

Abstract
Asymmetric stem cell division balances maintenance of the stem cell pool and generation of diverse cell types by simultaneously allowing one daughter progeny to maintain a stem cell fate and its sibling to acquire a progenitor cell identity. A progenitor cell possesses restricted developmental potential, and defects in the regulation of progenitor cell potential can directly impinge on the maintenance of homeostasis and contribute to tumor initiation. Despite their importance, the molecular mechanisms underlying the precise regulation of restricted developmental potential in progenitor cells remain largely unknown. We used the type II neural stem cell (neuroblast) lineage in Drosophila larval brain as a genetic model system to investigate how an intermediate neural progenitor (INP) cell acquires restricted developmental potential. We identify the transcription factor Klumpfuss (Klu) as distinguishing a type II neuroblast from an INP in larval brains. klu functions to maintain the identity of type II neuroblasts, and klu mutant larval brains show progressive loss of type II neuroblasts due to premature differentiation. Consistently, Klu protein is detected in type II neuroblasts but is undetectable in immature INPs. Misexpression of klu triggers immature INPs to revert to type II neuroblasts. In larval brains lacking brain tumor function or exhibiting constitutively activated Notch signaling, removal of klu function prevents the reversion of immature INPs. These results led us to propose that multiple mechanisms converge to exert precise control of klu and distinguish a progenitor cell from its sibling stem cell during asymmetric neuroblast division.
Keywords: Asymmetric cell division, Brain tumor, Intermediate neural progenitor cell, Klumpfuss, Neuroblast, Notch, Drosophila
INTRODUCTION
Asymmetric stem cell divisions provide an efficient mechanism for maintaining a steady stem cell pool while generating progenitor cells that give rise to differentiated progeny within the tissue where the stem cells reside (Morrison and Kimble, 2006; Pontious et al., 2008; Kriegstein and Alvarez-Buylla, 2009; Knoblich, 2010; Weng and Lee, 2011). Progenitor cells possess restricted developmental potential and function to protect the genomic integrity of stem cells by minimizing their proliferation. Since both daughter cells inherit the cellular content from their parental stem cell during asymmetric division, proper specification of sibling cell identity requires precise control of stem cell determinants. Failure to properly downregulate stem cell determinants in presumptive progenitor cells might allow them to acquire stem cell-like functional properties, and can perturb tissue homeostasis and contribute to tumor formation (Krivtsov et al., 2006; Wei et al., 2008). Thus, mechanistic insight into how the sibling cells assume distinct identities during asymmetric stem cell division is likely to advance our knowledge in stem cell biology, developmental biology and tumor biology.
In fly larval brains, two classes of neuroblast lineage can be unambiguously identified based on the expression of cell fate markers and the properties of their progeny (Chia et al., 2008; Doe, 2008; Egger et al., 2008; Knoblich, 2010; Weng and Lee, 2011). A type I neuroblast expresses Deadpan (Dpn) and Asense (Ase) and divides asymmetrically to self-renew and to generate a progenitor cell called a ganglion mother cell (GMC). By contrast, a type II neuroblast (Dpn+ Ase−) divides asymmetrically to self-renew and to generate an immature intermediate neural progenitor (INP) that lacks the expression of Dpn and Ase and undergoes maturation during which it acquires an INP identity (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008). Following maturation, an INP (Dpn+ Ase+) undergoes limited rounds of asymmetric division to regenerate and to produce GMCs. A key functional property that distinguishes these two neuroblast lineages rests on their dependence on Notch signaling for the maintenance of their identity (Bowman et al., 2008; Song and Lu, 2011; Weng et al., 2011). Although dispensable for the maintenance of a type I neuroblast, Notch signaling is crucial for the maintenance of type II neuroblasts (Haenfler et al., 2012).
In mitotic type II neuroblasts, polarization of the cell cortex allows the basal proteins, including Brain tumor (Brat) and Numb, to segregate into the cortex of the presumptive immature INP and promote the formation of INPs (Bello et al., 2006; Betschinger et al., 2006; Lee et al., 2006a; Lee et al., 2006c; Wang et al., 2006; Bowman et al., 2008; Wirtz-Peitz et al., 2008; Prehoda, 2009). Whereas a wild-type type II neuroblast is surrounded by three to five immature INPs and twenty to thirty INPs, a brat or numb mutant type II neuroblast is always surrounded by supernumerary neuroblasts at the expense of INPs. Thus, previous studies have proposed that brat and numb function in immature INPs, where these proteins promote the specification of an INP identity. However, the mechanisms by which brat and numb trigger an immature INP to assume the identity of an INP remain unknown.
In this study, we show that precise regulation of klu function is pivotal for distinguishing the self-renewing neuroblast from its sibling progenitor cell during asymmetric neuroblast division. Klu is necessary for the maintenance of type I and II brain neuroblasts, as klu mutant larvae showed progressive loss of both types of neuroblast. Klu is detected in all neuroblasts but is absent from their immediate daughter progenitor progeny. Misexpression of klu in immature INPs led to the formation of supernumerary type II neuroblasts. Importantly, removal of klu function prevented the reversion of immature INPs to type II neuroblasts triggered by the loss of brat function or constitutive activation of Notch signaling. Furthermore, overexpression of klu also exacerbated the reversion of GMCs to type I neuroblasts as triggered by the aberrant activation of Notch signaling. Together, we conclude that precise control of klu function by multiple signaling mechanisms distinguishes a neuroblast from a progenitor cell during asymmetric division of fly larval brain neuroblasts.
MATERIALS AND METHODS
Fly strains
Mutant and transgenic flies used include brat150 (Betschinger et al., 2006), numb2 (Skeath and Doe, 1998), kluR51 (Kaspar et al., 2008), erm-GAL4 (III) (Pfeiffer et al., 2008), wor-GAL4 (Lee et al., 2006b), UAS-klu-HA, UAS-klu1-583-HA and UAS-kluΔzf1-HA (Kaspar et al., 2008), UAS-Notchintra (Chung and Struhl, 2001) and UAS-cMyc (Benassayag et al., 2005). erm-GAL4 (II) was generously provided by Dr G. Rubin (HHMI). The following stocks were obtained from the Bloomington Drosophila Stock Center: Oregon R, bratDG19310, bratk06028 (Arama et al., 2000), brat11 (Arama et al., 2000), Notch55e11 (Artavanis-Tsakonas et al., 1984), klu09036, Df(H99) (White et al., 1994), UAS-mCD8-GFP, UAS-apoliner (Bardet et al., 2008), UAS-p35, UAS-GFP, FRT19A (Lee and Luo, 2001), FRT2A, hs-flp (Lee and Luo, 2001), Act-FRT-Stop-FRT-GAL4 (Pignoni and Zipursky, 1997), tub-GAL80 (Lee and Luo, 2001) and tub-GAL80ts (Bloomington Drosophila Stock Center). Transgenic fly lines UAS-brat-myc, UAS-HA-klu, UAS-HA-klu1-583, UAS-HA-kluΔzf1 and UAS-HA-kluΔzf4 were generated using the pUAST-attB vector for insertion into an identical docking site in the fly genome via ϕC31 integrase-mediated transgenesis (Bischof and Basler, 2008).
Immunofluorescent staining and antibodies
Larval brains were dissected in Schneider's medium (Sigma), fixed in 4% formaldehyde for 23 minutes and washed twice for 20 minutes each in 1× PBS containing 0.3% Triton X-100 (PBST). After washing, brains were incubated with primary antibodies in PBST for 3 hours at room temperature. Antibodies used include rat anti-Dpn (1:1000; this study), rabbit anti-Ase (1:400) (Weng et al., 2010), guinea pig anti-Ase (1:50; this study), mouse anti-Prospero (MR1A, 1:100) (Lee et al., 2006a), guinea pig anti-CycE (1:1000; T. Orr-Weaver, Massachusetts Institute of Technology, MA, USA), mouse anti-Dlg (1:50; Developmental Studies Hybridoma Bank), chicken anti-GFP (1:2000; Aves Labs), rabbit anti-Klu (1:200) (Yang et al., 1997), rat anti-Mira (1:100) (Lee et al., 2006a), guinea pig anti-Numb (1:1000; J. Skeath, Washington University, WA, USA), rabbit anti-aPKC (1:1000; Sigma), mouse anti-phosphohistone H3 (1:2000; Upstate Biotechnology), rabbit anti-PntP1 (1:600; J. Skeath) and rabbit anti-RFP (1:100; Rockland). Secondary antibodies were from Molecular Probes and Jackson Labs. We used Rhodamine phalloidin (1:100; Invitrogen) to visualize cortical actin. The confocal images were acquired on a Leica SP5 scanning confocal microscope.
Clonal analyses
Lineage clones were induced following the previously published method (Lee and Luo, 2001; Weng et al., 2010).
RESULTS
klu functions to maintain the identity of larval brain neuroblasts
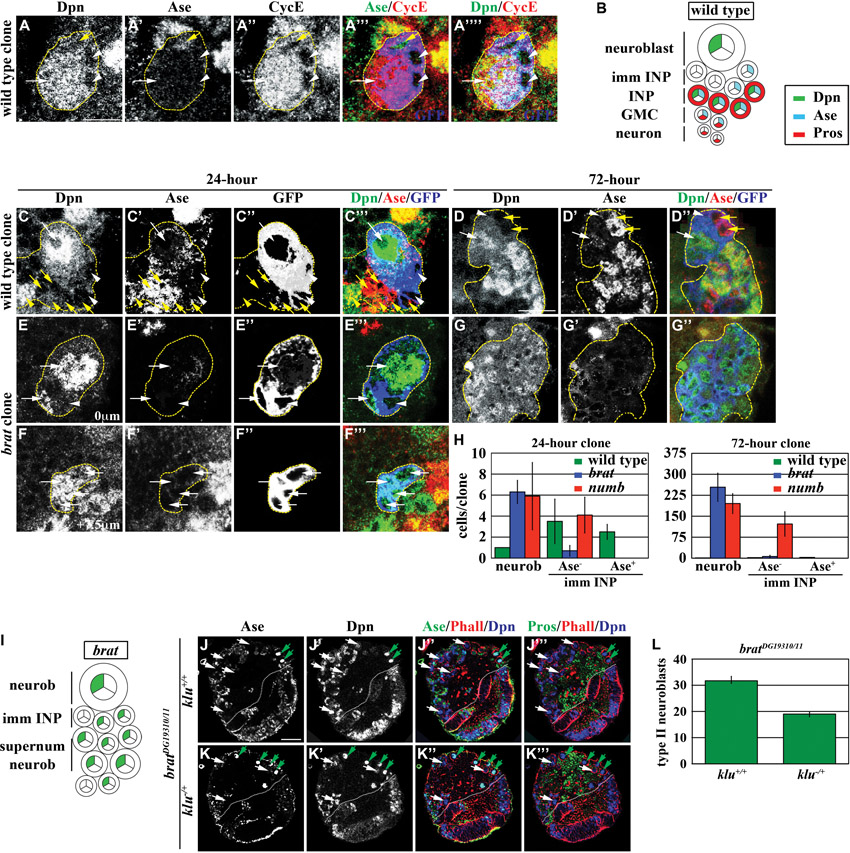
Brat is required cell-autonomously for the formation of INPs in larval brains (Betschinger et al., 2006; Lee et al., 2006c; Bowman et al., 2008). Thus, understanding how brat regulates the maturation of immature INPs will provide crucial insight into the mechanisms that distinguish the fates of sibling cells following the asymmetric division of type II neuroblasts. We assessed the identity of cells in the GFP-marked mosaic clones derived from a single wild-type or brat null mutant type II neuroblast using the onset of Ase expression as a marker for an intermediate stage during maturation (supplementary material Fig. S1A-B; see Discussion for more details). Each wild-type clone always contained one neuroblast surrounded by two to three Ase− immature INPs, two to three Ase+ immature INPs, INPs and GMCs (supplementary material Fig. S1C-D″,H; n=7 per stage). By contrast, a similarly staged brat mutant clone consisted of mostly neuroblasts, with very few Ase− immature INPs and never any Ase+ immature INPs or INPs (supplementary material Fig. S1E-I; n=7 per stage). These results led us to conclude that Brat functions during maturation to prevent an immature INP from acquiring a neuroblast fate while promoting it to assume an INP identity.
To elucidate the mechanisms by which Brat regulates the maturation of immature INPs, we screened for haploinsufficient loci in the fly genome that modify the supernumerary type II neuroblast phenotype in a sensitized bratDG19310/11 mutant genetic background (H.K. and C.-Y.L., unpublished). We identified klu as a genetic suppressor of brat, as heterozygosity of the klu locus strongly suppressed the formation of supernumerary neuroblasts in the brat-sensitized genetic background (supplementary material Fig. S1J-L; n=18 per genotype). Thus, we propose that Brat regulates the maturation of immature INPs by antagonizing klu.
To test whether Brat functions to prevent an immature INP from reacquiring a neuroblast fate or by promoting it to assume an INP identity, we first analyzed the expression of cell fate and cell proliferation markers in wild-type and klu mutant larval brains (Fig. 1A). In wild-type larvae, the total number of neuroblasts reached the plateau of almost 100 per brain hemisphere 72 hours after larval hatching (ALH) and remained at 100 per lobe at 96 hours ALH (Fig. 1B-B″,F; n=10 brains per stage). In similarly staged klu mutant larvae, total neuroblasts plateaued at ~80 per brain hemisphere at 72 hours ALH and decreased to less than 60 per lobe at 96 hours ALH (Fig. 1C-C″,F; n=10 brains per stage). Importantly, brain neuroblasts in wild-type or klu mutant larvae displayed similar proliferation profiles as indicated by the expression of Cyclin E (CycE) and EdU pulse-chase labeling (Fig. 1D,E; 100% of neuroblasts in the brain, n=10; data not shown). These results strongly suggest that klu is required for the maintenance of brain neuroblasts.
Fig. 1.

Neuroblasts prematurely differentiate in klu mutant brains. (A) Summary of the cell fate marker expression pattern in type I and II neuroblast lineages in Drosophila larval brains. GMC, ganglion mother cell; INP, intermediate neural progenitor; imm INP, immature INP; neurob, neuroblast; Pros, Prospero. (B-F) klu mutant brains show progressive loss of neuroblasts. (B-E) Brains were dissected from wild-type or kluR51/09036 mutant larvae at 96 hours ALH and stained for the markers indicated. The white dotted line separates the central brain (left) from the optic lobe (right). Discs large (Dlg) marks the cell cortex. (F) Average type I and II neuroblasts per brain lobe in larvae of genotypes and stages indicated. Error bars indicate s.e.m. (G-L) Neuroblasts show reduced cell diameter and are likely to prematurely differentiate in klu mutant brains. Larvae carrying GFP-marked klu+/+ or klu−/− mosaic neuroblast clones (outlined by the yellow dotted line) were aged for 110 hours after clone induction and larval brains were stained for the markers indicated. (G-H‴) Type I neuroblast clones. (I-K‴) Type II neuroblast clones. (L) The frequency of klu+/+ or klu−/− clones containing neuroblasts of the cell diameter indicated. The following are indicated: type I neuroblast (Dpn+ Ase+), green arrow; GMC (Dpn− Ase+), green arrowhead; type II neuroblast (Dpn+ Ase−), white arrow; Ase− immature INP (Dpn− Ase−), white arrowhead; Ase+ immature INP (Dpn− Ase+), yellow arrow; INP (Dpn+ Ase+), yellow arrowhead. Scale bars: 20 μm in B-E; 10 μm in G-K‴.
We next tested whether klu functions cell-autonomously to maintain brain neuroblasts by inducing GFP-marked mosaic clones derived from a single wild-type or klu mutant neuroblast. Although both wild-type and klu mutant type I neuroblast clones maintained a single neuroblast per clone, 36.7% of the klu mutant clones contained neuroblasts of reduced cell diameter (≤10 μm) (Fig. 1G-H‴,L; n=30 clones per genotype). Similarly, half of the klu mutant type II neuroblast clones also contained neuroblasts of reduced cell diameter (≤10 μm) (Fig. 1I-J‴,L; n=8 clones per genotype). Reduction in neuroblast diameter was previously shown to correlate with the onset of premature differentiation (Lee et al., 2006b; Song and Lu, 2011). Consistently, 12.5% of the klu mutant clones contained multiple INPs, GMCs and their progeny (Fig. 1K-L; n=8 clones). Together, these results led us to conclude that klu functions to maintain the identity of neuroblasts in larval brains and to propose that Brat is likely to prevent an immature INP from reacquiring a neuroblast fate by antagonizing Klu.
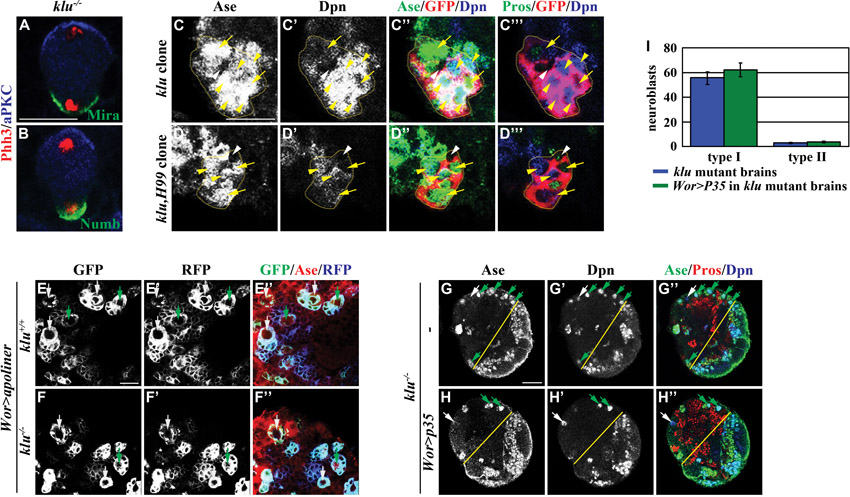
Defects in cell polarity or aberrant activation of cell death can lead to premature neuroblast loss in larval brain (Lee et al., 2006b; Bello et al., 2007), so we tested whether klu maintains neuroblast identity by regulating cell polarity or cell survival. To assess whether klu is required for polarization of the neuroblast cortex, we examined the localization of atypical Protein kinase C (aPKC), Miranda (Mira) and Numb (Albertson and Doe, 2003; Rolls et al., 2003; Lee et al., 2006a; Lee et al., 2006c) in telophase neuroblasts in klu mutant brains. We detected aPKC segregated exclusively into the cortex of the future neuroblast and Mira and Numb localized asymmetrically in the cortex of the future progenitor cell in klu mutant brains (supplementary material Fig. S2A,B). Thus, since mitotic klu mutant neuroblasts displayed asymmetric localization of the apical and basal proteins, it is unlikely that klu maintains the identity of neuroblasts by regulating polarization of the neuroblast cortex. To determine if klu is required for the maintenance of neuroblast survival, we examined whether blocking activation of apoptosis would prevent the premature loss of neuroblasts in klu mutant brains. We generated mosaic clones derived from a single type I or II neuroblast lacking klu alone or klu and the Df(3R)H99 locus. The H99 locus contains three crucial activators of apoptosis in the fly genome (White et al., 1994; Grether et al., 1995; Chen et al., 1996; White et al., 1996). However, removal of the H99 locus did not significantly decrease the occurrence of neuroblasts of reduced cell diameter (≤10 μm) or revert the absence of type II neuroblasts in klu mutant clones (supplementary material Fig. S2C-D‴; n=14 per genotype). Furthermore, we failed to detect aberrant activation of caspases in klu mutant brains, and blocking caspase activity did not prevent premature neuroblast loss in klu mutant brains (supplementary material Fig. S2E-I; n=15 per genotype). Thus, we conclude that Klu does not maintain the identity of neuroblasts by regulating cell polarity or cell survival.
Overexpression of klu induces massive expansion of type II neuroblasts
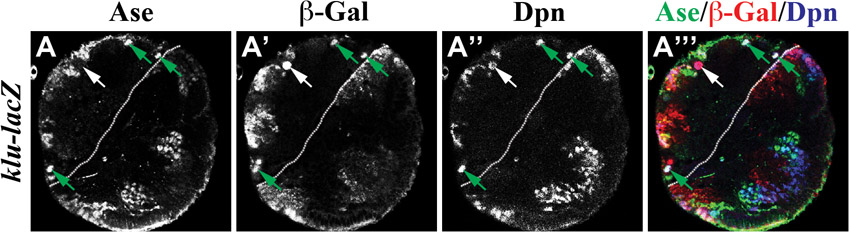
Phenotypic analyses of klu mutant brains led us to conclude that klu functions to maintain the identity of neuroblasts in larval brains, so we hypothesized that klu should be expressed in both type I and II neuroblasts. We first assessed the spatial expression pattern of the klu-lacZ enhancer trap line in larval brains. We detected lacZ expression in both type I and II neuroblasts as well as in their immediate progenitor progeny in larval brain (supplementary material Fig. S3; n=10). Since the half-life of the β-gal protein might be longer than that of endogenous Klu protein, we stained larval brains carrying GFP-marked lineage clones derived from a single wild-type type I or II neuroblast with an antibody specific for Klu protein. In the type I neuroblast lineage, Klu was detected in the neuroblast but undetectable in GMCs and their progeny (Fig. 2A-A″,C; n=9 clones). In the type II neuroblast lineage, Klu was present in the neuroblast and INPs but absent from immature INPs and GMCs (Fig. 2B-C; n=5 clones). Thus, we conclude that Klu is expressed in both types of neuroblast but is absent from their immediate progenitor progeny.
Fig. 2.

Overexpression of klu induces supernumerary type II neuroblasts. (A-C) Klu is detected in neuroblasts but is undetectable in their immediate progenitor progeny. (A-B‴) Drosophila larvae carrying GFP-marked wild-type type I or II neuroblast lineage clones (outlined by the yellow dotted line) were aged for 72 hours after clone induction and larval brains were stained for the markers indicated. (C) Summary of the Klu expression pattern in type I and II neuroblast lineages. (D-E″) Overexpression of klu induces excess type II neuroblasts. Larvae of the genotype indicated were raised at 31°C for 72 hours ALH and larval brains were stained for the markers indicated. The white dotted line separates the central brain (left) from the optic lobe (right). (F-G‴) Overexpression of klu specifically induces supernumerary neuroblasts in type II neuroblast lineage clones. Larvae carrying GFP-marked type I or II lineage clones (outlined by the yellow dotted line) overexpressing klu were aged for 24 hours after clone induction and brains were stained for the markers indicated. Abbreviations and arrows/arrowheads as Fig. 1. Scale bars: 10 μm in A-B‴,F-G‴; 20 μm in D-E‴.
The spatial expression pattern of Klu is consistent with its proposed function in the maintenance of neuroblast identity, so we tested whether increased function of klu can trigger the formation of supernumerary neuroblasts. We first overexpressed a UAS-klu transgene under the control of a pan-neuroblast wor-GAL4 driver in larval brains. Unexpectedly, we observed massive expansion of type II neuroblasts but did not detect any increase in type I neuroblasts (Fig. 2D-E″; n=7 per genotype). Similarly, lineage clones derived from a single type I neuroblast overexpressing klu driven by a constitutively active Actin-GAL4 driver reproducibly contained one neuroblast per clone (Fig. 2F-F‴; 100%, n=10 clones). By contrast, type II neuroblast clones overexpressing klu contained mostly neuroblasts (Fig. 2G-G‴; 100%, n=10 clones). Together, these results indicate that increased function of klu specifically leads to the expansion of type II neuroblasts.
Misexpression of klu in immature INPs leads to supernumerary type II neuroblasts
We next examined the cell type from which supernumerary neuroblasts arise in the type II neuroblast clones overexpressing Klu. We tested whether type II neuroblasts overexpressing Klu undergo symmetric division in telophase to generate supernumerary neuroblasts by analyzing the localization of aPKC, Mira and Numb. We observed that aPKC segregates into the cortex of the future neuroblast and Mira and Numb partition into the cortex of the future immature INP (Fig. 3B,C; n=15 per genotype). This result strongly suggests that a type II neuroblast overexpressing klu divides asymmetrically to generate a neuroblast and an immature INP. We reproducibly observed Ase− immature INPs in all type II neuroblast clones overexpressing klu (Fig. 2G-G‴). Thus, it is unlikely that type II neuroblasts overexpressing Klu undergo symmetric division to generate supernumerary neuroblasts. We next tested whether supernumerary neuroblasts arise from de-differentiation of INPs in type II neuroblast clones overexpressing Klu. The lineage clones derived from INPs overexpressing klu maintained a single INP per clone and contained GMCs and their progeny but never type II neuroblasts, indicating that overexpression of klu is not sufficient to trigger INPs to de-differentiate back into type II neuroblasts (Fig. 3A-A‴; 100%, n=8). Thus, it is unlikely that supernumerary neuroblasts in type II neuroblast clones overexpressing Klu originate from symmetric neuroblast division or de-differentiation of INPs.
Fig. 3.

Misexpression of klu triggers the reversion of immature INPs to type II neuroblasts. (A-A‴) Overexpression of klu is not sufficient to trigger de-differentiation of INPs. Drosophila larvae carrying GFP-marked INP lineage clones (outlined by the yellow dotted line) overexpressing klu were aged for 24 hours after clone induction and brains were stained for the markers indicated. (B,C) Telophase neuroblasts overexpressing klu show asymmetric localization of apical and basal proteins. Phh3, phosphorylated histone H3. (D-F) The activity of erm-GAL4 is first detected in immature INPs. (D-E‴) Larvae expressing GFP driven by erm-GAL4 (II) or erm-GAL4 (III) were aged for 72 hours and brains were stained for the markers indicated. PointedP1 (PntP1) marks type II neuroblasts and Ase− immature INPs. (F) Summary of the erm-GAL4 expression pattern in the type II neuroblast lineage. (G-J) Overexpression of klu in immature INPs leads to supernumerary type II neuroblasts. (G-I) Larvae overexpressing klu driven by erm-GAL4 were raised at 31°C for 72 hours ALH and brains were stained for the markers indicated. The white dotted line separates the central brain (left) from the optic lobe (right). (J) Average type II neuroblasts per brain lobe in larvae of the genotype indicated. 1×, 2× indicate the copy number of UAS-klu and erm-GAL4 (III) transgenes. Error bars indicate s.e.m. Abbreviations and arrows/arrowheads as Fig. 1. Scale bars: 10 μm in A-A‴,D-E‴; 5 μm in B,C; 20 μm in G-I.
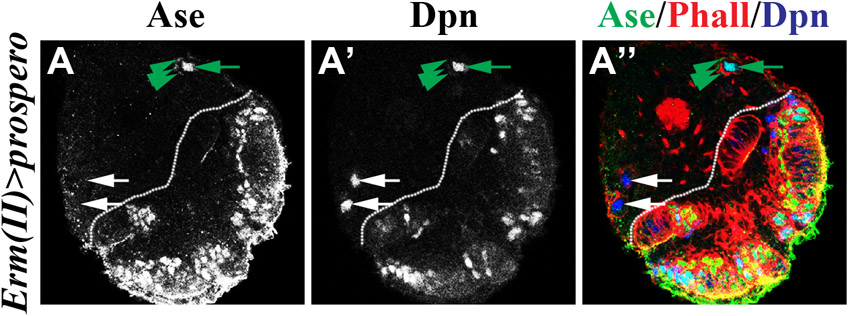
As an alternative, we tested whether overexpression of klu in neuroblasts indirectly leads to increased function of Klu in immature INPs, triggering them to acquire a neuroblast fate. We searched for GAL4 lines that can drive expression of the UAS transgene in immature INPs. The erm-GAL4 transgene inserted on the third chromosome (III) in the fly genome is sufficient to induce UAS transgene expression in INPs but not in type II neuroblasts (Pfeiffer et al., 2008; Weng et al., 2010). The identical erm-GAL4 transgene inserted on the second chromosome (II) (kindly provided by Dr G. Rubin, HHMI) showed a similar spatial expression pattern in larval brain, as ectopic expression of a UAS-prospero transgene driven by erm-GAL4 (II) induced premature loss of immature INPs and INPs without affecting the maintenance of type II neuroblasts (supplementary material Fig. S4; n=8). We next tested whether onset of the erm-GAL4 (II) and (III) activity occurs in immature INPs by colocalizing the expression of a UAS-GFP reporter transgene with Ase and PointedP1 (PntP1). We reproducibly detected GFP expression driven by erm-GAL4 (II) in both Ase− and Ase+ immature INPs (Fig. 3D-D‴,F; n=8). By contrast, the reporter expression driven by erm-GAL4 (III) was only first detected specifically in Ase+ immature INPs (Fig. 3E-F; n=8). We then tested whether increased function of klu in Ase− or Ase+ immature INPs can lead to the formation of supernumerary neuroblasts. Indeed, misexpression of klu driven by erm-GAL4 (II) led to a greater than 10-fold increase in type II neuroblasts per brain lobe compared with a similarly staged wild-type brain lobe (Fig. 3G,J and Fig. 1F; n=8). Although misexpression of one copy of UAS-klu driven by one copy of erm-GAL4 (III) failed to induce supernumerary type II neuroblasts, doubling the number of UAS-klu and erm-GAL4 (III) transgenes led to modest expansion of type II neuroblasts (Fig. 3H-J; n=12 per genotype). Together, these data strongly suggest that immature INPs can indeed revert to type II neuroblasts in response to misexpression of klu.
Promotion by Klu of supernumerary type II neuroblast formation is dependent on the zinc-finger motifs
klu, the fly ortholog of the mammalian Wilms tumor 1 (WT1) gene, encodes a putative transcriptional regulator characterized by four C2H2 zinc-finger motifs in the C-terminus (Klein and Campos-Ortega, 1997; Yang et al., 1997). Vertebrate studies have shown that WT1 requires its zinc-finger motifs to regulate transcription of its target genes (Roberts, 2005). To test whether Klu triggers supernumerary neuroblasts by acting as a transcriptional regulator, we ectopically expressed a series of UAS-klu transgenes in neuroblasts (Fig. 4A). We focused our analyses on the type II lineage as overexpression of the full-length Klu transgenic protein specifically led to the expansion of type II neuroblasts (Fig. 2E-E″).
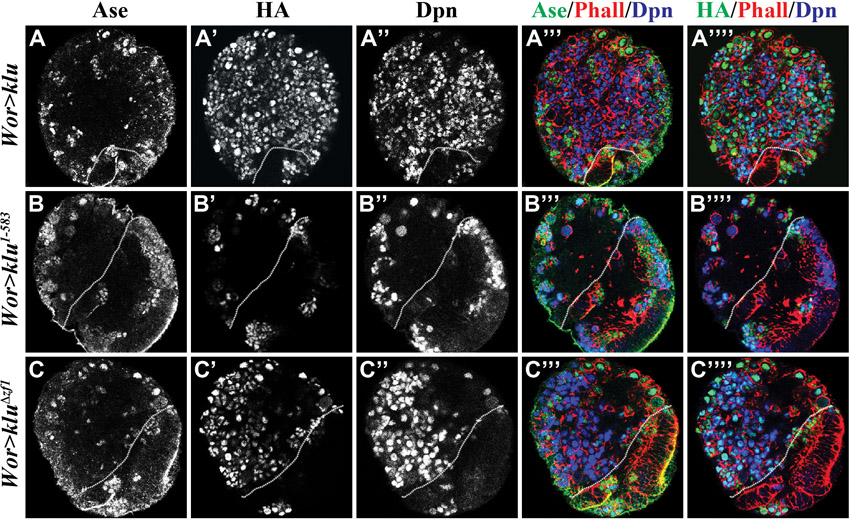
Fig. 4.

Induction of supernumerary type II neuroblasts by Klu is dependent on the zinc-finger motifs. (A) The klu transgenes used in this study. (B-D) Drosophila larvae overexpressing various klu transgenes were raised at 31°C for 72 hours ALH and brains were stained for the markers indicated. The white dotted line separates the central brain (left) from the optic lobe (right). Phalloidin (Phall) marks the cell cortex. Type II neuroblasts (Dpn+ Ase−) are indicated (arrows). Scale bar: 20 μm. (E) Average type II neuroblasts per brain lobe in larvae of the genotype indicated. Error bars indicate s.e.m.
Expression of the Klu1-583 transgenic protein (which lacks all four zinc-finger motifs) failed to induce supernumerary neuroblasts, indicating that the zinc-finger motifs are indispensable for Klu to promote the identity of type II neuroblasts (Fig. 4B,E; 100%, n=10 per genotype). Although expression of the KluΔzf1 transgenic protein (which lacks zinc-finger 1) was sufficient to induce supernumerary neuroblasts, it appeared to be less potent than expression of full-length Klu (Fig. 2E-E″ and Fig. 4C,E; 100%, n=10 per genotype). This result strongly suggests that zinc-finger 1 is necessary for the optimal function of Klu in promoting the formation of supernumerary neuroblasts. Significantly, expression of the KluΔzf4 transgenic protein (which lacks zinc-finger 4) completely failed to induce supernumerary neuroblasts, strongly suggesting that zinc-finger 4 is essential for Klu function (Fig. 4D,E; 100%, n=10).
Finally, we confirmed that expression levels of the various truncated Klu transgenic proteins under the above experimental conditions were indistinguishable from each other (supplementary material Fig. S5). Our data correlate well with a previously published domain analysis of the Klu protein in the developing sensory organ precursor cell (Kaspar et al., 2008). Thus, we propose that Klu promotes the identity of type II neuroblasts by regulating gene transcription.
Brat prevents the reversion of immature INPs to type II neuroblasts by antagonizing Klu
Our data thus far are consistent with our hypothesis that Brat distinguishes an immature INP from its sibling type II neuroblast in part by antagonizing the function of Klu. We directly tested whether removal of klu function can suppress the formation of supernumerary neuroblasts and restore INPs in brat11/k06028 strong hypomorphic mutant brains. The control type II neuroblast clones carrying both copies of the wild-type klu gene in brat11/k06028 mutant brains contained mostly neuroblasts and very few INPs (Fig. 5A-A‴⁗,C; 100%, n=10 clones). By contrast, klu mutant type II neuroblast clones in brat11/k06028 mutant brains contained a single neuroblast per clone and possessed INPs and GMCs (Fig. 5B-C; 92%, n=12 clones). These data strongly support our hypothesis that Brat distinguishes an immature INP from its sibling type II neuroblast by antagonizing Klu.
Fig. 5.

Brat suppresses reversion of immature INPs by antagonizing Klu. (A-C) Removal of klu function suppresses supernumerary type II neuroblasts and restores the formation of INPs and GMCs in brat strong hypomorphic mutant brains. (A-B⁗) brat11/k06028 mutant Drosophila larvae carrying GFP-marked control (klu+/+) and klu mutant type II neuroblast mosaic clones (outlined by the yellow dotted line) were aged for 72 hours after clone induction and brains were stained for the markers indicated. (C) Quantification of various cell types in the control and klu mutant clones in brat11/k06028 mutant brains. (D-F) Co-expression of Brat suppresses Klu-induced supernumerary type II neuroblasts. (D-E⁗) Larvae carrying GFP-marked type II neuroblast lineage clones (outlined by the yellow dotted line) overexpressing klu or klu and brat were aged for 72 hours after clone induction and brains were stained for the markers indicated. (F) Average type II neuroblasts per brain lobe in larvae of the genotype indicated. Error bars indicate s.e.m. Arrows/arrowheads as Fig. 1. Scale bars: 10 μm.
To confirm that Brat can indeed antagonize Klu in immature INPs, we induced genetic clones derived from a single type II neuroblast overexpressing klu alone or klu and brat simultaneously. The control clones overexpressing klu consisted of virtually all neuroblasts with very few Ase− immature INPs (Fig. 5D-D⁗,F; 62.5%, n=16 clones). Co-expression of brat but not an unrelated UAS transgene significantly suppressed the supernumerary neuroblast phenotype and restored the formation of Ase− and Ase+ immature INPs, INPs and GMCs in the type II neuroblast clones overexpressing klu (Fig. 5E-F; 100%, n=10 clones; data not shown). Finally, overexpression of brat alone did not alter cell fate specification in the type II neuroblast clones (data not shown). Together, these data led us to conclude that Brat antagonizes Klu in the immature INP, distinguishing it from its sibling type II neuroblast (Fig. 6H).
Fig. 6.

Aberrant activation of Notch signaling induces reversion of immature INPs through klu. (A-D) Removal of klu function suppresses supernumerary type II neuroblasts induced by constitutively activated Notch signaling. (A-C⁗) Drosophila larvae carrying GFP-marked wild-type (klu+/+) or klu−/− type II neuroblast mosaic clones (outlined by the yellow dotted line) overexpressing Notchintra were aged for 72 hours after clone induction and brains were stained for the markers indicated. (D) The frequency of clones containing one or more type II neuroblasts in larvae of the genotype indicated. (E-G) Overexpression of klu prevents Notch mutant type II neuroblasts from premature differentiation. (E-F⁗) Larvae carrying GFP-marked Notch mutant type II neuroblast mosaic clones (outlined by the yellow dotted line) alone or overexpressing klu were aged for 72 hours after clone induction and brains were stained for the markers indicated. (G) The frequency of clones containing one or no type II neuroblasts in larvae of the genotype indicated. (H) Model: Brat or Numb prevent the reversion of immature INPs to type II neuroblasts by antagonizing Klu. Abbreviations and arrows/arrowheads as Fig. 1. Scale bars: 10 μm.
Aberrant activation of Notch signaling promotes the reversion of immature INPs through klu
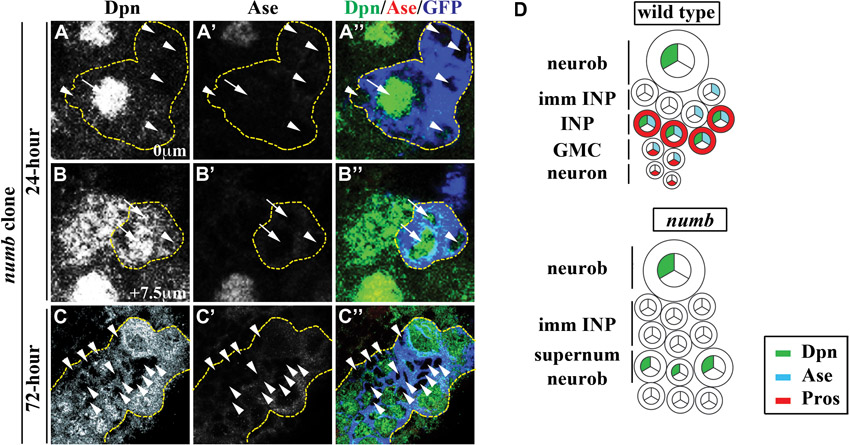
The basal protein Numb, which is an evolutionarily conserved inhibitor of Notch signaling, is also necessary for the formation of INPs in larval brain, but how Numb regulates maturation of immature INPs has never been characterized (Rhyu et al., 1994; Guo et al., 1996; Bowman et al., 2008). We first investigated the role of Numb during maturation by assessing the identity of cells in the GFP-marked clones derived from a single numb null mutant type II neuroblast. Whereas a 24-hour numb mutant clone contained 5.9±3.2 neuroblasts, 4.7±1.7 Ase− immature INPs and no Ase+ immature INPs, a 72-hour numb clone possessed 195.4±35.4 neuroblasts, 122.2±43.6 Ase− immature INPs and no Ase+ immature INPs (supplementary material Fig. S1H and Fig. S6; n=9 per stage). Indistinguishable from the numb mutant clones, the type II neuroblast clones expressing a constitutively activated form of Notch (Notchintra) also contained neuroblasts and Ase− immature INPs but never Ase+ immature INPs and INPs (Fig. 6A-A⁗,D; 100%, n=15). Numb thereby functions to prevent an immature INP from acquiring a neuroblast fate and instead promotes it to assume an INP identity most likely through inhibition of Notch signaling.
We next tested whether aberrant activation of Notch signaling induces the reversion of immature INPs to type II neuroblasts via a Klu-dependent mechanism. Removal of klu function significantly reduced supernumerary neuroblasts and restored INPs and GMCs in half of the clones derived from type II neuroblasts overexpressing Notchintra (Fig. 6B-B‴,D; n=18 clones). Most significantly, 33.3% of these clones possessed a single neuroblast per clone (Fig. 6C-D; n=18 clones). Thus, aberrant activation of Notch signaling in immature INPs leads to the formation of supernumerary neuroblasts via a Klu-dependent mechanism.
We directly tested whether klu acts downstream of Notch signaling to maintain type II neuroblasts by assessing the identity of cells in the mosaic clones derived from Notch mutant type II neuroblasts and those overexpressing klu. Whereas most Notch mutant clones did not contain neuroblasts, overexpression of klu completely suppressed the premature loss of type II neuroblasts in the Notch mutant clones (Fig. 6E-G; 100%, n=8 clones). This result strongly suggests that Notch signaling maintains the identity of type II neuroblasts via a klu-dependent mechanism. Interestingly, overexpression of klu in Notch mutant type II neuroblast clones failed to induce the formation of supernumerary neuroblasts (Fig. 6F-G; 100%, n=8 clones). Thus, we propose that aberrant activation of Notch signaling induces the reversion of immature INPs to type II neuroblasts by activating multiple downstream genes including klu (Fig. 6H).
Aberrant activation of Notch signaling promotes reversion of GMCs through klu
Although klu is necessary for the maintenance of type I neuroblasts, overexpression of klu did not lead to an increase in type I neuroblasts. One plausible reason is that additional fate determinants might function redundantly in the specification of GMC identity, leading us to identify Notch signaling as an excellent candidate (Bowman et al., 2008; Wirtz-Peitz et al., 2008; Kaspar et al., 2008). We tested this hypothesis by first overexpressing klu in the numb mutant clones. Whereas the numb mutant clones possessed an average of three neuroblasts per clone, overexpression of klu tripled the number of neuroblasts in the same genetic background (supplementary material Fig. S7; n=10 per genotype). This indicates that increased function of klu can trigger a further increase in supernumerary type I neuroblasts in the absence of Numb.
We next tested whether Klu can exacerbate the formation of supernumerary type I neuroblasts induced by activated Notch signaling by examining the identity of cells in the clones derived from a single type I neuroblast ectopically expressing Notchintra alone or Notchintra and klu simultaneously. Although the type I neuroblast clones overexpressing Notchintra contained an average of six neuroblasts per clone, only 60% of these clones contained more than one neuroblast per clone (Fig. 7B-B⁗,D; n=10 per genotype). By contrast, the type I neuroblast clones co-expressing Notchintra and klu contained an average of 18 neuroblasts per clone, and 100% of the clones displayed the supernumerary neuroblast phenotype (Fig. 7C-D; n=10 per genotype). Since the clones derived from neuroblasts overexpressing Notchintra alone or Notchintra and klu contained GMCs and their progeny, it is unlikely that the supernumerary neuroblasts arose from symmetric neuroblast division. Instead, increased function of klu most likely further enhances the reversion of GMCs to type I neuroblasts induced by aberrant activation of Notch signaling. To test whether activated Notch signaling promotes the reversion of GMCs to type I neuroblasts via a klu-dependent mechanism, we induced type I neuroblast clones overexpressing Notchintra with or without klu function. Removal of klu function significantly reduced the average number of supernumerary neuroblasts per clone as well as the frequency of clones containing greater than one neuroblast compared with the control clones (Fig. 7E-G; n=20 clones per genotype). Thus, we propose that aberrant activation of Notch signaling induces the reversion of GMCs to type I neuroblasts by activating multiple downstream genes including klu (Fig. 7H).
Fig. 7.

Aberrant activation of Notch signaling induces reversion of GMCs in part through klu. (A-D) Co-expression of klu further exacerbates the formation of supernumerary type I neuroblasts induced by constitutively activated Notch signaling. (A-C⁗) Drosophila larvae carrying GFP-marked type I neuroblast lineage clones (outlined by the yellow dotted line) overexpressing klu, Notchintra or klu and Notchintra were aged for 48 hours after clone induction and brains were stained for the markers indicated. (D) Average type I neuroblasts per clone and the frequency of clones containing one or more type I neuroblasts in larvae of the genotype indicated. (E-G) Removal of klu function suppresses supernumerary type I neuroblasts induced by constitutively activated Notch signaling. (E-F⁗) Larvae carrying GFP-marked klu+/+ or klu−/− type I neuroblast mosaic clones (outlined by the yellow dotted line) overexpressing Notchintra were aged for 72 hours after clone induction and brains were stained for the markers indicated. (G) Average type I neuroblasts per clone and the frequency of clones containing one or more type I neuroblasts in larvae of the genotype indicated. (H) Model: Numb prevents the reversion of GMCs to type I neuroblasts by antagonizing Klu. Abbreviations and arrows/arrowheads as Fig. 1. Scale bars: 10 μm.
DISCUSSION
Asymmetric stem cell division provides an efficient mechanism to preserve a steady stem cell pool while generating differentiated progeny within the tissue where the stem cells reside. Precise spatial control of the stem cell determinants inherited by both sibling cells in every asymmetric cell division ensures that a daughter cell maintains the stem cell characteristics while the sibling progeny acquires the progenitor cell identity. In mitotic type II neuroblasts, the basal proteins Brat and Numb segregate into immature INPs and are required for the formation of INPs (Bello et al., 2006; Betschinger et al., 2006; Lee et al., 2006a; Lee et al., 2006c; Wang et al., 2006; Bowman et al., 2008; Wirtz-Peitz et al., 2008). Our study significantly extends the findings from previous studies and showed that Brat and Numb function in immature INPs to prevent them from acquiring a neuroblast fate while promoting the INP identity (supplementary material Figs S1, S6). Identification and characterization of the klu gene led us to propose that Brat and Numb converge to exert precise control of Klu to distinguish an immature INP from its sibling type II neuroblast (Fig. 6H). Numb also prevents a GMC from reverting to a type I neuroblast by inhibiting Notch signaling in the type I neuroblast lineage (Fig. 7 and supplementary material Fig. S7). Interestingly, although overexpression of klu was insufficient to induce supernumerary type I neuroblasts, increased function of klu can drastically enhance the reversion of GMCs to type I neuroblasts in the presence of activated Notch signaling (Fig. 7). Thus, we propose that aberrant activation of Notch signaling induces reversion of GMCs by activating multiple downstream genes including klu. Together, our data led us to conclude that precise regulation of klu by multiple signaling mechanisms distinguishes a progenitor cell from its sibling stem cell during asymmetric stem cell division.
Regulation of INP maturation
The essential role of Brat and Numb in regulating the formation of INPs is well established, but lack of insight into maturation has hindered investigation into the mechanisms by which these two proteins distinguish an immature INP from its sibling type II neuroblast (Bello et al., 2006; Betschinger et al., 2006; Lee et al., 2006a; Lee et al., 2006c; Wang et al., 2006; Bowman et al., 2008; Wirtz-Peitz et al., 2008). A previous study defined immature INPs by the following criteria: (1) being immediately adjacent to the parental type II neuroblast, (2) lacking Dpn expression and (3) displaying a very low level of CycE expression (Bowman et al., 2008). Based on these criteria, analyses of the spatial expression pattern of various cell fate markers in the type II neuroblast lineage clones in wild-type brains revealed that onset of Ase expression correlates with an intermediate stage of maturation (supplementary material Fig. S1A-A⁗). In the 16-hour clones, we reproducibly observed one type II neuroblast (Dpn+ Ase− CycE+), two to three Ase− immature INPs (Dpn− Ase− CycE−), two to three Ase+ immature INPs (Dpn− Ase+ CycE−) and INPs (Dpn+ Ase+ CycE+) (supplementary material Fig. S1A-B). Furthermore, we showed that Ase− immature INPs maintain expression of the type II neuroblast-specific marker PntP1, whereas Ase+ immature INPs showed virtually undetectable PntP1 expression (Fig. 3F-H). Thus, onset of Ase expression should serve as a useful marker for an intermediate stage during maturation.
Our data led us to propose that Brat distinguishes an immature INP from its sibling type II neuroblast by indirectly antagonizing the function of Klu based on the following evidence. First, Klu was undetectable in Ase− immature INPs in the brat single-mutant or brat and numb double-mutant type II neuroblast clones (data not shown). Thus, a Brat-independent mechanism must exist to downregulate Klu in immature INPs. Second, overexpression of a truncated Brat transgenic protein lacking the NHL domain, which is required for repression of mRNA translation (Sonoda and Wharton, 2001), completely suppresses the formation of supernumerary neuroblasts (H.K. and C.-Y.L., unpublished). Thus, it is unlikely that downregulation of Klu in immature INPs occurs via a Brat-dependent translational repression of klu mRNA. We propose that Brat might suppress the expression of a co-factor necessary for the function of Klu, just as WT1 requires co-factors in order to regulate the expression of its target genes in vertebrates (Roberts, 2005). Further investigation will be necessary to discern how Brat establishes restricted developmental potential in immature INPs by antagonizing the function of Klu.
The role of Klu in promoting neuroblast identity
WT1 requires its zinc-finger motifs to regulate transcription of its target genes and can function as an activator or a repressor of transcription in a context-dependent manner (Roberts, 2005). A previous study showed that overexpression of Klu can partially suppress the expression of a lacZ reporter transgene containing the cis-regulatory elements from the even-skipped gene, a putative direct target of Klu, in the fly embryonic central nervous system (McDonald et al., 2003). Since Klu and WT1 display extensive homology in zinc-fingers 2-4, Klu is likely to recognize a similar DNA binding sequence as WT1 (Klein and Campos-Ortega, 1997; Yang et al., 1997; McDonald et al., 2003). The even-skipped cis-regulatory element contains three putative WT1 binding sites, but nucleotide substitutions in these sites that were predicted to abolish Klu binding failed to render the lacZ reporter transgene unresponsive to overexpression of klu (McDonald et al., 2003). These data led us to speculate that Klu might recognize a distinct consensus DNA binding sequence to WT1. To test this hypothesis, we generated two UAS-WT1 transgenes that encode the two most prevalent isoforms of the WT1 protein, WT1 −KTS and WT1 +KTS. Interestingly, neither WT1 transgene, when overexpressed by wor-GAL4, triggered the formation of supernumerary type II neuroblasts in larval brain (data not shown). This is consistent with Klu recognizing a distinct consensus DNA binding sequence to WT1. However, we cannot rule out the possibility that the inability of the WT1 transgenic protein to induce supernumerary type II neuroblasts is simply due to the absence of necessary co-factors in the fly, as repression of target gene transcription by WT1 requires additional co-factors in vertebrates (Shervington et al., 2006). More studies will be necessary to elucidate the molecular function of Klu in promoting type II neuroblast identity.
Progressive restriction of developmental potential during maturation of immature INPs
Restricted developmental potential functionally defines progenitor cells and allows them to generate differentiated progeny through limited rounds of cell division without impinging on the homeostatic state of the stem cell pool (Zon, 2008; Knoblich, 2010; Weng and Lee, 2011). Despite their importance, the molecular mechanisms by which progenitor cells acquire restricted developmental potential remain experimentally inaccessible in most stem cell lineages. However, studies from various groups have paved the way for using fly larval brain neuroblast lineages as an in vivo model system for investigating how progenitor cells acquire restricted developmental potential (Bello et al., 2008; Boone and Doe, 2008; Bowman et al., 2008; Bayraktar et al., 2010; Weng et al., 2010).
In this study, we describe the expression pattern of additional molecular markers that allow us to unambiguously identify two distinct populations of immature INPs. Furthermore, we provide experimental evidence strongly suggesting that these two groups of immature INPs possess distinct functional properties. More specifically, Ase− immature INPs readily revert to type II neuroblasts in response to misexpression of Klu, whereas Ase+ immature INPs appear much less responsive to Klu. These data led us to propose that the genome in immature INPs becomes reprogrammed during maturation such that these cells become progressively less responsive to neuroblast fate determinants such as Klu. As a consequence, an INP becomes completely unresponsive to Klu following maturation. Further experiments will be required to validate this model in the future.
Supplementary Material
Acknowledgements
We thank Drs C. Doe, A. Gould, T. Klein, T. Orr-Weaver, G. Rubin, J. Skeath, G. Struhl and X. Yang for fly stocks and antibody reagents; Dr M. Mindren for WT1 cDNA clones; the Bloomington Drosophila Stock Center and the Developmental Studies Hybridoma Bank for antibodies; Krista L. Golden for technical assistance throughout the course of this work; and the members of the C.-Y.L. laboratory for reading the manuscript and providing critical comments.
Footnotes
Funding
H.K. was supported by a fellowship from the Japan Society for the Promotion of Science. C.-Y.L. is supported by the University of Michigan Start-Up Fund, the Burroughs Wellcome Fund Career Award in the Biomedical Sciences [1006160.01], a Sontag Foundation Distinguished Scientist Award and the National Institutes of Health [R01-GM092818, R01-NS077914]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.081687/-/DC1
References
- Albertson R., Doe C. Q. (2003). Dlg, Scrib and Lgl regulate neuroblast cell size and mitotic spindle asymmetry. Nat. Cell Biol. 5, 166-170 [DOI] [PubMed] [Google Scholar]
- Arama E., Dickman D., Kimchie Z., Shearn A., Lev Z. (2000). Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 19, 3706-3716 [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S., Grimwade B. G., Harrison R. G., Markopoulou K., Muskavitch M. A., Schlesinger-Bryant R., Wharton K., Yedvobnick B. (1984). The Notch locus of Drosophila melanogaster: a molecular analysis. Dev. Genet. 4, 233-254 [Google Scholar]
- Bardet P. L., Kolahgar G., Mynett A., Miguel-Aliaga I., Briscoe J., Meier P., Vincent J. P. (2008). A fluorescent reporter of caspase activity for live imaging. Proc. Natl. Acad. Sci. USA 105, 13901-13905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar O. A., Boone J. Q., Drummond M. L., Doe C. Q. (2010). Drosophila type II neuroblast lineages keep Prospero levels low to generate large clones that contribute to the adult brain central complex. Neural Dev. 5, 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello B., Reichert H., Hirth F. (2006). The brain tumor gene negatively regulates neural progenitor cell proliferation in the larval central brain of Drosophila. Development 133, 2639-2648 [DOI] [PubMed] [Google Scholar]
- Bello B., Holbro N., Reichert H. (2007). Polycomb group genes are required for neural stem cell survival in postembryonic neurogenesis of Drosophila. Development 134, 1091-1099 [DOI] [PubMed] [Google Scholar]
- Bello B. C., Izergina N., Caussinus E., Reichert H. (2008). Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural Dev. 3, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benassayag C., Montero L., Colombié N., Gallant P., Cribbs D., Morello D. (2005). Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 25, 9897-9909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J., Mechtler K., Knoblich J. A. (2006). Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell 124, 1241-1253 [DOI] [PubMed] [Google Scholar]
- Bischof J., Basler K. (2008). Recombinases and their use in gene activation, gene inactivation, and transgenesis. Methods Mol. Biol. 420, 175-195 [DOI] [PubMed] [Google Scholar]
- Boone J. Q., Doe C. Q. (2008). Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev. Neurobiol. 68, 1185-1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. K., Rolland V., Betschinger J., Kinsey K. A., Emery G., Knoblich J. A. (2008). The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 14, 535-546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Nordstrom W., Gish B., Abrams J. M. (1996). grim, a novel cell death gene in Drosophila. Genes Dev. 10, 1773-1782 [DOI] [PubMed] [Google Scholar]
- Chia W., Somers W. G., Wang H. (2008). Drosophila neuroblast asymmetric divisions: cell cycle regulators, asymmetric protein localization, and tumorigenesis. J. Cell Biol. 180, 267-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H. M., Struhl G. (2001). Nicastrin is required for Presenilin-mediated transmembrane cleavage in Drosophila. Nat. Cell Biol. 3, 1129-1132 [DOI] [PubMed] [Google Scholar]
- Doe C. Q. (2008). Neural stem cells: balancing self-renewal with differentiation. Development 135, 1575-1587 [DOI] [PubMed] [Google Scholar]
- Egger B., Chell J. M., Brand A. H. (2008). Insights into neural stem cell biology from flies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 39-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether M. E., Abrams J. M., Agapite J., White K., Steller H. (1995). The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 9, 1694-1708 [DOI] [PubMed] [Google Scholar]
- Guo M., Jan L. Y., Jan Y. N. (1996). Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron 17, 27-41 [DOI] [PubMed] [Google Scholar]
- Haenfler J. M., Kuang C., Lee C. Y. (2012). Cortical aPKC kinase activity distinguishes neural stem cells from progenitor cells by ensuring asymmetric segregation of Numb. Dev. Biol. 365, 219-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar M., Schneider M., Chia W., Klein T. (2008). Klumpfuss is involved in the determination of sensory organ precursors in Drosophila. Dev. Biol. 324, 177-191 [DOI] [PubMed] [Google Scholar]
- Klein T., Campos-Ortega J. A. (1997). klumpfuss, a Drosophila gene encoding a member of the EGR family of transcription factors, is involved in bristle and leg development. Development 124, 3123-3134 [DOI] [PubMed] [Google Scholar]
- Knoblich J. A. (2010). Asymmetric cell division: recent developments and their implications for tumour biology. Nat. Rev. Mol. Cell Biol. 11, 849-860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A., Alvarez-Buylla A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A. V., Twomey D., Feng Z., Stubbs M. C., Wang Y., Faber J., Levine J. E., Wang J., Hahn W. C., Gilliland D. G., et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818-822 [DOI] [PubMed] [Google Scholar]
- Lee C. Y., Andersen R. O., Cabernard C., Manning L., Tran K. D., Lanskey M. J., Bashirullah A., Doe C. Q. (2006a). Drosophila Aurora-A kinase inhibits neuroblast self-renewal by regulating aPKC/Numb cortical polarity and spindle orientation. Genes Dev. 20, 3464-3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. Y., Robinson K. J., Doe C. Q. (2006b). Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 439, 594-598 [DOI] [PubMed] [Google Scholar]
- Lee C. Y., Wilkinson B. D., Siegrist S. E., Wharton R. P., Doe C. Q. (2006c). Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self-renewal. Dev. Cell 10, 441-449 [DOI] [PubMed] [Google Scholar]
- Lee T., Luo L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251-254 [DOI] [PubMed] [Google Scholar]
- McDonald J. A., Fujioka M., Odden J. P., Jaynes J. B., Doe C. Q. (2003). Specification of motoneuron fate in Drosophila: integration of positive and negative transcription factor inputs by a minimal eve enhancer. J. Neurobiol. 57, 193-203 [DOI] [PubMed] [Google Scholar]
- Morrison S. J., Kimble J. (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068-1074 [DOI] [PubMed] [Google Scholar]
- Pfeiffer B. D., Jenett A., Hammonds A. S., Ngo T. T., Misra S., Murphy C., Scully A., Carlson J. W., Wan K. H., Laverty T. R., et al. (2008). Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. USA 105, 9715-9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignoni F., Zipursky S. L. (1997). Induction of Drosophila eye development by decapentaplegic. Development 124, 271-278 [DOI] [PubMed] [Google Scholar]
- Pontious A., Kowalczyk T., Englund C., Hevner R. F. (2008). Role of intermediate progenitor cells in cerebral cortex development. Dev. Neurosci. 30, 24-32 [DOI] [PubMed] [Google Scholar]
- Prehoda K. E. (2009). Polarization of Drosophila neuroblasts during asymmetric division. Cold Spring Harb. Perspect. Biol. 1, a001388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyu M. S., Jan L. Y., Jan Y. N. (1994). Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell 76, 477-491 [DOI] [PubMed] [Google Scholar]
- Roberts S. G. (2005). Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 15, 542-547 [DOI] [PubMed] [Google Scholar]
- Rolls M. M., Albertson R., Shih H. P., Lee C. Y., Doe C. Q. (2003). Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epithelia. J. Cell Biol. 163, 1089-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shervington A., Cruickshanks N., Wright H., Atkinson-Dell R., Lea R., Roberts G., Shervington L. (2006). Glioma: what is the role of c-Myc, hsp90 and telomerase? Mol. Cell. Biochem. 283, 1-9 [DOI] [PubMed] [Google Scholar]
- Skeath J. B., Doe C. Q. (1998). Sanpodo and Notch act in opposition to Numb to distinguish sibling neuron fates in the Drosophila CNS. Development 125, 1857-1865 [DOI] [PubMed] [Google Scholar]
- Song Y., Lu B. (2011). Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 25, 2644-2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J., Wharton R. P. (2001). Drosophila Brain Tumor is a translational repressor. Genes Dev. 15, 762-773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Somers G. W., Bashirullah A., Heberlein U., Yu F., Chia W. (2006). Aurora-A acts as a tumor suppressor and regulates self-renewal of Drosophila neuroblasts. Genes Dev. 20, 3453-3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J., Wunderlich M., Fox C., Alvarez S., Cigudosa J. C., Wilhelm J. S., Zheng Y., Cancelas J. A., Gu Y., Jansen M., et al. (2008). Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 13, 483-495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Lee C. Y. (2011). Keeping neural progenitor cells on a short leash during Drosophila neurogenesis. Curr. Opin. Neurobiol. 21, 36-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Golden K. L., Lee C. Y. (2010). dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev. Cell 18, 126-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Haenfler J. M., Lee C. Y. (2011). Changes in Notch signaling coordinates maintenance and differentiation of the Drosophila larval optic lobe neuroepithelia. Dev. Neurobiol., doi: 10.1002/dneu.20995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K., Grether M. E., Abrams J. M., Young L., Farrell K., Steller H. (1994). Genetic control of programmed cell death in Drosophila. Science 264, 677-683 [DOI] [PubMed] [Google Scholar]
- White K., Tahaoglu E., Steller H. (1996). Cell killing by the Drosophila gene reaper. Science 271, 805-807 [DOI] [PubMed] [Google Scholar]
- Wirtz-Peitz F., Nishimura T., Knoblich J. A. (2008). Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell 135, 161-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Bahri S., Klein T., Chia W. (1997). Klumpfuss, a putative Drosophila zinc finger transcription factor, acts to differentiate between the identities of two secondary precursor cells within one neuroblast lineage. Genes Dev. 11, 1396-1408 [DOI] [PubMed] [Google Scholar]
- Zon L. I. (2008). Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 453, 306-313 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}