on levels of plasma cholesterol and would complement current clinical treatment strategies, thereby also suggesting potential applications for this long-established drug, and its related compounds, beyond the treatment of dyslipidemia.
Furthermore, an increasing understanding of the pharmacology of niacin and its mechanisms of action suggest that some of the beneficial effects may lie beyond lipoprotein modulation. Importantly, the ability of niacin to bind GPR109A and signal via β-Arrestin 1,29 a central modulator of G-protein signal transduction and β-Arrestin–dependent signaling, represents multiple potential cellular targets with which to develop biased ligands. In the future, new agents may be able to develop pleiotropic anti-inflammatory effects and avoid the intrusive side effects that have hampered the routine use of niacin in clinical practice.
Supplementary Material
Acknowledgments
The authors thank Professor Edward A. Fisher and Dr Gemma White for insights and valuable discussion. The laboratory management by Phil Townsend is gratefully acknowledged. Robin P. Choudhury, Janet E. Digby, Neil Ruparelia, and David R. Greaves acknowledge the support of the BHF Centre of Research Excellence, Oxford. Robin P. Choudhury is a Wellcome Trust Senior Research Fellow in Clinical Science. This study was supported by the Oxford Comprehensive Biomedical Research Centre, NIHR funding scheme. Work in the laboratory of David R. Greaves was supported by BHF grant PG/09/076.
Footnotes
Disclosures Robin P. Choudhury has received fees for consultancy/speaking engagements from Merck, Roche, Astra Zeneca, GSK, and Sanofi Aventis, and research funds from Merck, GSK, and Novartis.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.111.241836/-/DC1.
References
- 1.Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in coronary drug project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–1255. doi: 10.1016/s0735-1097(86)80293-5. [DOI] [PubMed] [Google Scholar]
- 2.Lee JM, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I, Digby JE, Bannister T, Handa A, Wiesmann F, Durrington PN, Channon KM, Neubauer S, Choudhury RP. Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function: a randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol. 2009;54:1787–1794. doi: 10.1016/j.jacc.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 3.Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, Weissman NJ, Turco M. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361:2113–2122. doi: 10.1056/NEJMoa0907569. [DOI] [PubMed] [Google Scholar]
- 4.Plaisance EP, Lukasova M, Offermanns S, Zhang Y, Cao G, Judd RL. Niacin stimulates adiponectin secretion through the GPR109a receptor. Am J Physiol Endocrinol Metab. 2009;296:E549–E558. doi: 10.1152/ajpendo.91004.2008. [DOI] [PubMed] [Google Scholar]
- 5.Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-g and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–355. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- 6.Digby JE, McNeill E, Dyar OJ, Lam V, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotinic acid in adipocytes demonstrated by suppression of fractalkine, RANTES, and MCP-1 and upregulation of adiponectin. Atherosclerosis. 2010;209:89–95. doi: 10.1016/j.atherosclerosis.2009.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knowles HJ, Poole RT, Workman P, Harris AL. Niacin induces PPARγ expression and transcriptional activation in macrophages via HM74 and HM74a-mediated induction of prostaglandin synthesis pathways. Biochem Pharmacol. 2006;71:646–656. doi: 10.1016/j.bcp.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Maciejewski-Lenoir D, Richman JG, Hakak Y, Gaidarov I, Behan DP, Connolly DT. Langerhans cells release prostaglandin D2 in response to nicotinic acid. J Invest Dermatol. 2006;126:2637–2646. doi: 10.1038/sj.jid.5700586. [DOI] [PubMed] [Google Scholar]
- 9.Kostylina G, Simon D, Fey MF, Yousefi S, Simon HU. Neutrophil apoptosis mediated by nicotinic acid receptors (GPR109a) Cell Death Differ. 2008;15:134–142. doi: 10.1038/sj.cdd.4402238. [DOI] [PubMed] [Google Scholar]
- 10.Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: Macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;2:309–315. doi: 10.1038/ncpcardio0195. [DOI] [PubMed] [Google Scholar]
- 11.Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109a expressed by immune cells. J Clin Invest. 2011;121:1163–1173. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jefferson A, Wijesurendra RS, McAteer MA, Digby JE, Douglas G, Bannister T, Perez-Balderas F, Bagi Z, Lindsay AC, Choudhury RP. Molecular imaging with optical coherence tomography using ligand-conjugated microparticles that detect activated endothelial cells: Rational design through target quantification. Atherosclerosis. 2011;219:579–587. doi: 10.1016/j.atherosclerosis.2011.07.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang FX, Kirschning CJ, Mancinelli R, Xu XP, Jin Y, Faure E, Mantovani A, Rothe M, Muzio M, Arditi M. Bacterial lipopolysaccharide activates nuclear factor-κB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J Biol Chem. 1999;274:7611–7614. doi: 10.1074/jbc.274.12.7611. [DOI] [PubMed] [Google Scholar]
- 14.Ortego M, Bustos C, Hernandez-Presa MA, Tunon J, Diaz C, Hernandez G, Egido J. Atorvastatin reduces NF-κB activation and chemokine expression in vascular smooth muscle cells and mono-nuclear cells. Atherosclerosis. 1999;147:253–261. doi: 10.1016/s0021-9150(99)00193-8. [DOI] [PubMed] [Google Scholar]
- 15.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 17.Thorp E, Subramanian M, Tabas I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol. 2011;41:2515–2518. doi: 10.1002/eji.201141719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Capuzzi DM, Guyton JR, Morgan JM, Goldberg AC, Kreisberg RA, Brusco OA, Brody J. Efficacy and safety of an extended-release niacin (niaspan): A long-term study. Am J Cardiol. 1998;82:74U–81U. doi: 10.1016/s0002-9149(98)00731-0. discussion 85U–86U. [DOI] [PubMed] [Google Scholar]
- 19.Digby JE, Lee JM, Choudhury RP. Nicotinic acid and the prevention of coronary artery disease. Curr Opin Lipidol. 2009;20:321–326. doi: 10.1097/MOL.0b013e32832d3b9d. [DOI] [PubMed] [Google Scholar]
- 20.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 21.Bursill CA, Choudhury RP, Ali Z, Greaves DR, Channon KM. Broad-spectrum CC-chemokine blockade by gene transfer inhibits macrophage recruitment and atherosclerotic plaque formation in apolipoprotein e-knockout mice. Circulation. 2004;110:2460–2466. doi: 10.1161/01.CIR.0000145122.58420.CO. [DOI] [PubMed] [Google Scholar]
- 22.Dansky HM, Barlow CB, Lominska C, Sikes JL, Kao C, Weinsaft J, Cybulsky MI, Smith JD. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21:1662–1667. doi: 10.1161/hq1001.096625. [DOI] [PubMed] [Google Scholar]
- 23.Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, Lobb RR. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 1990;60:577–584. doi: 10.1016/0092-8674(90)90661-w. [DOI] [PubMed] [Google Scholar]
- 24.Alon R, Kassner PD, Carr MW, Finger EB, Hemler ME, Springer TA. The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J Cell Biol. 1995;128:1243–1253. doi: 10.1083/jcb.128.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 26.Chigaev A, Waller A, Zwartz GJ, Buranda T, Sklar LA. Regulation of cell adhesion by affinity and conformational unbending of α4β1 integrin. The J Immunol. 2007;178:6828–6839. doi: 10.4049/jimmunol.178.11.6828. [DOI] [PubMed] [Google Scholar]
- 27.Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, Teissier E, Minnich A, Jaye M, Duverger N, Brewer HB, Fruchart JC, Clavey V, Staels B. Ppar-α and ppar-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med. 2001;7:53–58. doi: 10.1038/83348. [DOI] [PubMed] [Google Scholar]
- 28.Rubic T, Trottmann M, Lorenz RL. Stimulation of cd36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochemical Pharmacology. 2004;67:411–419. doi: 10.1016/j.bcp.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, Chen JR, Muehlbauer MJ, Whalen EJ, Lefkowitz RJ. Beta-arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J Clin Invest. 2009;119:1312–1321. doi: 10.1172/JCI36806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanson J, Gille A, Zwykiel S, Lukasova M, Clausen BE, Ahmed K, Tunaru S, Wirth A, Offermanns S. Nicotinic acid- and monomethyl fumarate-induced flushing involves GPR109a expressed by keratinocytes and cox-2-dependent prostanoid formation in mice. J Clin Invest. 2010;120:2910–2919. doi: 10.1172/JCI42273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paolini JF, Mitchel YB, Reyes R, Kher U, Lai E, Watson DJ, Norquist JM, Meehan AG, Bays HE, Davidson M, Ballantyne CM. Effects of laropiprant on nicotinic acid-induced flushing in patients with dyslipidemia. Am J Cardiol. 2008;101:625–630. doi: 10.1016/j.amjcard.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 32.Maccubbin D, Koren MJ, Davidson M, Gavish D, Pasternak RC, Macdonell G, Mallick M, Sisk CM, Paolini JF, Mitchel Y. Flushing profile of extended-release niacin/laropiprant versus gradually titrated niacin extended-release in patients with dyslipidemia with and without ischemic cardiovascular disease. Am J Cardiol. 2009;104:74–81. doi: 10.1016/j.amjcard.2009.02.047. [DOI] [PubMed] [Google Scholar]
- 33.Gosset P, Pichavant M, Faveeuw C, Bureau F, Tonnel AB, Trottein F. Prostaglandin D2 affects the differentiation and functions of human dendritic cells: impact on the T cell response. Eur J Immunol. 2005;35:1491–1500. doi: 10.1002/eji.200425319. [DOI] [PubMed] [Google Scholar]
- 34.Meyers CD, Liu P, Kamanna VS, Kashyap ML. Nicotinic acid induces secretion of prostaglandin D2 in human macrophages: an in vitro model of the niacin flush. Atherosclerosis. 2007;192:253–258. doi: 10.1016/j.atherosclerosis.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Carlson LA, Hanngren A. Initial distribution in mice of 3H-labeled nicotinic acid studied with autoradiography. Life Sci. 1964;3:867–871. doi: 10.1016/0024-3205(64)90149-3. [DOI] [PubMed] [Google Scholar]
- 36.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- 37.Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, Kaul S, Arditi M. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 38.Monaco C, Gregan SM, Navin TJ, Foxwell BM, Davies AH, Feldmann M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation. 2009;120:2462–2469. doi: 10.1161/CIRCULATIONAHA.109.851881. [DOI] [PubMed] [Google Scholar]
- 39.Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, Davies AH, Flavell RA, Feldmann M, Monaco C. Unexpected protective role for toll-like receptor 3 in the arterial wall. Proceedings of the National Academy of Sciences. 2011;108:2372–2377. doi: 10.1073/pnas.1018515108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ganji SH, Qin S, Zhang L, Kamanna VS, Kashyap ML. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis. 2009;202:68–75. doi: 10.1016/j.atherosclerosis.2008.04.044. [DOI] [PubMed] [Google Scholar]
- 41.Wu BJ, Yan L, Charlton F, Witting P, Barter PJ, Rye KA. Evidence that niacin inhibits acute vascular inflammation and improves endothelial dysfunction independent of changes in plasma lipids. Arterioscler Thromb Vasc Biol. 2010;30:968–975. doi: 10.1161/ATVBAHA.109.201129. [DOI] [PubMed] [Google Scholar]
- 42.Soga T, Kamohara M, Takasaki J, Matsumoto S, Saito T, Ohishi T, Hiyama H, Matsuo A, Matsushime H, Furuichi K. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun. 2003;303:364–369. doi: 10.1016/s0006-291x(03)00342-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.