

Abstract
Glial cells are essential for the development and function of the nervous system. In the mammalian brain, vast numbers of glia of several different functional types are generated during late embryonic and early foetal development. However, the molecular cues that instruct gliogenesis and determine glial cell type are poorly understood. During post-embryonic development, the number of glia in the Drosophila larval brain increases dramatically, potentially providing a powerful model for understanding gliogenesis. Using glial-specific clonal analysis we find that perineural glia and cortex glia proliferate extensively through symmetric cell division in the post-embryonic brain. Using pan-glial inhibition and loss-of-function clonal analysis we find that Insulin-like receptor (InR)/Target of rapamycin (TOR) signalling is required for the proliferation of perineural glia. Fibroblast growth factor (FGF) signalling is also required for perineural glia proliferation and acts synergistically with the InR/TOR pathway. Cortex glia require InR in part, but not downstream components of the TOR pathway, for proliferation. Moreover, cortex glia absolutely require FGF signalling, such that inhibition of the FGF pathway almost completely blocks the generation of cortex glia. Neuronal expression of the FGF receptor ligand Pyramus is also required for the generation of cortex glia, suggesting a mechanism whereby neuronal FGF expression coordinates neurogenesis and cortex gliogenesis. In summary, we have identified two major pathways that control perineural and cortex gliogenesis in the post-embryonic brain and have shown that the molecular circuitry required is lineage specific.
Keywords: Drosophila, FGF, Gliogenesis, Insulin signalling, TOR
INTRODUCTION
Glia play a variety of crucial roles in nervous system development. They are not only integrated into the developing nervous system, but are also required to provide targets for axonal growth cones and to insulate developing neurons and axon fascicles (Chotard and Salecker, 2007; Lemke, 2001; Parker and Auld, 2006). In the adult nervous system, glia act both functionally, in neurotransmitter homeostasis, and structurally to enwrap axons and maintain the blood-brain barrier. Specialised glial populations have also been shown to act as neural stem cells, providing the primary progenitors of the neuronal and glial lineages (Kriegstein and Alvarez-Buylla, 2009).
In the mammalian CNS neurogenesis generally precedes gliogenesis, when the same progenitor cells switch from generating neurons early in development to generating glia at later stages (Freeman, 2010; Rowitch and Kriegstein, 2010). This switch is governed by both cell-intrinsic factors and extracellular cues in the environment. In the forebrain, pathways such as Notch and JAK/STAT promote gliogenic competence in neural precursor cells during the temporal transition from a neurogenic to a gliogenic state (Freeman, 2010; Rowitch and Kriegstein, 2010). However, the instructive mechanisms that specify glial cell fate and control the proliferation of specific glial subtypes in vivo are poorly understood.
Nervous system development in Drosophila occurs in two phases: embryonic and post-embryonic. Development of the embryonic nervous system is completed just prior to larval hatching, whereas the post-embryonic nervous system develops in the larval and early pupal stages, during which time the majority of neurons and glia found in the adult fly are generated. The Drosophila CNS contains several different glial types (Freeman and Doherty, 2006; Parker and Auld, 2006). Surface glia, which are divided into perineural and sub-perineural glia, are found immediately below the neural lamella and function as the blood-brain barrier (Stork et al., 2008). Cortex or cell body glia ensheath neuronal cell bodies and form a supportive ‘trophospongium’ throughout the developing brain (Dumstrei et al., 2003). Neuropil glia wrap processes around axon fascicles in the neuropil and therefore function in a manner analogous to astrocytes and oligodendrocytes, although they do not generate myelin.
In contrast to embryonic gliogenesis, which has been well characterised (Giesen et al., 1997; Scholz et al., 1997), the genesis of glia in the Drosophila post-embryonic nervous system is poorly understood, particularly in the brain. Previous studies have shown that precursors of neuropil glia are located at the base of the embryonic brain primordium and spread along the extending neuropil during late embryogenesis. Precursors of brain surface and cortex glia originate during late embryogenesis from a small number of discrete clusters that migrate outwards to populate the entire brain (Hartenstein et al., 2008). After embryogenesis, the CNS remains in a largely quiescent state until the end of the first instar when neuroblasts begin to generate precursors that differentiate to expand the neuronal content of the nervous system (Sousa-Nunes et al., 2010). Gliogenesis restarts during the second instar and increases exponentially throughout the third instar (Awasaki et al., 2008; Pereanu et al., 2005). During this period, glia are generated both from neuroglioblast precursors and by glial cell division. Nascent cortex and neuropil glia migrate to reach their final positions and send out processes that ensheath neuronal cell bodies, axons and axon fascicles (Colonques et al., 2007; Pereanu et al., 2005). Although some types of post-embryonic gliogenesis have been characterised, including elegant studies in the visual system (Chotard et al., 2005; Chotard and Salecker, 2007), the mode and molecular pathways that control the genesis of the majority of glia during this stage have not been elucidated.
In this study we have investigated the genesis of distinct glial populations in the Drosophila post-embryonic brain. We find that glial proliferation occurs extensively in the larval brain. Using glial-specific mosaic analysis with a repressible cell marker [MARCM (Lee and Luo, 1999)] analysis we show that perineural glia and cortex glia divide in the larval CNS. The Insulin-like receptor (InR)/Target of rapamycin (TOR) pathway is required for the proliferation of perineural glia, where it acts synergistically with fibroblast growth factor (FGF) signalling. FGF signalling controls the generation of both perineural and cortex glia, but its requirement in each glial type differs. In addition, cortex, but not perineural, gliogenesis requires the expression of FGF in neurons. Our data suggest a model that describes the molecular control of cortex and perineural gliogenesis in the post-embryonic brain.
MATERIALS AND METHODS
Fly strains
Mutant stocks were: dilp668, dilp641 (Gronke et al., 2010); FRT82B,Ras85DΔC40B (Tseng et al., 2007); FRT82B,htlAB42 (Gisselbrecht et al., 1996); FRT82B,pntΔ88 (Klambt, 1993); pyr02915, ths02026 (Stathopoulos et al., 2004); FRT82B,dof1 (Vincent et al., 1998); FRT82B,InR31 (Brogiolo et al., 2001); FRT82B,Dp110A (Weinkove et al., 1999); FRT82B,Rheb2D1 (Garami et al., 2003); and FRT82B,Tsc1Q600X (Gao and Pan, 2001). Transgenic stocks were: UAS-pyr (Stathopoulos et al., 2004); a UAS-htl-IR line carrying two copies of the dsRNA insertion (Franzdottir et al., 2009) (the dsRNA has no predicted off-target effects); UAS-pyr-IR (Franzdottir et al., 2009) [this dsRNA has no predicted off-target effects and was used to generate the data described, while the line UAS-pyr-IR (VDRC 36523) gave similar phenotypes (data not shown)]; UAS-Dp110DN, UAS-Dp110CAAX (Weinkove et al., 1999); a line carrying two copies of UAS-Rheb (Garami et al., 2003); and repo-MARCM (Stork et al., 2008). UAS-htlDN, UAS-htlACT, UAS-InR, UAS-TorDN (TorTED), tub-Gal80ts, repo-Gal4, elavc155 (elav-Gal4), elavc155;UAS-Dcr2, UAS-p35 and UAS-cdc2-IR (36117) were all from the Bloomington Stock Center. NP6293-Gal4 (perineural glia specific), NP2222-Gal4 (cortex glia specific) and NP1079-Gal4 (dilp6-Gal4) were from the National Institute of Genetics Stock Center. The repo-MARCM stock genotype was repo-flp,repo-Gal4,UAS-actinGFP; FRT82B,tub-Gal80. The elav-MARCM stock genotype was hs-flp,elavc155; UAS-mCD8GFP,UAS-nlacZ; FRT82B,tub-Gal80. The heat-shock flp-MARCM stock genotype was hs-flp; tub-Gal4, UAS-mCD8GFP; FRT82B,tub-Gal80.
RNAi and overexpression studies
To enhance RNAi and overexpression phenotypes for all experiments (except pyr overexpression experiments and cdc2 RNAi) embryos were laid over 24 hours at 25°C and larvae were moved to 29°C 24 hours after egg laying. Clonal loss-of-function experiments were also performed in this way so that the conditions were identical to RNAi and overexpression experiments. Overexpression of pyr with repo-Gal4 or elav-Gal4 caused embryonic lethality; therefore, tub-Gal80ts was used to prevent expression until late first/early second instar (embryos were laid at 18°C and kept at this temperature for 3 days, then moved to 29°C for 3 days before dissection).
Immunohistochemistry and in situ hybridisation
Tissue preparation and staining were performed as described (Kaul and Bateman, 2009). Antibodies were: rat anti-PntP2 (see below, 1/500); mouse anti-Repo (DSHB, 1/100); mouse anti-β-Gal (Promega, 1/1000); rabbit anti-GFP (Molecular Probes, 1/1000); mouse anti-BrdU (DSHB, 1/100); rabbit anti-Htl (from A. Michelson, Harvard University, MA, USA, 1/2000); rabbit anti-PH3 (Upstate, 1/50); mouse anti-Miranda (from G. Tear, King's College London, London, 1/10); and 22C10 (DSHB, 1/100). Secondary antibodies were FITC donkey anti-mouse, Cy3 donkey anti-rat and Cy5 donkey anti-mouse (Jackson Immunolabs).
BrdU labelling was performed either by feeding third instar larvae food containing 3 mg/ml BrdU for 18 hours, or by incubating dissected brains in 75 μg/ml BrdU in PBS for 2 hours, then fixing and staining for Repo before incubating in 3M HCl for 30 minutes and staining for BrdU.
Imaging was performed on a Zeiss LSM 710 microscope and images were processed in Adobe Photoshop.
In situ hybridisations were performed as described previously (Silva et al., 2006).
PntP2 antibody
To generate the PntP2 antibody the PNT/SAM domain from pntP2 was amplified from BAC clone BACR01E13 using primers 5′-GAGGAGGGATCCCATGGATGCAAGGATCTGTG-3′ and 5′-AGACCGCTCGAGTGGTGGCCGACTCAAAGGAATTG-3′ and cloned into pGEX-4T-2 (Amersham) using BamHI and XhoI (underlined) to generate an N-terminal GST fusion construct. Purified GST-PNT protein was then used to generate antibodies.
Quantification of glia
To quantify superficial glia, 3-μm projections (z-projections of three 1-μm optical sections) of the dorsal and ventral surfaces of third instar larval brain hemispheres stained for Repo were used and quantified automatically using ImageJ (NIH). Dorsal and ventral values were combined to give the total superficial glia number per hemisphere, and six to eight hemispheres were quantified for each genotype. To quantify cortex glia, the total number of Repo/PntP2 co-expressing glia throughout each hemisphere were manually quantified in ImageJ. Clone sizes were quantified manually by counting the number of β-Gal-expressing nuclei in ImageJ. Glia were counted as associated with a clone if they were in contact with a GFP-positive cell or cells. Neuronal clone volumes were measured using Volocity (PerkinElmer).
Statistics
Significant differences were determined using Student's t-test.
RESULTS
Mitotic glial populations in the larval brain
Gliogenesis in the post-embryonic brain occurs mainly during the second and third larval instars but is poorly understood (Awasaki et al., 2008; Colonques et al., 2007; Pereanu et al., 2005). To probe for dividing glia we used BrdU incorporation and phospho-histone H3 (PH3) expression in the brain that colocalised with cells expressing the glial-specific protein Repo (Xiong et al., 1994). Similar to previous studies (Colonques et al., 2007; Pereanu et al., 2005; Read et al., 2009), we found frequent BrdU incorporation and occasional PH3 staining in glia in the superficial layer of the larval brain (Fig. 1A, supplementary material Fig. S1). The superficial layer of the larval brain contains three glial types: perineural, sub-perineural and cortex glia (Fig. 1B) (Awasaki et al., 2008; Stork et al., 2008). Perineural glia overlay sub-perineural glia, which function as the blood-brain barrier, whereas cortex glia ensheath neuronal cell bodies and neurites as they begin to project towards the neuropil. These data suggest that glia actively proliferate in the superficial layer of the brain during larval development.
Fig. 1.
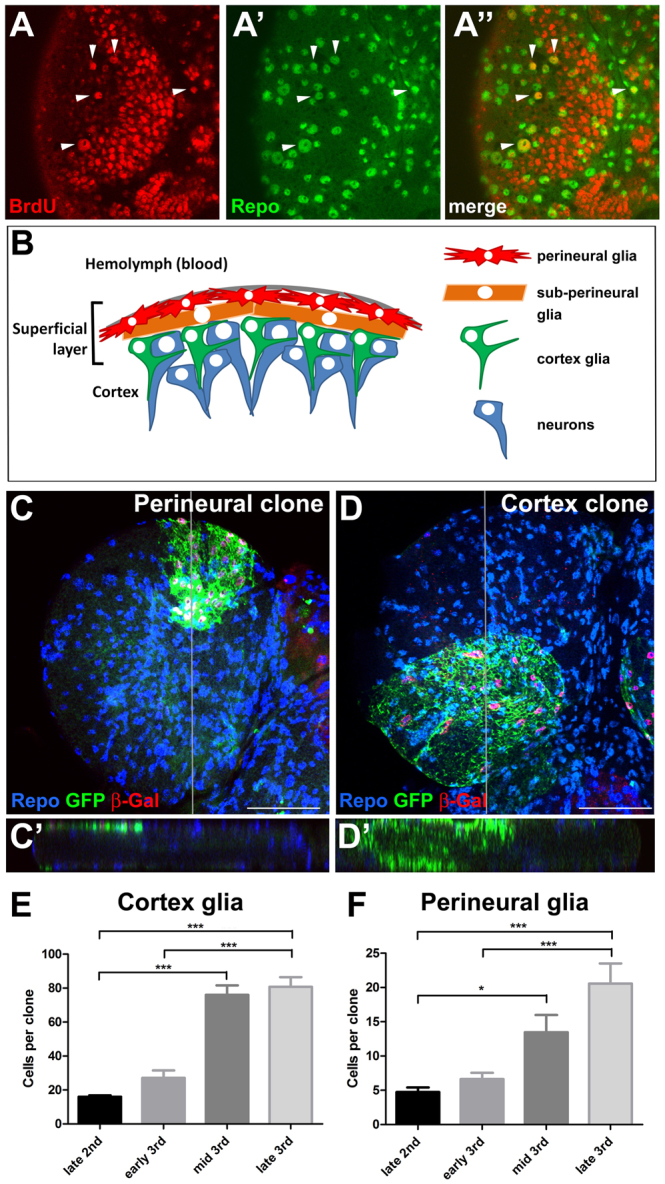
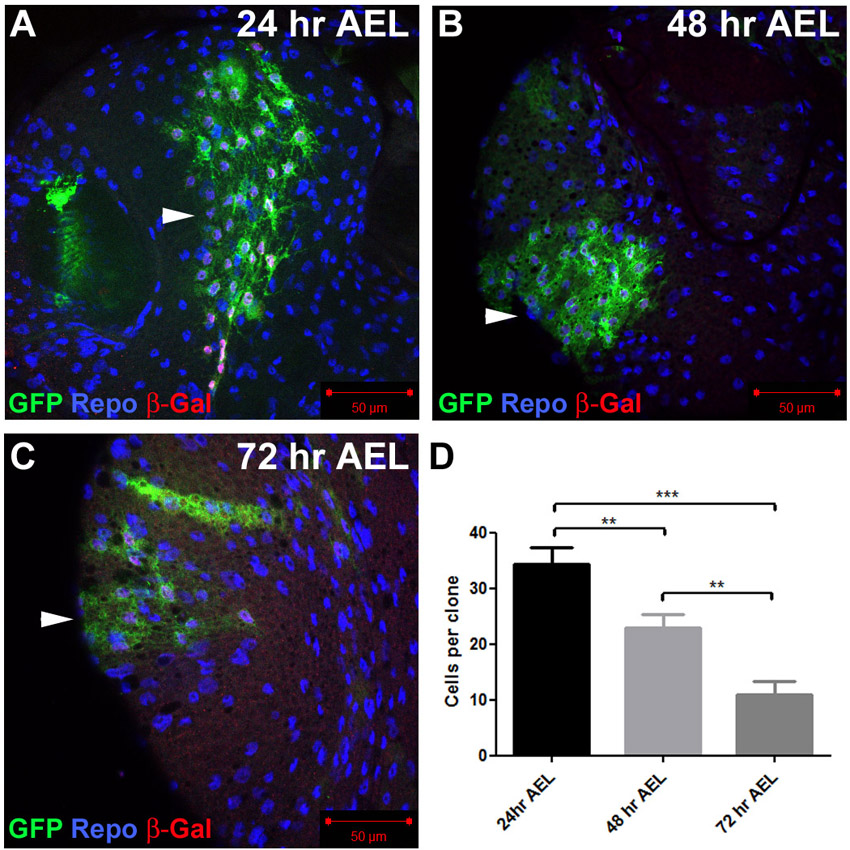
Glial proliferation in the Drosophila larval brain. (A-A″) Ventral superficial layer of a third instar larval brain stained for BrdU incorporation (red) and Repo expression (green) in glia. Arrowheads indicate glia that have incorporated BrdU. (B) Schematic of the superficial layer of the larval brain. Below the dense extracellular matrix, known as the neural lamella (grey), lie perineural glia (red) that are star-shaped and extend thin protrusions laterally but do not contact neurons (blue). Below the perineural layer are large sub-perineural cells (orange) connected by septate junctions required for the integrity of the blood-brain barrier. Cortex glia (green), which are found below the sub-perineural layer and throughout the brain, send out fine processes that ensheath neuronal cell bodies and neurites as they project between cell bodies towards the neuropil (supplementary material Fig. S2). (C) Perineural repo-MARCM clone in the dorsal surface of the larval brain stained for Repo (blue), GFP (green) and β-Gal (red) expression. (C′) Orthogonal view shows GFP expression in the perineural layer. (D) Cortex repo-MARCM clone in the ventral surface of the larval brain. (D′) Orthogonal view shows cortex glia processes extending into the brain. Note the difference in appearance to the perineural clone in C. Grey lines indicate positions of the orthogonal sections. (E,F) Average clone size of cortex glia (E) and perineural glia (F) during larval development. Error bars indicate s.e.m. *P<0.05; ***P<0.001. Scale bars: 50 μm.
To determine the specific mitotic glial populations we used the repo-MARCM system (Stork et al., 2008), which uses repo-flp to generate glial-specific MARCM clones. We identified two classes of glia in the superficial layer of the brain that divide extensively during the larval stage. First, clones of perineural glia were found exclusively on the surface of the optic lobe and central brain (Fig. 1B,C). Second, clones of cortex glia were observed with cell bodies both in the superficial layer and throughout the brain and with processes that enwrap neuronal cell bodies and projecting neurites (Fig. 1B,D, supplementary material Fig. S2). Clones of sub-perineural glia were rarely seen and contained only a few very large cells (data not shown). Thus, repo-MARCM analysis confirms that glial proliferation occurs in the superficial layer of the larval brain and identifies perineural and cortex glia as two distinct highly proliferative glial populations.
The proliferation of perineural glia has been shown previously to occur throughout the larval stages (Awasaki et al., 2008). To determine whether cortex glia actively divide during the larval stages we quantified the size of repo-MARCM clones during the late second and third instar stages. To accurately quantify the number of cells in repo-MARCM clones and avoid effects of cell migration, we introduced a nuclear-localised lacZ-expressing transgene so that the cell number could be determined by counting nuclei (for examples see Fig. 1C,D). This analysis showed that the average clone size of cortex glia increased over fourfold between late second and late third instar (Fig. 1E). Thus, similar to perineural glia (Fig. 1F) (Awasaki et al., 2008), cortex glia proliferate extensively during the post-embryonic larval stages. To ensure that clones were generated from a single progenitor cell, we also induced MARCM clones using heat-shock flp (hs-MARCM), which confirmed that perineural glia proliferate throughout the larval stage (supplementary material Fig. S3). Similar to Awasaki et al. (Awasaki et al., 2008), we did not observe cortex clones using hs-MARCM. The reason for this is not clear, but cortex glia clones occur much less frequently than perineural clones using repo-MARCM, suggesting that cortex glia proliferation begins from a smaller pool of progenitor cells.
To determine whether there was any regional effect on perineural or cortex gliogenesis we quantified repo-MARCM clones separately in the central brain (CB) and optic lobe (OL). We did not find any significant difference in size between CB and OL clones or clones that spanned these regions for perineural clones, or between cortex OL clones and cortex clones that spanned the OL and CB (supplementary material Fig. S4). We did not find cortex clones that were exclusively within the CB (supplementary material Fig. S4), suggesting that cortex clones always derive from cells within the OL. These data suggest that perineural and cortex glia proliferate at a similar rate throughout the larval brain.
InR/TOR signalling is required for the proliferation of perineural glia
TOR is a large serine/threonine kinase that typically acts downstream of InR and PI3K (supplementary material Fig. S6A), and InR/TOR signalling has been implicated in glial cancer and in a Drosophila glioma model (Furnari et al., 2007; Read et al., 2009). In Drosophila there are seven insulin-like peptides (DILPs; Ilp1-7 – FlyBase) (Gronke et al., 2010). Immunostaining for DILP2 or analysis of enhancer trap lines showed that neither dilp2 nor dilp3 is expressed in glia in late third instar larvae (data not shown). However, dilp6 has recently been shown to be expressed in glia in the second instar CNS (Sousa-Nunes et al., 2011). We found by in situ hybridisation analysis that dilp6 was strongly expressed throughout the CNS in the early third instar (supplementary material Fig. S5), although expression was barely detectable by the end of the third instar (not shown). Moreover, a Gal4 enhancer in the dilp6 locus drove expression in the majority of glia in the larval brain (Fig. 2A). Thus, InR/TOR signalling may be active in gliogenesis during the third instar.
Fig. 2.
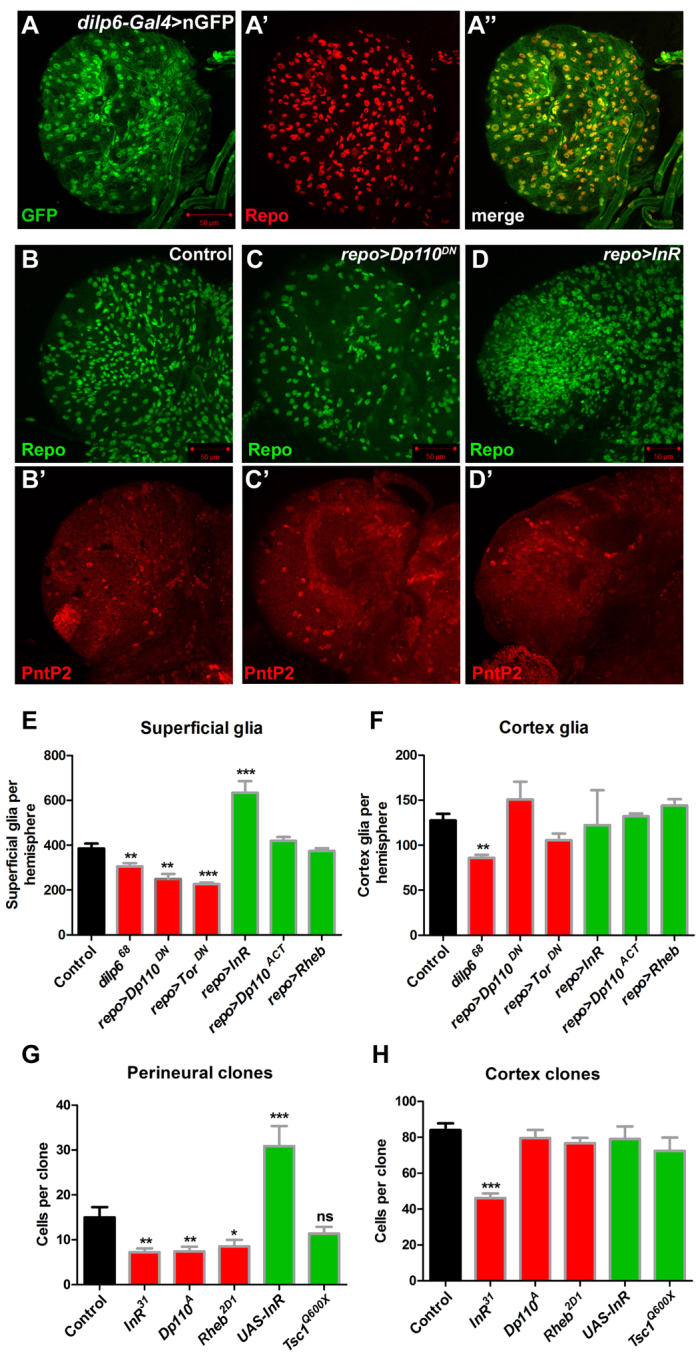
InR/TOR signalling is required for perineural gliogenesis. (A-A″) dilp6-Gal4 driving nuclear GFP (green) colocalises with Repo expression (red) in glia in the Drosophila larval brain. (B-D′) The ventral superficial layer of larval brain stained for Repo (green) or PntP2 (red) from control (B,B′), or repo-Gal4 driving expression of (C,C′) a dominant-negative form of Dp110 (repo>Dp110DN) or (D,D′) InR (repo>InR). (E,F) Quantification of superficial (E) or cortex (F) glia from homozygous dilp6 mutant larvae (dilp668) or brains expressing inhibitors (red bars) or activators (green bars) of the InR/TOR pathway in glia using repo-Gal4; control is repo-Gal4/+ (G) Quantification of perineural repo-MARCM clone size for FRT82B controls (n=71), InR31 (n=40), Dp110A (n=38), Rheb2D1 (n=53), UAS-InR (n=52) or Tsc1Q600X (n=46). (H) Quantification of cortex repo-MARCM clone size for FRT82B controls (n=15), InR31 (n=19), Dp110A (n=16), Rheb2D1 (n=10), UAS-InR (n=13) or Tsc1Q600X (n=7). Error bars indicate s.e.m. *P<0.05; **P<0.01; ***P<0.001; ns, not significant.
To test whether InR/TOR signalling is required in superficial glia we inhibited this pathway using mutants in dilp6, or by expressing a dominant-negative form of the catalytic subunit of PI3K, Dp110 (Pi3K92E – FlyBase) (Dp110DN), or a dominant-negative form of TOR (TorDN) in glia using repo-Gal4. Since no perineural-specific markers have been identified we quantified Repo-expressing glia in 3-μm dorsal and ventral z-projections of the superficial layer of the larval brain (see Materials and methods). Quantification using a perineural-specific Gal4 driver (NP6293-Gal4) to express nGFP showed that the majority [74±9% (±s.d.), n=4 brain hemispheres] of Repo-expressing glia imaged using this method were perineural glia (supplementary material Fig. S6B). Loss-of-function (LOF) mutants in dilp6 (Fig. 2E; data not shown) or expression of either Dp110DN or TorDN in glia caused a significant decrease in the number of superficial glia (Fig. 2C,E). Moreover, overexpression of InR with repo-Gal4 caused a dramatic increase in the number of superficial glia (Fig. 2D,E). Surprisingly, expression of an activated form of Dp110 (Dp110CAAX), or overexpression of the small GTPase Rheb, which activates TOR, did not alter the number of superficial glia (Fig. 2E). Therefore, the InR/TOR pathway is required for perineural gliogenesis, but only overexpression of the receptor can promote perineural glia proliferation.
Cortex glia were quantified by counting the total number of glia throughout the brain expressing a specific isoform of the ETS domain transcription factor Pointed, Pointed P2 (PntP2) (see Materials and methods), which is expressed in cortex glia and some neurons (see below and Fig. 3C,D). Only loss of dilp6 caused a decrease in cortex glia numbers, and overexpression of InR or of downstream components of the InR/TOR pathway did not significantly alter the number of PntP2-expressing cortex glia (Fig. 2C′,D′,F). Thus, cortex glia require the ligand but not intracellular components of the InR/TOR pathway.
Fig. 3.
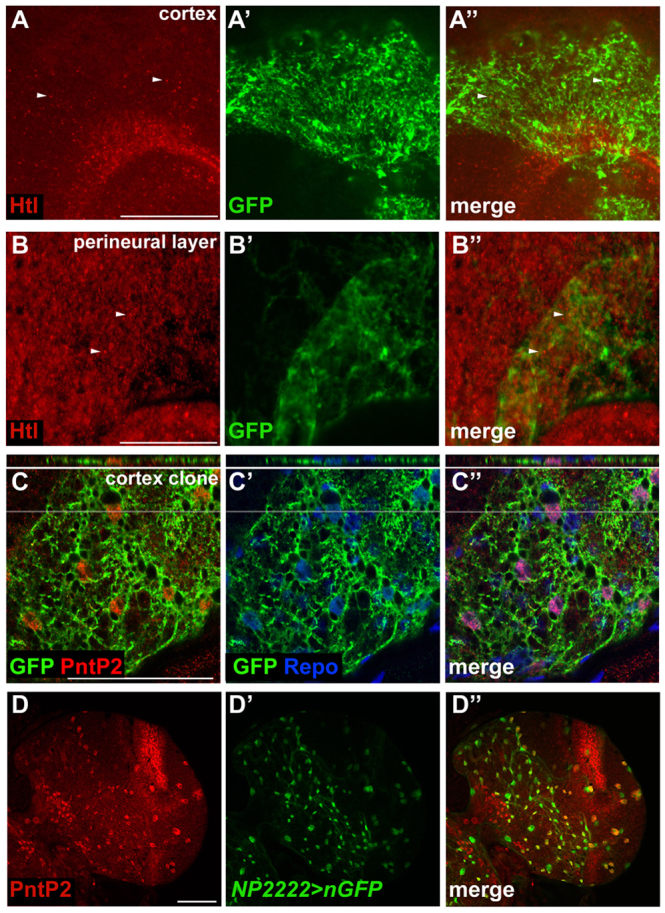
Expression of FGF pathway components in the larval brain. (A-B″) Htl expression (red) in glia in the cortex (A-A″) or perineural (B-B″) layers of the Drosophila larval brain. repo-MARCM clones expressing GFP (green) were used to identify the respective glial types. (C-C″) Third instar larval brain hemisphere showing a repo-MARCM cortex clone stained for GFP (green), PntP2 (red) and Repo (blue) expression. Note that PntP2-positive cortex glia colocalise with GFP in the orthogonal view (top; grey lines indicate the position of orthogonal section). (D-D″) Cortex glia-specific NP2222-Gal4 driving UAS-nGFP (green) showing colocalisation with PntP2 expression (red). Scale bars: 50 μm.
Clonal analysis demonstrates different requirements for InR/TOR signalling in cortex and perineural glia
To test more specifically whether the InR/TOR pathway is required for perineural glia proliferation, LOF repo-MARCM clones were generated for InR (InR31), Dp110 (Dp110A) and Rheb (Rheb2D1), thereby inhibiting the pathway, or for the inhibitor Tsc1 (Tsc1Q600X), resulting in pathway activation. The size of mutant clones was compared with that of wild-type (control) clones to determine the requirement of each of these genes for glial proliferation. The size of perineural clones was reduced by about half upon loss of either InR, Dp110 or Rheb (Fig. 2G), confirming that the InR/TOR pathway is required for the proliferation of perineural glia. In accordance with the pan-glial activation of the TOR pathway using repo-Gal4, Tsc1 mutant perineural clones were not significantly different to controls (Fig. 2G), confirming that activation of the TOR pathway is insufficient to increase perineural gliogenesis. However, overexpression of InR caused a doubling in perineural clone size (Fig. 2G), suggesting that the receptor can activate perineural glia proliferation through an alternative pathway.
We also quantified the size of cortex clones to determine whether the requirement for InR/TOR signalling was specific to perineural glia. Cortex clones mutant for Dp110, Rheb or Tsc1 were similar in size to control clones (Fig. 2H), so PI3K/TOR signalling is not required in these cells. By contrast, clones mutant for InR were about half the size of control clones (Fig. 2H). InR is therefore required in part for cortex glia proliferation, whereas the downstream PI3K/TOR pathway is not.
Together, these data demonstrate that the proliferation of perineural glia is reduced, but not completely blocked, by inhibition of InR/TOR signalling. In cortex glia, only InR is required for proliferation and its loss abrogates, but does not completely block, proliferation. We therefore hypothesised that a second pathway might also regulate the genesis of perineural and cortex glia.
FGF signalling is necessary and sufficient for superficial gliogenesis
FGF signalling (supplementary material Fig. S7A) has been shown to specify oligodendrocyte precursors and to regulate proliferation, migration and differentiation in the Drosophila eye imaginal disc (Chandran et al., 2004; Franzdottir et al., 2009; Kessaris et al., 2004). There are two FGF receptors (FGFRs) in Drosophila, Heartless (Htl) and Breathless (Btl). btl is expressed exclusively in trachea in the larval brain (data not shown). We used an antibody against Htl to determine its expression in cortex and perineural glia. Staining with this antibody was strongly reduced in brains in which Htl expression in glia was knocked down by RNAi (supplementary material Fig. S8). Htl was widely expressed in the larval brain in both cortex and perineural glia and, similar to a previous study (Wilson et al., 2004), was seen in punctuate structures (Fig. 3A,B). In contrast to the pan-glial expression of Htl, PntP2, which acts downstream in the FGF pathway (Ghabrial et al., 2003), was expressed in cortex glia (where it colocalised with GFP expression in repo-MARCM cortex clones or with GFP expression driven by the cortex-specific NP2222-Gal4 driver), but not in perineural glia (Fig. 3C,D; data not shown). There are two FGF ligands in Drosophila, Pyramus (Pyr) and Thisbe (Ths), which have overlapping functions in embryonic mesoderm specification (Stathopoulos et al., 2004). In third instar larvae, the pyr transcript is expressed throughout the OLs and CB, but not in the ventral nerve cord (VNC) (supplementary material Fig. S7B). ths transcript was expressed in the OL and CB, but unlike pyr, was also expressed in cell bodies in the VNC (supplementary material Fig. S7C). Together, these data demonstrate that several FGF pathway components are expressed in the larval brain, suggesting that FGF signalling might be active during this stage.
The requirement for the Htl receptor in glia was tested by RNAi of htl or overexpression of htlDN using repo-Gal4. Both of these manipulations caused a significant reduction in the number of superficial glia (Fig. 4B,G). The RNAi phenotype was confirmed with an independent non-overlapping dsRNA (data not shown). Conversely, overexpression of an activated form of Htl (htlACT) caused dramatic overproliferation of superficial glia (Fig. 4C,G). These data suggest that the FGFR Htl is necessary and sufficient for the genesis of superficial glia.
Fig. 4.

FGF signalling is necessary and sufficient for glial proliferation. (A-C′) Ventral superficial layer of Drosophila larval brain stained for Repo (green) and PntP2 (red, to visualise cortex glia) expressing the following transgenes in glia: (A) repo-Gal4/+ control, (B) dsRNA against htl (repo>htl-IR), (C) an activated form of the Htl receptor (repo>htlACT). (D,D′) pyr02915 homozygous larval brain stained as in A. (E-F′) The ventral superficial layer of larval brains expressing either dsRNA against pyr (elav>pyr-IR) (E), or wild-type pyr (elav>pyr) (F) in neurons, stained as in A. (G) Superficial glia numbers from larvae with repo-Gal4 driving the expression of a dsRNA against htl (repo>htl-IR), a dominant-negative form of Htl (repo>htlDN), an activated form of Htl (repo>htlACT), a dsRNA against pyr (repo>pyr-IR), or wild-type pyr (repo>pyr), or in mutants for pyr (pyr02915) or ths (ths02026). (H) Quantification of cortex glia numbers in the same brains as in G. (I) Quantification of superficial glia numbers in larvae with elav-Gal4 driving the expression of a dsRNA against pyr (elav>pyr-IR), a dsRNA against htl (elav>htl-IR), wild-type pyr (elav>pyr), dsRNA against cdc2 (elav>cdc2-IR), or combined cdc2 dsRNA and pyr (elav>cdc2-IR,pyr) in neurons. (J) Quantification of cortex glia numbers in the same brains as in I. Green bars indicate pathway activation, red bars indicate pathway inhibition. Error bars indicate s.e.m. *P<0.05; **P<0.01; ***P<0.001.
We next sought to determine whether either of the canonical FGF ligands is required for superficial gliogenesis. Superficial glia were quantified in LOF mutants for both pyr and ths. pyr02915 homozygous larvae had significantly reduced numbers of superficial glia (Fig. 4D,G), whereas superficial glia numbers in ths02026 homozygous larval brains were similar to those in the control (Fig. 4G). To confirm the requirement for pyr, a dsRNA against pyr was expressed using repo-Gal4. These larvae had moderately reduced numbers of superficial glia (Fig. 4G). This was confirmed using an independent non-overlapping dsRNA (data not shown; see Materials and methods). Moreover, overexpression of pyr with repo-Gal4 (see Material and methods) caused significant overproliferation of superficial glia (Fig. 4G). Therefore, Pyr is necessary and sufficient to activate superficial glia proliferation in the larval brain.
The requirement for FGF signalling in cortex gliogenesis was determined by quantifying PntP2-expressing cortex glia. Loss or inhibition of htl caused an almost complete loss of cortex glia (Fig. 4B′,H), whereas overexpression of htlACT caused strong overproliferation of cortex glia throughout the brain (Fig. 4C′,H). As with perineural glia, pyr, but not ths, was required in cortex glia, as pyr02915 homozygous larvae had a near-complete loss of cortex glia (Fig. 4D′,H), whereas ths02026 homozygous larval brains were similar to wild type (Fig. 4H). This was confirmed by knockdown of Pyr in glia, which caused a dramatic reduction in the number of cortex glia (Fig. 4H). Similar to overexpression of the receptor, overexpression of pyr caused a substantial increase in cortex gliogenesis (Fig. 4F′,H).
We next addressed whether there is an autocrine requirement for pyr in glia by knockdown of Pyr in repo-MARCM clones. However, both cortex and perineural pyr RNAi clones were similar in size to controls (see Fig. 7A,B), suggesting that Pyr is not required cell-autonomously for glial proliferation. In accordance with this, pyr-overexpressing repo-MARCM clones were similar in size to control clones (see Fig. 7A,B), whereas glia surrounding the clones overproliferated (Fig. 5B,D, supplementary material Fig. S9). This non-cell-autonomous overproliferation was restricted by glial subtype, as perineural clones overexpressing pyr did not induce cortex glia overproliferation and cortex glia pyr expression clones did not induce perineural glia proliferation (Fig. 5D; data not shown). These data suggest that FGF signalling controls gliogenesis through a paracrine mechanism in which Pyr activates the Htl receptor non-cell-autonomously on glia of the same subtype.
Fig. 7.
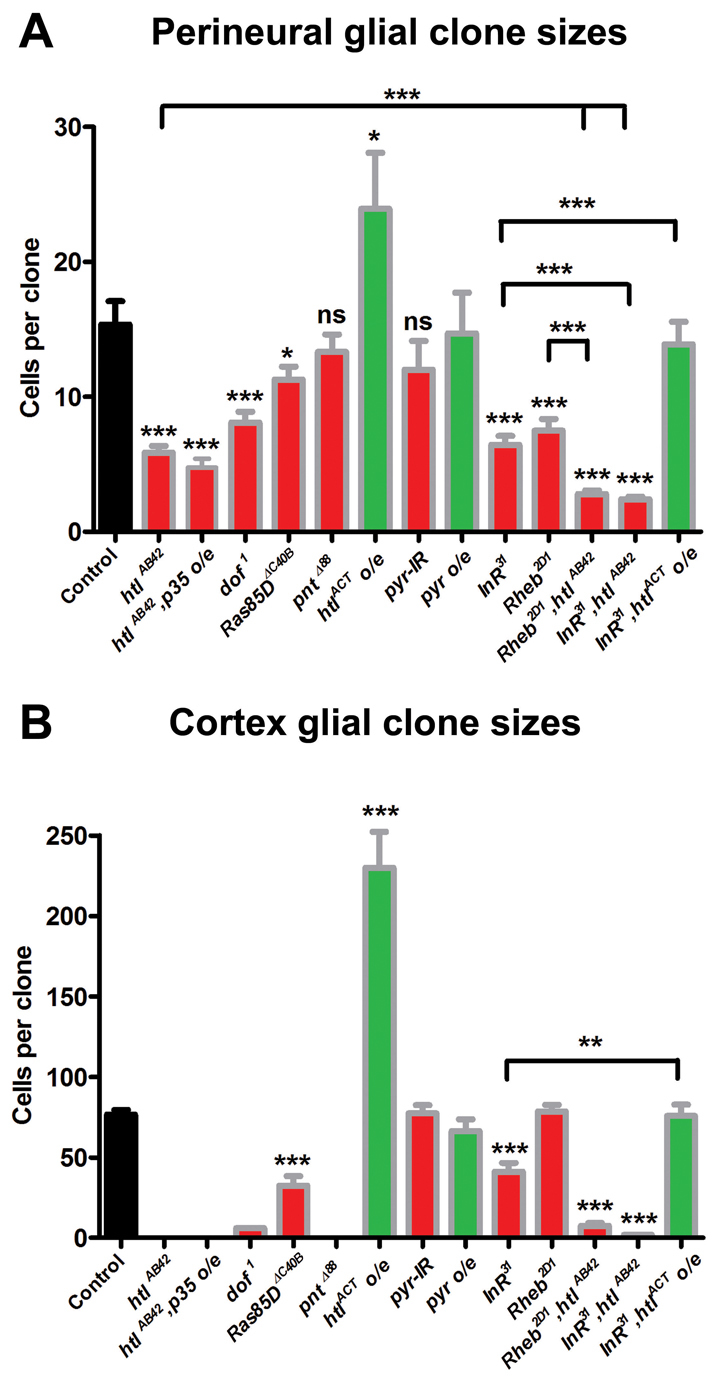
FGF signalling acts together with InR/TOR signalling to control the genesis of perineural and cortex glia. (A) Quantification of perineural repo-MARCM clone size. Average clone size of FRT82B control clones (n=71), htlAB42 (n=28), htlAB42,p35 overexpression (htlAB42,p35 o/e, n=28), dof1 (n=63), Ras85DΔC40B (n=88), pntΔ88 (n=67), htlACT overexpression (htlACT o/e, n=23), pyr RNAi (pyr-IR, n=31), pyr overexpression (pyr o/e, n=20), InR31 (n=37), Rheb2D1 (n=66), Rheb2D1, htlAB42 (n=74), InR31, htlAB42 (n=54), and InR31,htlACT overexpression (InR31,htlACT o/e, n=41). (B) Quantification of cortex repo-MARCM clone size. FRT82B control clones (n=13), dof1 (n=1), Ras85DΔC40B (n=10), htlACT overexpression (htlACT o/e, n=5), pyr-IR (n=4), pyr overexpression (pyr o/e, n=7), InR31 (n=12), Rheb2D1 (n=15), Rheb2D1, htlAB42 (n=8), InR31, htlAB42 (n=5) and InR31,htlACT overexpression (InR31,htlACT o/e, n=7). No cortex clones were observed for htlAB42 (in over 100 hemispheres), htlAB42,p35 overexpression (in 60 hemispheres) or pntΔ88 (in 56 hemispheres). One six-cell clone was observed in 42 hemispheres for dof1. Green bars indicate pathway activation, red bars indicate pathway inhibition. Error bars indicate s.e.m. *P<0.05; **P<0.01; ***P<0.001; ns, not significant.
Fig. 5.
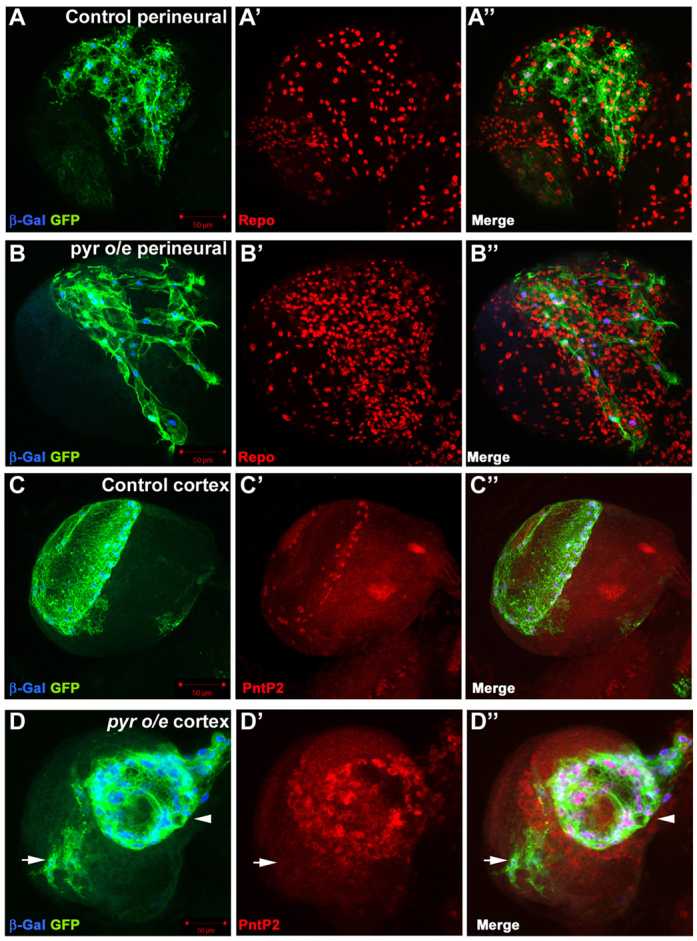
pyr overexpression in glia causes non-cell-autonomous glial proliferation. (A-B″) Control (A-A″) or pyr-overexpressing (B-B″) repo-MARCM perineural clones stained for β-Gal (blue) and Repo (red) expression. Note the extended processes (B) and the increased number of glial cells surrounding the clone (B′,B″). (C-D″) Control (C-C″) or pyr-overexpressing (arrowhead in D,D″) repo-MARCM cortex clones stained for β-Gal (blue) and PntP2 (red) expression. Note the non-cell-autonomous proliferation of PntP2-expressing cortex glia, clustered cell bodies and shortened processes in the pyr-overexpressing cortex clone (arrowhead in D,D″). Note also that the adjacent small perineural clone overexpressing pyr (arrow in D-D″) does not cause cortex glia proliferation.
pyr overexpression did have a strong cell-autonomous effect on perineural glia morphology, inducing the formation of elongated processes (Fig. 5B) that were not observed in control clones (Fig. 5A) or in clones expressing htlACT (data not shown). This phenotype might reflect the role of FGF signalling in glial cell migration (Franzdottir et al., 2009). Surprisingly, pyr overexpression had the opposite effect on cortex clones, causing shortened processes and inducing cortex glia cell bodies to cluster together (Fig. 5D) more than in control clones (Fig. 5C).
Neuronal pyr expression is required for cortex gliogenesis
We hypothesised that neurogenesis and cortex gliogenesis might be coordinated via FGF signalling. To test this we knocked down Pyr in neurons using elav-Gal4. Interestingly, RNAi of pyr did not cause a significant reduction in the number of superficial glia (Fig. 4E,I), but did result in a dramatic decrease in the number of cortex glia in the larval brain (Fig. 4E′,J). To determine whether this phenotype was a secondary consequence of a requirement for FGF signalling in neurons we knocked down Htl in neurons. RNAi of htl with elav-Gal4 did not affect superficial glia or cortex glia numbers (Fig. 4I,J), demonstrating that inhibition of FGF signalling in neurons does not affect gliogenesis. If gliogenesis is mediated by neuronal FGF expression then overexpression of FGF in neurons should increase glial numbers. Neuronal overexpression of pyr using elav-Gal4 resulted in a dramatic increase in the number of cortex glia (Fig. 4F′,J), most of which were located below the superficial layer (not shown), but caused no significant change in the number of superficial glia (Fig. 4F,I).
The effect of neuronal pyr overexpression on glia could be indirect. To test whether neuronal pyr expression could affect gliogenesis directly we generated elav-MARCM clones that overexpressed pyr in neurons. These clones were not significantly larger than control clones (Fig. 6C), but had dramatically increased numbers of glia clustered around the pyr-overexpressing neurons (Fig. 6B,D). Furthermore, knockdown of Cdc2 using elav-Gal4 caused a reduction in the number of neuroblasts (supplementary material Fig. S10) and cortex glia (Fig. 4J, supplementary material Fig. S10), but knockdown of Cdc2 combined with pyr overexpression still resulted in overproliferation of cortex glia (Fig. 4J, supplementary material Fig. S10). Taken together, these data suggest that developing cortical neurons signal directly to adjacent cortex glia by secreting Pyr to control cortex gliogenesis via the FGF pathway.
Fig. 6.
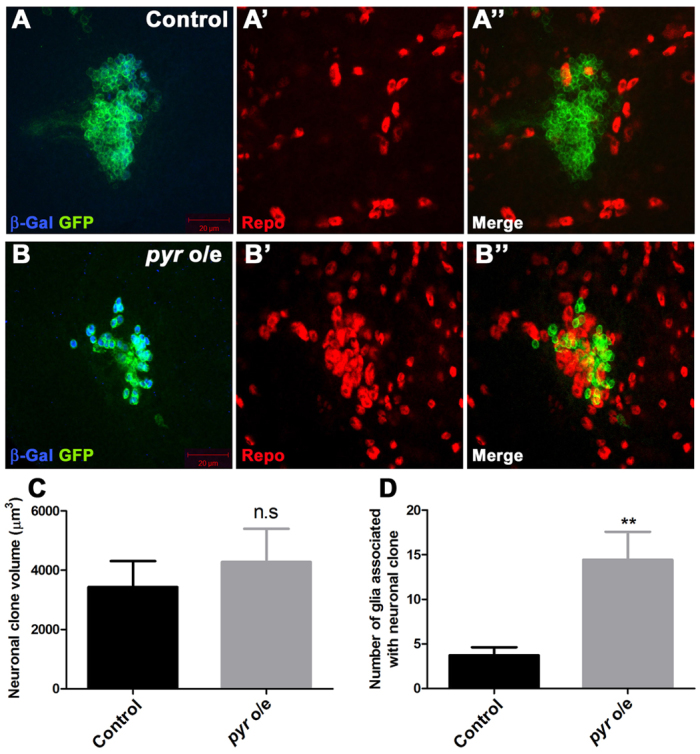
pyr overexpression in neurons induces gliogenesis. (A-B″) Control (A-A″) or pyr-overexpressing (B-B″) elav-MARCM clones stained for β-Gal (blue) and Repo (red) expression. Note the cluster of glial cells surrounding the clone in B″. (C) Quantification of neuronal clone volume for control or pyr-overexpressing neuronal clones (pyr o/e) (n=10 clones per genotype). (D) Quantification of glia associated with neuronal clones for control or pyr-overexpressing neuronal clones (pyr o/e) (n=10 clones per genotype). Error bars indicate s.e.m. **P<0.01; n.s, not significant.
Clonal analysis shows that FGF signalling is partially required in perineural glia and absolutely required in cortex glia
repo-MARCM analysis was used to further elucidate the contribution of FGF signalling to the generation of perineural and cortex glia. LOF mutant perineural clones for the FGFR htl (htlAB42) or downstream of FGF (stumps – FlyBase) (dof1), an essential intracellular FGF pathway component, were about half the size of control clones, whereas Ras85D LOF (Ras85DΔC40B) perineural clones were ~75% the size of controls (Fig. 7A), suggesting that, like InR/TOR signalling, FGF signalling is only partially required for normal levels of proliferation in these cells. To investigate whether FGF signalling is required to maintain perineural glia by preventing apoptosis, the caspase inhibitor p35 was expressed in htl perineural clones. Expression of p35 did not rescue the phenotype (Fig. 7A), suggesting that FGF signalling regulates proliferation rather than cell death. LOF perineural clones for the ETS transcription factor pnt (pntΔ88) were not significantly different to controls (Fig. 7A), as would be expected from the lack of PntP2 expression in perineural glia (data not shown). Activation of FGF signalling in perineural glia was sufficient to increase their proliferation, as expression of htlACT in perineural glia significantly increased clone size (Fig. 7A). These data show that FGF signalling is sufficient, but only partially required, for perineural glia proliferation.
In contrast to perineural glia, repo-MARCM analysis demonstrated an almost absolute requirement for FGF signalling in cortex glia. LOF mutant cortex clones for either htl or pnt were not observed (Fig. 7B). One LOF dof mutant clone was obtained, but this was dramatically smaller than control clones (Fig. 7B). Therefore, loss of FGF signalling almost entirely prevents the gliogenesis and/or maintenance of cortex glia. LOF mutant cortex clones for Ras85D were obtained, but were significantly smaller than controls (Fig. 7B), demonstrating that Ras85D is required for cortex glia proliferation or maintenance but might function semi-redundantly. As with perineural clones, expression of p35 in htl cortex clones did not rescue the phenotype (Fig. 7B), suggesting that htl is required either for the early genesis of cortex glia or to prevent a type of cell death that is not blocked by p35 expression. We also analysed repo-MARCM htl cortex clones earlier in development (in early third instar brains), but did not observe cortex clones at this stage (data not shown), suggesting that htl is required for proliferation from the early stages of cortex gliogenesis. To determine whether FGF signalling is sufficient for cortex glia proliferation, repo-MARCM clones were generated expressing htlACT. Cortex clones expressing htlACT were, on average, three times the size of control clones (Fig. 7B), confirming that FGF signalling is sufficient to drive the proliferation of cortex glia. These data confirm that FGF signalling is essential for the proliferation of cortex glia in the larval brain.
Combined InR/TOR and FGF signalling control perineural and cortex gliogenesis through different mechanisms
To examine the relationship between the InR/TOR and FGF pathways we generated double mutants for components of both pathways and compared the size of these with single repo-MARCM mutant perineural or cortex clones. Proliferation was almost completely inhibited in htl,Rheb or htl,InR perineural clones (Fig. 7A), suggesting that the FGF and InR/TOR pathways act synergistically and are wholly responsible for the proliferation of perineural glia. By contrast, htl,Rheb and htl,InR double-mutant cortex clones were rare and when observed were extremely small, a characteristic of the loss of FGF signalling (Fig. 7B).
To investigate the interaction of the two pathways further, repo-MARCM clones were generated that were mutant for InR and overexpressed htlACT. The reduced proliferation caused by loss of InR was ameliorated in perineural and cortex clones by expression of htlACT, so that clones were similar in size to control clones, but were smaller than clones expressing htlACT alone (Fig. 7A,B). These epistatic analyses suggest that InR/TOR and FGF signalling act in parallel in both perineural and cortex glia and that these two cell types use distinct signalling circuits to drive proliferation during larval development.
DISCUSSION
The correct control of gliogenesis is crucial to CNS development and the Drosophila post-embryonic nervous system is a powerful model for elucidating the molecular players that control this process. We have identified two separate glial populations that proliferate extensively and have defined the key molecular players that control their genesis and proliferation. Perineural and cortex glia both use insulin and FGF signalling in a concerted manner, but the requirements for these pathways are different in each glial type. Our data suggest a model that describes the molecular requirements for post-embryonic gliogenesis in each of these glial types in the brain (Fig. 8).
Fig. 8.
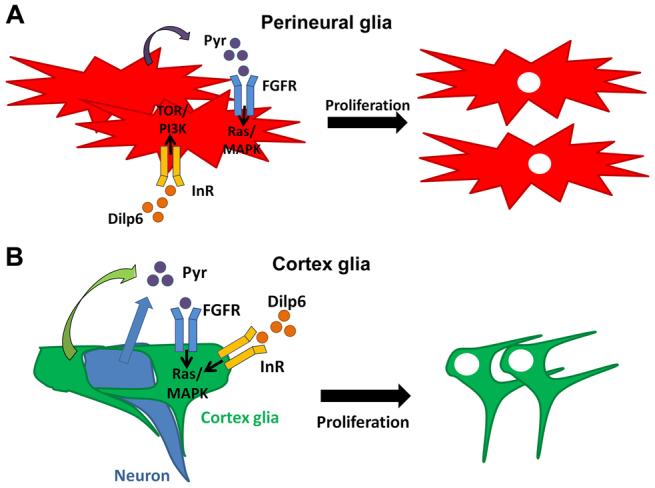
A model for the control of perineural and cortex glia proliferation in the Drosophila larval brain. (A) Pyr is expressed by perineural glia to activate FGF signalling in adjacent glia and acts in parallel to InR/TOR signalling (activated by the expression of Dilp6). These two pathways act synergistically to generate the correct complement of perineural glia. (B) Cortex glia proliferation is controlled by FGF signalling through FGFR (Htl) and the Ras/MAPK pathway. Pyr expression is required from both glia (green) and neurons (blue) and acts non-cell-autonomously. Neuronal Pyr expression activates the FGFR on adjacent cortex glia, thereby coordinating neurogenesis and glial proliferation. InR is also partially required in cortex glia and is likely to signal through the Ras/MAPK pathway.
Using both pan-glial inhibition and LOF clonal analysis we showed that the InR/TOR pathway is required for perineural glia proliferation. InR/TOR signalling has widespread roles in nervous system development (Bateman and McNeill, 2006) and we have previously demonstrated a role for this pathway in the temporal control of neurogenesis (Bateman and McNeill, 2004; McNeill et al., 2008). InR can be activated by any one of seven DILPs encoded by the Drosophila genome (Brogiolo et al., 2001), which can act redundantly by compensating for each other (Gronke et al., 2010). We find that dilp6 is expressed in most glia during larval development, including perineural and cortex glia, and that dilp6 mutants have reduced gliogenesis (Fig. 2). The dilp6 phenotype is weaker than that associated with the inhibition of downstream components of the InR/TOR pathway, suggesting that other DILPs might be able to compensate for the absence of dilp6 expression in glia (Gronke et al., 2010). Pan-glial inhibition and clonal analysis also demonstrated that the FGF pathway is required for normal levels of perineural glia proliferation. FGF signalling is activated in perineural glia by paracrine expression of Pyr. Inhibition of either the InR/TOR or FGF pathway reduced perineural glia proliferation by about half, so we tested whether these two pathways act together. Our data demonstrate that inhibition of both pathways simultaneously has a synergistic effect, suggesting that these two pathways act in parallel, rather than sequentially, and that their combined activities generate the large numbers of perineural glia found in the adult brain (Fig. 8A).
Cortex glia employ a molecular mechanism distinct from that of perineural glia to regulate their proliferation (Fig. 8B). Cortex glia have a clear requirement for InR, as InR mutant cortex clones are significantly reduced in size. The early events in post-embryonic gliogenesis are poorly understood, but FGF signalling is likely to be required during this stage as LOF clones for components of this pathway almost completely block cortex gliogenesis. Our data suggest that InR acts in parallel to FGF signalling in these cells, as loss of InR combined with activation of FGF signalling only partially rescues the InR phenotype. Interestingly, the PI3K/TOR pathway is not required in cortex glia, suggesting that InR signals through the Ras/MAPK pathway to control cortex glia proliferation.
The FGF pathway in cortex glia responds to paracrine Pyr expression from both glia and neurons. Expression from both glia and neurons is required to activate the pathway and stimulate cortex gliogenesis (Fig. 8B). Neuronal regulation of glial FGF signalling enables cortical neurogenesis to modulate the rate of gliogenesis, so that the requisite number of glia are generated to correctly enwrap and support developing cortical neurons. Recent studies have also identified a mechanism by which DILP secretion by glia controls neuroblast cell-cycle re-entry in the Drosophila early post-embryonic CNS (Chell and Brand, 2010; Sousa-Nunes et al., 2011). Thus, neurons and glia mutually regulate each other's proliferation to coordinate correct brain development.
We have shown that two major glial populations in the larval brain, perineural and cortex glia, are generated by glial proliferation rather than differentiation from neuroglioblast or glioblast precursors. Differentiation of most embryonic glia from neuroglioblasts in the VNC requires the transcription factor glial cells missing (gcm) (Hosoya et al., 1995; Jones et al., 1995), which is both necessary and sufficient for glial cell fate. In the larval brain the role of gcm is much more restricted and it is not expressed in, nor required for, generation of perineural glia (Awasaki et al., 2008; Colonques et al., 2007). Thus, the developmental constraints on gliogenesis in the embryonic and larval CNS are distinct. The larval brain undergoes a dramatic increase in size during the third instar, which might favour a proliferative mode, rather than continuous differentiation from a progenitor cell type.
Glial dysfunction is a major contributor to human disease. The release of toxic factors from astrocytes has been suggested to be a contributory factor in amyotrophic lateral sclerosis and astrocytes might also play a role in the clearance of toxic Aβ in Alzheimer's disease (Nagai et al., 2007; Nicoll and Weller, 2003). Rett syndrome is an autism spectrum disorder caused by LOF of the transcription factor methyl-CpG-binding protein 2 (MeCP2) (Chahrour and Zoghbi, 2007). Astrocytes from MeCP2-deficient mice proliferate slowly and have been suggested to cause aberrant neuronal development (Maezawa et al., 2009). This hypothesis was recently confirmed by astrocyte-specific re-expression of Mecp2 in MeCP2-deficient mice, which improved the neuronal morphology, lifespan and behavioural phenotypes associated with Rett syndrome (Lioy et al., 2011). Characterisation of the molecular control of gliogenesis during development might lead to a better understanding of such diseases.
Supplementary Material
Acknowledgements
We thank Christian Klämbt for the repo-MARCM stock; Alex Gould and Rita Sousa-Nunes for valuable advice and stocks; and Tzumin Lee, Alan Michelson, Angelike Stathopoulos, K. VijayRaghavan and the Bloomington, National Institute of Genetics and Vienna Drosophila RNAi stock centres for providing stocks.
Footnotes
Funding
This work was funded by King's College London and the Wellcome Trust [WT088460MA, WT089622MA]. Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.074179/-/DC1
References
- Awasaki T., Lai S. L., Ito K., Lee T. (2008). Organization and postembryonic development of glial cells in the adult central brain of Drosophila. J. Neurosci. 28, 13742-13753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman J. M., McNeill H. (2004). Temporal control of differentiation by the insulin receptor/tor pathway in Drosophila. Cell 119, 87-96 [DOI] [PubMed] [Google Scholar]
- Bateman J. M., McNeill H. (2006). Insulin/IGF signalling in neurogenesis. Cell. Mol. Life Sci. 63, 1701-1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogiolo W., Stocker H., Ikeya T., Rintelen F., Fernandez R., Hafen E. (2001). An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 11, 213-221 [DOI] [PubMed] [Google Scholar]
- Chahrour M., Zoghbi H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422-437 [DOI] [PubMed] [Google Scholar]
- Chandran S., Compston A., Jauniaux E., Gilson J., Blakemore W., Svendsen C. (2004). Differential generation of oligodendrocytes from human and rodent embryonic spinal cord neural precursors. Glia 47, 314-324 [DOI] [PubMed] [Google Scholar]
- Chell J. M., Brand A. H. (2010). Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 143, 1161-1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotard C., Salecker I. (2007). Glial cell development and function in the Drosophila visual system. Neuron Glia Biol. 3, 17-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotard C., Leung W., Salecker I. (2005). glial cells missing and gcm2 cell autonomously regulate both glial and neuronal development in the visual system of Drosophila. Neuron 48, 237-251 [DOI] [PubMed] [Google Scholar]
- Colonques J., Ceron J., Tejedor F. J. (2007). Segregation of postembryonic neuronal and glial lineages inferred from a mosaic analysis of the Drosophila larval brain. Mech. Dev. 124, 327-340 [DOI] [PubMed] [Google Scholar]
- Dumstrei K., Wang F., Hartenstein V. (2003). Role of DE-cadherin in neuroblast proliferation, neural morphogenesis, and axon tract formation in Drosophila larval brain development. J. Neurosci. 23, 3325-3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzdottir S. R., Engelen D., Yuva-Aydemir Y., Schmidt I., Aho A., Klambt C. (2009). Switch in FGF signalling initiates glial differentiation in the Drosophila eye. Nature 460, 758-761 [DOI] [PubMed] [Google Scholar]
- Freeman M. R. (2010). Specification and morphogenesis of astrocytes. Science 330, 774-778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M. R., Doherty J. (2006). Glial cell biology in Drosophila and vertebrates. Trends Neurosci. 29, 82-90 [DOI] [PubMed] [Google Scholar]
- Furnari F. B., Fenton T., Bachoo R. M., Mukasa A., Stommel J. M., Stegh A., Hahn W. C., Ligon K. L., Louis D. N., Brennan C., et al. (2007). Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 21, 2683-2710 [DOI] [PubMed] [Google Scholar]
- Gao X., Pan D. (2001). TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 15, 1383-1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami A., Zwartkruis F. J., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S. C., Hafen E., Bos J. L., Thomas G. (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457-1466 [DOI] [PubMed] [Google Scholar]
- Ghabrial A., Luschnig S., Metzstein M. M., Krasnow M. A. (2003). Branching morphogenesis of the Drosophila tracheal system. Annu. Rev. Cell Dev. Biol. 19, 623-647 [DOI] [PubMed] [Google Scholar]
- Giesen K., Hummel T., Stollewerk A., Harrison S., Travers A., Klambt C. (1997). Glial development in the Drosophila CNS requires concomitant activation of glial and repression of neuronal differentiation genes. Development 124, 2307-2316 [DOI] [PubMed] [Google Scholar]
- Gisselbrecht S., Skeath J. B., Doe C. Q., Michelson A. M. (1996). heartless encodes a fibroblast growth factor receptor (DFR1/DFGF-R2) involved in the directional migration of early mesodermal cells in the Drosophila embryo. Genes Dev. 10, 3003-3017 [DOI] [PubMed] [Google Scholar]
- Gronke S., Clarke D. F., Broughton S., Andrews T. D., Partridge L. (2010). Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 6, e1000857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenstein V., Spindler S., Pereanu W., Fung S. (2008). The development of the Drosophila larval brain. In Brain Development in Drosophila melanogaster, vol. 628 (ed. Technau G. M.), pp. 1-31 Austin, TX: Landes Bioscience; [DOI] [PubMed] [Google Scholar]
- Hosoya T., Takizawa K., Nitta K., Hotta Y. (1995). glial cells missing: a binary switch between neuronal and glial determination in Drosophila. Cell 82, 1025-1036 [DOI] [PubMed] [Google Scholar]
- Jones B. W., Fetter R. D., Tear G., Goodman C. S. (1995). glial cells missing: a genetic switch that controls glial versus neuronal fate. Cell 82, 1013-1023 [DOI] [PubMed] [Google Scholar]
- Kaul A. K., Bateman J. M. (2009). A novel RFP reporter to aid in the visualization of the eye imaginal disc in Drosophila. J. Vis. Exp., 34, e1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N., Jamen F., Rubin L. L., Richardson W. D. (2004). Cooperation between sonic hedgehog and fibroblast growth factor/MAPK signalling pathways in neocortical precursors. Development 131, 1289-1298 [DOI] [PubMed] [Google Scholar]
- Klambt C. (1993). The Drosophila gene pointed encodes two ETS-like proteins which are involved in the development of the midline glial cells. Development 117, 163-176 [DOI] [PubMed] [Google Scholar]
- Kriegstein A., Alvarez-Buylla A. (2009). The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T., Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451-461 [DOI] [PubMed] [Google Scholar]
- Lemke G. (2001). Glial control of neuronal development. Annu. Rev. Neurosci. 24, 87-105 [DOI] [PubMed] [Google Scholar]
- Lioy D. T., Garg S. K., Monaghan C. E., Jacob Raber J., Foust K. D., Kaspar B. K., Hirrlinger P. G., Kirchhoff F., Bissonnette J. M., Ballas N., et al. (2011). A role for glia in the progression of Rett's syndrome. Nature 475, 497-500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I., Swanberg S., Harvey D., LaSalle J. M., Jin L. W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051-5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H., Craig G. M., Bateman J. M. (2008). Regulation of neurogenesis and epidermal growth factor receptor signalling by the insulin receptor/target of rapamycin pathway in Drosophila. Genetics 179, 843-853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M., Re D. B., Nagata T., Chalazonitis A., Jessell T. M., Wichterle H., Przedborski S. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615-622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll J. A., Weller R. O. (2003). A new role for astrocytes: beta-amyloid homeostasis and degradation. Trends Mol. Med. 9, 281-282 [DOI] [PubMed] [Google Scholar]
- Parker R. J., Auld V. J. (2006). Roles of glia in the Drosophila nervous system. Semin. Cell Dev. Biol. 17, 66-77 [DOI] [PubMed] [Google Scholar]
- Pereanu W., Shy D., Hartenstein V. (2005). Morphogenesis and proliferation of the larval brain glia in Drosophila. Dev. Biol. 283, 191-203 [DOI] [PubMed] [Google Scholar]
- Read R. D., Cavenee W. K., Furnari F. B., Thomas J. B. (2009). A Drosophila model for EGFR-Ras and PI3K-dependent human glioma. PLoS Genet. 5, e1000374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch D. H., Kriegstein A. R. (2010). Developmental genetics of vertebrate glial-cell specification. Nature 468, 214-222 [DOI] [PubMed] [Google Scholar]
- Scholz H., Sadlowski E., Klaes A., Klambt C. (1997). Control of midline glia development in the embryonic Drosophila CNS. Mech. Dev. 64, 137-151 [DOI] [PubMed] [Google Scholar]
- Silva E., Tsatskis Y., Gardano L., Tapon N., McNeill H. (2006). The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr. Biol. 16, 2081-2089 [DOI] [PubMed] [Google Scholar]
- Sousa-Nunes R., Cheng L. Y., Gould A. P. (2010). Regulating neural proliferation in the Drosophila CNS. Curr. Opin. Neurobiol. 20, 50-57 [DOI] [PubMed] [Google Scholar]
- Sousa-Nunes R., Yee L. L., Gould A. P. (2011). Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature 471, 508-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulos A., Tam B., Ronshaugen M., Frasch M., Levine M. (2004). pyramus and thisbe: FGF genes that pattern the mesoderm of Drosophila embryos. Genes Dev. 18, 687-699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork T., Engelen D., Krudewig A., Silies M., Bainton R. J., Klambt C. (2008). Organization and function of the blood-brain barrier in Drosophila. J. Neurosci. 28, 587-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng A. S., Tapon N., Kanda H., Cigizoglu S., Edelmann L., Pellock B., White K., Hariharan I. K. (2007). Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr. Biol. 17, 728-733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S., Wilson R., Coelho C., Affolter M., Leptin M. (1998). The Drosophila protein Dof is specifically required for FGF signaling. Mol. Cell 2, 515-525 [DOI] [PubMed] [Google Scholar]
- Weinkove D., Neufeld T. P., Twardzik T., Waterfield M. D., Leevers S. J. (1999). Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr. Biol. 9, 1019-1029 [DOI] [PubMed] [Google Scholar]
- Wilson R., Battersby A., Csiszar A., Vogelsang E., Leptin M. (2004). A functional domain of Dof that is required for fibroblast growth factor signaling. Mol. Cell. Biol. 24, 2263-2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W. C., Okano H., Patel N. H., Blendy J. A., Montell C. (1994). repo encodes a glial-specific homeo domain protein required in the Drosophila nervous system. Genes Dev. 8, 981-994 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}