

Abstract
Lesions in the epithelially expressed human gene FRAS1 cause Fraser syndrome, a complex disease with variable symptoms, including facial deformities and conductive hearing loss. The developmental basis of facial defects in Fraser syndrome has not been elucidated. Here we show that zebrafish fras1 mutants exhibit defects in facial epithelia and facial skeleton. Specifically, fras1 mutants fail to generate a late-forming portion of pharyngeal pouch 1 (termed late-p1) and skeletal elements adjacent to late-p1 are disrupted. Transplantation studies indicate that fras1 acts in endoderm to ensure normal morphology of both skeleton and endoderm, consistent with well-established epithelial expression of fras1. Late-p1 formation is concurrent with facial skeletal morphogenesis, and some skeletal defects in fras1 mutants arise during late-p1 morphogenesis, indicating a temporal connection between late-p1 and skeletal morphogenesis. Furthermore, fras1 mutants often show prominent second arch skeletal fusions through space occupied by late-p1 in wild type. Whereas every fras1 mutant shows defects in late-p1 formation, skeletal defects are less penetrant and often vary in severity, even between the left and right sides of the same individual. We interpret the fluctuating asymmetry in fras1 mutant skeleton and the changes in fras1 mutant skeletal defects through time as indicators that skeletal formation is destabilized. We propose a model wherein fras1 prompts late-p1 formation and thereby stabilizes skeletal formation during zebrafish facial development. Similar mechanisms of stochastic developmental instability might also account for the high phenotypic variation observed in human FRAS1 patients.
Keywords: fras1, Zebrafish, Craniofacial, Fraser syndrome, Developmental instability
INTRODUCTION
Human FRAS1 lesions cause Fraser syndrome (McGregor et al., 2003; Slavotinek et al., 2006; van Haelst et al., 2008), a complex disorder with numerous variably expressed symptoms, including ear defects and other craniofacial birth defects (Fraser, 1962; Gattuso et al., 1987; Slavotinek and Tifft, 2002; Thomas et al., 1986; van Haelst et al., 2007). The symptoms of Fraser syndrome vary extensively in both presence and severity from one patient to another. Fraser syndrome variation has been a topic of intense interest to clinicians since the syndrome was first described (Fraser, 1962), and yet the causes of this variability have remained unclear. Models have been proposed in which genetic modifiers control the degree of symptomatic variation (e.g. Slavotinek and Tifft, 2002). Subsequent studies revealed that other members of the FRAS1 protein complex (the Fraser complex) can cause Fraser syndrome (Jadeja et al., 2005; Shafeghati et al., 2008) or related diseases (Alazami et al., 2009; Slavotinek et al., 2011), explaining some of the causes of the different Fraser spectrum diseases. However, a wide range of symptoms are present even in patients carrying genetic lesions that are likely to severely disrupt FRAS1 function (van Haelst et al., 2008). For instance, siblings can show life and death differences in symptomatic expressivity (Prasun et al., 2007). Other models focus on stochastic sources of variation in Fraser syndrome, noting that symptoms (cryptophthalmos) can vary between left and right sides of patients at frequencies predicted by chance (Cavalcanti et al., 2007).
Similar to the epithelial abnormalities frequently found in Fraser syndrome (e.g. Slavotinek and Tifft, 2002), severe epithelial defects are present in Fras1 mutant mice (McGregor et al., 2003; Vrontou et al., 2003) and fras1 mutant zebrafish (Carney et al., 2010). In mouse (McGregor et al., 2003; Vrontou et al., 2003) and zebrafish (Gautier et al., 2008), Fras1/fras1 mRNA is expressed by epithelial cells, including endoderm and ectoderm lining the pharyngeal arches. Fras1 encodes a transmembrane protein containing motifs implicated in signaling and adhesion (Gautier et al., 2008; McGregor et al., 2003). The large extracellular portion of Fras1 is cleaved and released into basal lamina underlying epithelia (Carney et al., 2010; Chiotaki et al., 2007; Kiyozumi et al., 2006; Petrou et al., 2007). Hence, a layer of Fras1 protein surrounds the neural crest-derived skeletogenic portion of pharyngeal arches. This layer of Fras1 protein might signal to arches or physically connect epithelia and arch mesenchyme during processes such as endodermal pouch formation.
In this study we use zebrafish to model craniofacial symptoms of Fraser syndrome, particularly conductive hearing loss. In humans, derivatives of the first endodermal pouch (pouch 1, or p1) contribute to the Eustachian tube, whereas neural crest-derived cells in the first two arches contribute to the jaw and middle ear skeletons. Here we describe a previously unrecorded late-forming portion of the first pharyngeal pouch (termed late-p1) in zebrafish. We show that zebrafish fras1 is required for the formation of late-p1 and that fras1 mutants often show defects in skeletal elements which, in wild-type (WT) fish, form near late-p1. We find that WT endoderm rescues both epithelial and skeletal defects in fras1 mutants, indicating that epithelial fras1 non-autonomously sculpts nearby facial skeleton. Using time-lapse and timecourse analysis, we find that the late-p1 defects in fras1 mutants arise alongside some cartilage defects, but prior to others. The physical relationship between late-p1 and second arch cartilages suggests that the late-forming second arch cartilage fusions may occur in fras1 mutants because late-p1 is unable to physically stabilize cartilage formation. For instance, late-p1 might physically prevent fusion between symplectic and ceratohyal cartilages. Late-p1 might also help pull apart first arch cartilages, or bring signaling cues to the presumptive first arch joint that ensure joint formation. fras1 mutants show fluctuating left-right asymmetries in all skeletal defects, suggesting that stochastic developmental instability might govern the degree of phenotypic defect in the zebrafish fras1 mutant skeleton. We propose a model wherein fras1 acts in endoderm to generate late-p1 and to stabilize the development of nearby skeletal elements.
MATERIALS AND METHODS
Fish maintenance, husbandry, morpholinos and strains
Fish were raised as described (Kimmel et al., 1995; Westerfield, 2007). Mutant lines were maintained on the genetic backgrounds shown in Table 1. We identified fras1 mutants using previously described fully penetrant tail fin blisters or previously described PCR genotyping protocols (Carney et al., 2010). Unless stated otherwise, the b1048 allele of fras1 is used. b1048 causes a premature stop codon prior to the vital transmembrane domain of Fras1; similar to previous reports using fras1te262 (Carney et al., 2010), we find that anti-Fras1 label is lost in fras1b1048 (Fig. 1A,B), confirming that b1048 causes a strong loss of fras1 function. Furthermore, this Fras1 antibody was produced using an epitope prior to the b1048 and te262 lesions, so the loss of anti-Fras1 label observed in te262 (Carney et al., 2010) and b1048 (Fig. 1A′,B′) mutants suggests that the entire Fras1 protein is lost in fish homozygous for fras1te262 or fras1b1048. A transgenic line formally named sox9azc81Tg (see https://www.facebase.org/fishface/home) expresses EGFP in cartilage cells under the likely control of a sox9a enhancer; for clarity, we refer to the line as sox9a:EGFP. sox10:mRFP (Kirby et al., 2006) and her5:GFP (Tallafuss and Bally-Cuif, 2003) are described elsewhere.
Table 1.
The early-stop fras1 alleles cause similar skeletal defects

Fig. 1.

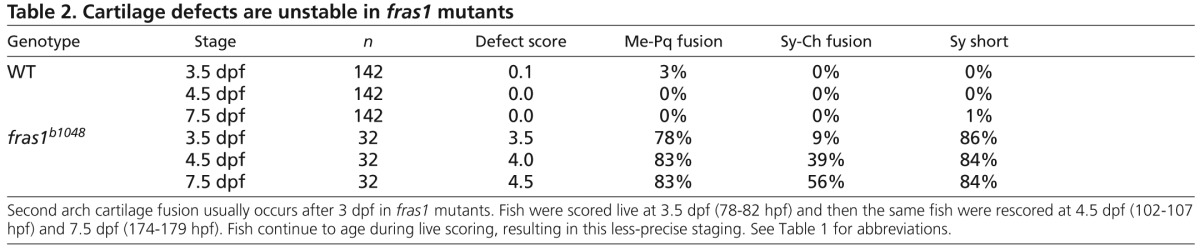
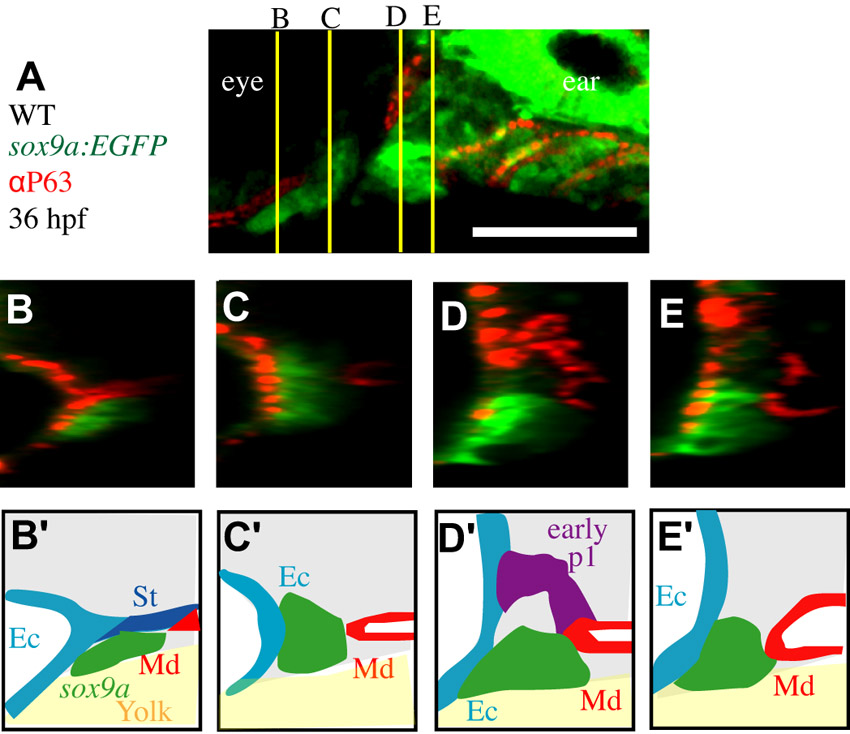
Zebrafish fras1 mutants show specific late-p1 defects. (A-B′) Transverse sections of antibody-stained tissues, oriented lateral to left, dorsal up. (B) At the section level of late-p1, fras1 mutant endoderm does not extend as far laterally (asterisk) as WT endoderm at 72 hpf. (A′,B′) High-resolution detail near trabecula (Tb) from A and B shows that (A′) WT Fras1 is deposited basal to the epithelial nuclei marker and (B′) anti-Fras1 label is absent from fras1 mutants. Because these insets are from the roof of the mouth, basal is up and lateral is to the left. (C-E) By contrast, early pouches appear normal in fras1 mutants. (C,D) Confocal section of (C) WT and (D) fras1 mutant epithelia at 36 hpf, oriented anterior to left, dorsal up. Note that the early forming portion of pouch 1 is only mildly misshapen in fras1 mutants and posterior pouches appear normal. (E) Diagram of WT pouch structure at 36 hpf, viewed with anterior to left, dorsal up. Medial endoderm (orange dashed lines) lies beneath the plane of section shown in G and H (see supplementary material Fig. S2). Yolk is in yellow and somatic tissue is gray. (F) Diagram of pouch derivatives at 72 hpf, viewed with anterior to left, dorsal up. Between 36 and 72 hpf, late-p1 has protruded laterally, early-p1 has been covered by cartilages, and pouch 2 (p2) has extended posteriorly, covering posterior pouches. (G-N) RNA in situ hybridization on tissue sections, shown lateral to left, dorsal up. Epithelial fras1 expression lines pharyngeal arches during late-p1 formation. (G-J) Sections through early-p1 show that this early forming pouch is eventually covered by the hyomandibular cartilage, whereas (K-N) late-p1 protrudes laterally, anterior to early-p1. (O,P) Transverse sections of antibody-stained tissues, oriented lateral to left, dorsal up. Endodermal pouching defect persists in fras1 mutants at 7 dpf (asterisk). (Q,R) Illustrations of 7-dpf tissue sections, indicating the location of late-p1. Endoderm abbreviations: early-p1, early forming portion of pouch 1; late-p1, late-forming portion of pouch 1; p3, pouch 3; p4, pouch 4; p5, pouch 5; St, stomadeum. Cartilage abbreviations: Pq, palatoquadrate; Ch, ceratohyal; Bh, basihyal; Ep, ethmoid plate; Hm, hyomandibular. Scale bars: 50 μm (in A,A′,C,O for B,B′,D,P, respectively; in N for G-N).
Tissue labeling
RNA in situ hybridization was performed as described (Rodriguez-Mari et al., 2005) using fras1 probe (Carney et al., 2010). For RNA in situ hybridization, embryos were raised in 0.0015% 1-phenyl 2-thiourea (PTU) to inhibit melanogenesis (Westerfield, 2007). Alcian Blue and Alizarin Red staining were performed as described (Walker and Kimmel, 2007). Epithelia were labeled with antibodies to human TP63 (P63 4A4, sc-8431, Santa Cruz Biotechnology) and zebrafish Fras1 (Carney et al., 2010), using a protocol available at ZFIN (https://wiki.zfin.org/x/XACiAQ). Image processing utilized LSM (Carl Zeiss), Volocity (PerkinElmer), ImageJ (NIH) and MetaMorph (Molecular Devices) software.
Time-lapse microscopy
When imaged on a Zeiss Pascal LSM 5 laser-scanning microscope, mounting was essentially as described (Westerfield, 2007). An inverted microscope (Leica SD6000 spinning-disk confocal microscope with Borealis Illumination Technology) allowed concurrent imaging of multiple fish. For this latter set-up, fish were mounted in 0.4% agarose in glass-bottom dishes and covered with embryo medium containing 80 mg/l clove oil (Hilltech). After imaging, all fish were raised for several days, then re-examined for skeletal morphology and to confirm health.
Cartilage scoring
Fish were visually examined for overt mutant phenotypes. Any ectopic connection of skeletal elements (cartilage fusion) was scored as ‘fused’. Symplectic cartilages were scored as ‘short’ if they were less than two-thirds the mean WT length. Symplectic length was scored independently of symplectic fusion whenever possible. Phenotypic penetrance is the proportion of fish expressing a given defect. Phenotypes scored on fixed Alcian Blue-stained preparations (Table 1) showed similar penetrances to live scoring using sox9a:EGFP expression (Table 2). Overall defects are represented by a ‘defect score’: the average sum of Meckel's to palatoquadrate fusion, symplectic to ceratohyal fusion, short symplectic phenotypes, including both sides of a fish. Statistical analyses used JMP 9.0 (SAS Institute, Cary, NC, USA) software. Fluctuating asymmetry was analyzed following the prescribed guidelines (Palmer and Strobeck, 2003).
Table 2.
Cartilage defects are unstable in fras1 mutants
Endoderm transplantation
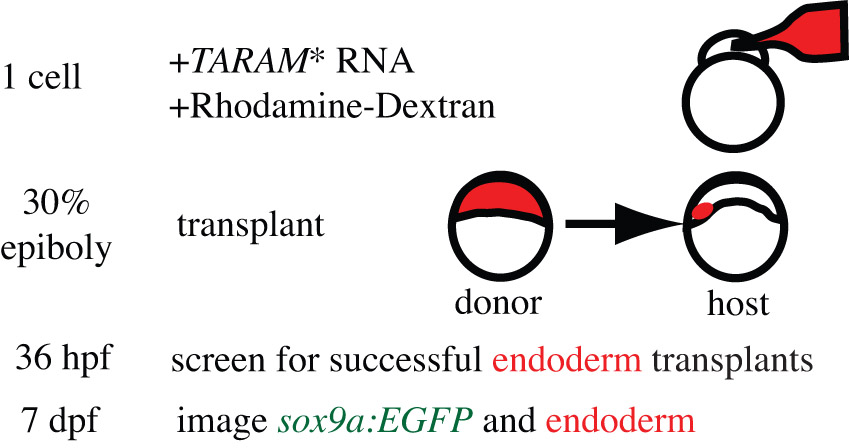
Endoderm transplantation experiments were performed (supplementary material Fig. S1) essentially as described (Crump et al., 2004a; Crump et al., 2004b; Walker et al., 2007). In brief, donors were injected at early one-cell stage with TARAM* RNA and 1% Rhodamine-dextran. At 3-4 hours post-fertilization (hpf), 20-30 donor cells were transferred to host embryos near the yolk margin. At 34-38 hpf, hosts were selected on the basis of strong labeling of medial endoderm underlying the first two arches and early pouch 1. Hosts were raised to 7 days post-fertilization (dpf) and imaged. Host tails (not labeled with donor) and donor embryos were PCR genotyped.
RESULTS
fras1 mutation consistently disrupts late-p1 formation
Because fras1 is expressed in the basal lamina of facial epithelia (Fig. 1A-B′) and is known to sculpt epithelia, we assayed the shape of facial epithelia in fras1 mutants. We find that an anterior portion of epithelia is consistently misshapen in all fras1 mutants (Fig. 1A,B). In WT fish, anterior endoderm is closely juxtaposed with anterior ectoderm at 72 hpf (Fig. 1A). However, in fras1 mutants, anterior endoderm is situated medially at 72 hpf (Fig. 1A,B).
Pharyngeal pouches form via lateral protrusion of endoderm, connecting medial endoderm to ectoderm; successive pouches segment the arches along the anterior-posterior (A-P) axis (reviewed by Graham et al., 2005). The fras1-dependent endodermal protrusion is the most anterior portion of endoderm to abut with ectoderm, indicating that it is a portion of the first pharyngeal pouch (pouch 1, or p1). Although fras1 mutants show pronounced pouch 1 defects by 72 hpf (Fig. 1A,B), the early forming and well-known dorsal portion of the first pouch (termed ‘early-p1’; Fig. 1, supplementary material Fig. S2) appears normal in fras1 mutants at 36 hpf (Fig. 1C-E). Early-p1 appears to contribute primarily to a dorsal structure, medial to the hyomandibular cartilage (Fig. 1F-J).
fras1-expressing endoderm, situated between early-p1 and the stomadeum, continues to migrate laterally between 36 and 72 hpf (Fig. 1K-N). Ectoderm remains flat over arches between 36 and 72 hpf (Fig. 1G-N), indicating that late-p1 formation occurs primarily via endoderm protrusion rather than ectodermal intrusion. The pouch 1 defect persists in fras1 mutants until at least 168 hpf (Fig. 1O-R), while the rest of the fish develops normally, indicating that anterior endoderm defects are not simply caused by developmental delay. Serial sections reveal that only this most anterior pouch is affected, both in fras1b1048 (n=8/8; Fig. 2A-G) and fras1te262 (n=8/8) mutants at 72 hpf, consistent with the normal formation of posterior pouches at 36 hpf (Fig. 1). Hence, we conclude that fras1 is specifically required for epithelial morphogenesis that generates a late-forming portion of pouch 1, hereafter referred to as ‘late-p1’.
Fig. 2.
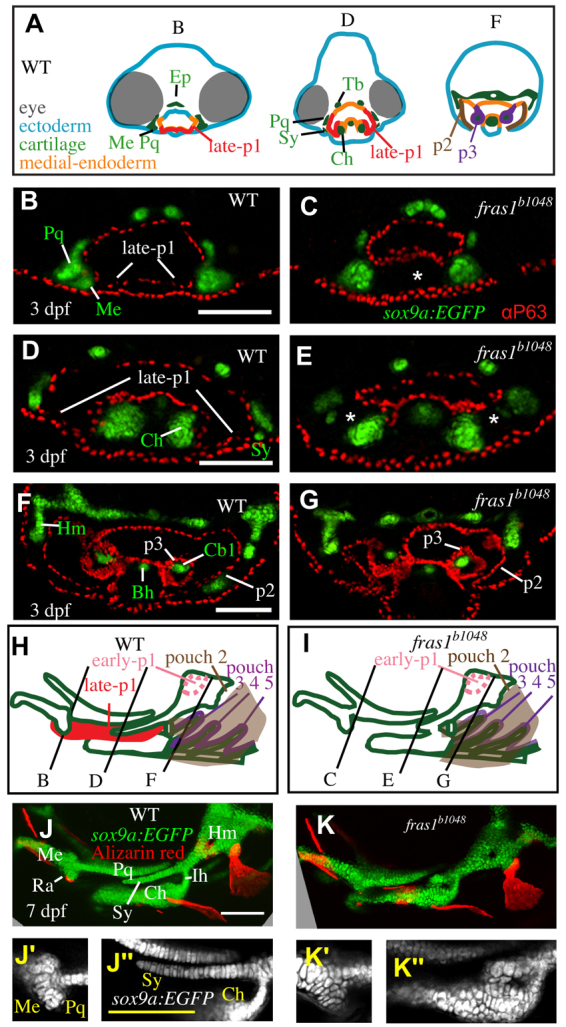
Skeletal elements near late-p1 are affected by fras1 mutation. (A) Illustrations of WT epithelia and cartilage on transverse sections, shown dorsal up. Ep, ethmoid plate; Tb, trabecula. (B-G) Tissue sections labeled with anti-P63 and sox9a:EGFP, taken from the section level of (B,C) Meckel's-palatoquadrate joint, (D,E) symplectic cartilage, and (F,G) opercular flap. By 72 hpf, fras1 mutants exhibit loss (asterisk) of late-p1 (B-E) but not p2 or p3 (F,G) derivatives. (H,I) Illustrations of 7-dpf cartilaginous skeletons, showing how pouch derivatives intersect with cartilages and how the sections shown in B-G map onto 7-dpf skeletons; shown anterior to left, dorsal up. (J-K″) Confocal projections of live-imaged fish showing bone (Alizarin Red staining) and cartilage (sox9a:EGFP expression). Dermal bone morphology typically resembles WT (J) in fras1 mutants (K), although mild opercle bone defects are sometimes found in fras1 mutants (not shown). Inset confocal sections highlight morphology at first arch-derived (J′,K′) and second arch-derived (J″,K″) joint regions. Fully expressive fras1 mutants display distinct cartilage defects including (K′) M-Pq fusion and (K″) Sy-Ch fusion with short Sy. Arch 1-derived cartilages: Me, Meckel's cartilage; Ra, retroarticular process of Meckel's cartilage; Pq, palatoquadrate. Arch 2-derived cartilages: Cb1, first ceratobranchial cartilage; Ch, ceratohyal; Ih, interhyal; Hm, hyomandibular; Sy, symplectic. Scale bars: 100 μm (applicable to each row).
Skeletal elements near late-p1 are often disrupted in fras1 mutants
Skeletal elements near late-p1 are frequently abnormal in fras1 mutants (Fig. 2). In 72-hpf WT fish, late-p1 separates the ceratohyal cartilage from Meckel's, palatoquadrate and symplectic cartilages (Fig. 2A-H). fras1 mutants show three major skeletal defects, all in cartilages adjacent to late-p1 (Fig. 2H-K″): Meckel's to palatoquadrate fusion (Me-Pq fusion; Table 1, Fig. 2J′,K′); shortened symplectic length (short Sy; Table 1, Fig. 2J″,K″); and symplectic to ceratohyal fusion (Sy-Ch fusion; Table 1, Fig. 2J″,K″). All three early-stop fras1 alleles show the same skeletal phenotypes with similar penetrance (Table 1). Skeletal defects near late-p1 are also present, although rare, in a hypomorphic fras1 allele (tm95b; Table 1). By contrast, skeletal elements further from late-p1, such as the interhyal cartilage, hyomandibular cartilage and the opercle bone, are relatively normal in fras1 mutants (Fig. 2J-K″, Table 1). Hence, we hypothesize that fras1 functions in late-p1 to non-autonomously shape nearby cartilage elements during development.
WT endoderm rescues fras1 mutants
fras1 is expressed in epithelia, not skeleton, so we hypothesize that fras1 functions non-cell-autonomously to sculpt the facial skeleton. We tested this hypothesis with reciprocal transplants between WT and fras1 mutant fish (Fig. 3). Labeled WT endoderm transplanted into unlabeled WT hosts (WT→WT transplants) marks late-p1 (19/19 transplants; Fig. 3A,A′), confirming the endodermal origin of this structure. As expected, fras1 mutant endoderm is unable to produce late-p1 when transplanted into fras1 mutant hosts (6/6 transplants; Fig. 3B,B′), and fras→fras1 transplants show skeletal defects with the variation expected for non-mosaic fras1 mutants (Table 1). However, when WT endoderm is transplanted into fras1 mutant hosts (7/7 transplants; Fig. 3C,C′), both late-p1 and cartilage shapes appear similar to those of WT→WT transplants, indicating that WT endodermal fras1 expression rescues facial development in fish otherwise mutant for fras1. For example, in WT→fras1 transplants the symplectic cartilage is elongated normally and no cartilage fusions are seen (Fig. 3C,C′). When fras1 mutant endoderm is transplanted into WT hosts, late-p1 defects are sometimes seen in the second pharyngeal arch (6/12 hosts; Fig. 3D,D′), but even when present, neither the late-p1 nor skeletal defects are as pronounced as in non-mosaic fras1 mutants (see Discussion). Our transplantation experiments support the hypothesis that endodermally expressed fras1 sculpts both endoderm and skeleton.
Fig. 3.

Endodermal fras1 sculpts epithelia and skeleton. Confocal transverse sections taken at a section level midway through Sy length at 7 dpf. Medial to right, dorsal up. (A) In mosaic fish in which WT endoderm has been transplanted into WT hosts (WT→WT mosaics), late-p1 appears normal (+) and is labeled as endoderm. (B) In fras1te262→fras1te262 mosaic fish, late-p1 does not form, and anterior endoderm remains medial to cartilages (asterisk). (C) WT endoderm transplanted into fras1te262 mutants rescues the late-p1 defects of these hosts. (D) Half of the fras1te262→WT mosaics examined exhibited defective endoderm medial to Sy and Ch. (A′-D′) Rendered confocal stacks show endoderm and skeletal morphology; oriented anterior to the left, dorsal up. Image rendering makes both tissues opaque, allowing visualization of late-p1 covering separated Sy and Ch cartilages (circled). In WT→WT (A′) and WT→fras1te262 (C′) mosaics, late-p1 covers part of Ch. However, in fras1te262→fras1te262 mosaics, fused cartilages cover the medial endoderm (arrowhead). Half of the fras1te262→WT mosaics show late-p1 defects, but cartilage defects are more subtle than in non-mosaic fras1te262 mutants, perhaps owing to the presence of late-p1 in the first pharyngeal arch (tilde). Scale bar: 100 μm.
fras1-dependent cartilage morphogenesis proceeds concurrently with late-p1 formation
In addition to occurring in the same space as late-p1 formation, fras1-dependent skeletal elements develop during the same time as late-p1. In fras1 mutants, late-p1 formation fails between 36 and 72 hpf (Figs 1, 4). The lateral migration of WT late-p1 can be quantified by the dramatic decrease in the distance between anterior endoderm and anterior ectoderm that occurs between 36 and 72 hpf (Fig. 4A-J). By contrast, all fras1 mutants, lacking late-p1 morphogenesis, maintain large endoderm to ectoderm distances from 36-72 hpf (Fig. 4A-J). This same 36-72 hpf time window is a period of dramatic skeletal morphogenesis (supplementary material Movie 1). For instance, the symplectic cartilage protrudes anteriorly between 36 and 72 hpf, and symplectic protrusion is frequently reduced in fras1 mutants slightly after late-p1 formation initiates (Fig. 4K-M). By contrast, fras1 mutant hyomandibular length increases normally during this time interval (supplementary material Fig. S3), indicating that the fras1 mutant symplectic length defect is not the result of a general developmental delay.
Fig. 4.

fras1-dependent symplectic extension occurs concurrently with late-p1 formation. (A-C) Schematics of the transverse sections shown in D-I, illustrating ectoderm (blue), endoderm (orange), cartilage (green), somatic tissue (gray) and eye (dark gray). (D-I) Transverse view of confocal stacks, showing WT late-p1 formation between 36 and 72 hpf (D-F, arrowhead), which fails (asterisk) in fras1 mutants (G-I). (J,K) Measurements of (J) endoderm-ectoderm distances and (K) Sy lengths, taken from the same fish. (J) Minimum distance between endoderm and ectoderm measured as illustrated (blue lines in D-I) on randomly selected fish. Endoderm-ectoderm distance decreases in WT fish but remains relatively constant in fras1 mutants. (K) Symplectic length, as measured from above the center of interhyal to the anterior tip of the symplectic (orange lines in L,M). At 36 hpf, symplectic cartilage precursors do not protrude beyond the bulk of the second arch sox9a:EGFP expression, so 36-hpf ‘Sy length’ is measured from the anterior edge of second arch sox9a:EGFP expression to its center. (L,M) Confocal section of 72-hpf symplectic cartilage, illustrating Sy length measurements (orange lines); oriented anterior to left, dorsal up. Error bars show mean ± 1.96 times the s.e. Scale bars: 100 μm (in I for A-I; in L for L,M).
Two-color time-lapse microscopy of WT cartilage and endoderm development confirms that WT late-p1 moves laterally into the space between palatoquadrate and ceratohyal cartilages, concurrent with symplectic extension (Fig. 5). In WT fish, cells that form the symplectic migrate out of the path that endoderm will follow during late-p1 formation (supplementary material Movie 2), with the tip of the symplectic moving anteriorly, immediately dorsal to late-p1 (Fig. 5, supplementary material Movie 2). Similarly, Meckel's cartilage separates from palatoquadrate cartilage between 36 and 72 hpf in WT fish (supplementary material Movie 3). In fras1 mutants, Meckel's cartilage often fails to separate from palatoquadrate cartilage during this time interval (supplementary material Movie 3). Hence, many fras1-dependent aspects of skeletal morphogenesis occur at a very similar time as fras1-dependent late-p1 formation, further indicating a connection between fras1 function in skeletal and epithelial morphogenesis.
Fig. 5.
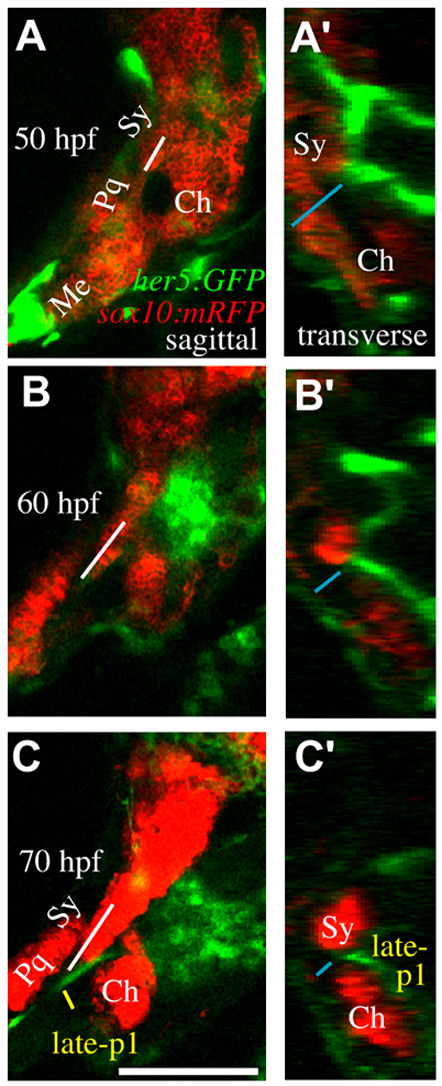
Time-lapse microscopy reveals concurrent late-p1 and symplectic outgrowth. (A-C′) sox10:mRFP labels cartilages and her5:GFP labels late-p1 and some neural cells in these sections. Still images from time-lapse microscopy showing symplectic growth (white line in A-C) and endoderm-ectoderm distance (blue line in A′-C′). (A-C) Sagittal confocal section, anterior to left, dorsal up. By 70 hpf, symplectic tip extends out of the plane of section. (A′-C′) Transverse view, lateral to left, dorsal up, constructed from the same confocal stacks. Between 50 and 70 hpf, late-p1 moves laterally through a region between the symplectic and ceratohyal cartilages. By 70 hpf, development is slightly delayed, resulting in a mild reduction of late-p1 and cartilage formation. Scale bar: 100 μm.
Symplectic to ceratohyal fusion often occurs long after late-p1 formation
Although in fras1 mutants some skeletal defects begin to develop during the same time interval as late-p1 defects, we often observe fusion between symplectic and ceratohyal cartilages long after 72 hpf (Fig. 6A-B′, Table 2). Often, individual fras1 mutant fish lacking Sy-Ch fusions at 3.5 dpf will show Sy-Ch fusions when examined 1 day later (Table 2), and the probability of fusion increases further by 7.5 dpf (Fig. 6A-B′, Table 2). Both the Me-Pq (Fig. 6C-D′) and Sy-Ch (not shown) fusions persist once they form.
Fig. 6.

Me-Pq fusion occurs by 72 hpf, whereas Sy-Ch fusion often occurs later. (A-B′) Confocal sections of second arch-derived zebrafish skeleton at 3.5 dpf (A,B) and the same skeletal elements reimaged later in development (A′,B′). Anterior to left, dorsal up. By 3.5 dpf, a large gap is present between WT symplectic and ceratohyal cartilages (A), and this gap persists through larval development (A′). In fras1 mutants, although there is often a space between symplectic and ceratohyal cartilage (B, circled) at 3.5 dpf, these skeletal elements are typically found fused together when examined later in larval development (B′, yellow arrow). (C-D′) Confocal projections of first arch joint region, shown anterior to left and dorsal up. By 3 dpf, WT embryos have formed a cleft between Meckel's and palatoquadrate cartilages (C), which persists as the embryos develop (C′). When fras1 mutants show skeletal fusions in the first arch (D, asterisk) they are always visible by 3 dpf, and persist when re-examined later in larval development (D′). Scale bars: 100 μm.
The difference in timing between Me-Pq and Sy-Ch fusions might be caused by different mechanisms of fusion. ‘Short’ mutant symplectic cartilages resemble younger, unextended WT symplectic cartilages. Similarly, Me-Pq fusions resemble unseparated WT cartilages (supplementary material Movie 3) due to failure of cleft formation between fras1 mutant Meckel's and palatoquadrate cartilages. However, symplectic to ceratohyal fusions are never seen during WT development (supplementary material Movies 1, 2, 4). Instead, time-lapse microscopy of cartilage development in fras1 mutants (supplementary material Movie 4) reveals that second arch fusions in fras1 mutants occur after symplectic extension, via ectopic attachment of symplectic to ceratohyal cartilages. These developmental differences between joint fusions in the first and second arches of fras1 mutants (failure to separate versus ectopic fusion) indicate that fras1 uses different mechanisms to shape the first versus the second pharyngeal arch.
fras1 stabilizes skeletal development
Consistent with epithelial fras1 function, all fras1 mutants examined have defects in late-p1 formation, whereas fras1 mutant skeletal defects are frequently less severe, falling within the WT range (Fig. 7A). Hence, our dataset combining WT and fras1 mutants shows a strong global correlation [correlation (C)=−0.76, P<0.0001] between symplectic length and endoderm-ectoderm distance (Fig. 7A). This correlation is also seen among WT fish (C=−0.40, P<0.0007); however, in fras1 mutants endoderm defects are always severe (compared with WT) and the correlation between symplectic length and endoderm-ectoderm distance is lost (C=0.04, P<0.86; Fig. 7A).
Fig. 7.

Skeletal phenotypes fluctuate asymmetrically in fras1 mutants. (A) Plot of 72-hpf right symplectic length versus right endoderm to ectoderm distance, measured as in Fig. 4, with grouped 95% density ellipses. Linear regression reveals that significant correlation (asterisk) between endoderm-ectoderm distances and symplectic length exists in the grouped data, and in WT fish, but not fras1 mutants. All fras1 mutant fish show endoderm-ectoderm distance defects; however, fras1 mutant symplectic lengths are sometimes within the WT range. (B,C) Symplectic length analysis on fish live-imaged at 7.5 dpf. For each side, symplectic length was measured twice, then averaged. (B) Plot of left versus right symplectic length, with grouped 95% density ellipses. In fras1 mutants, linear regression does not reveal a significant correlation between left and right symplectic lengths. (C) fras1 mutants show fluctuating asymmetry, twofold higher than WT. This increase in fluctuating asymmetry is much larger than can be accounted by measurement error. Asymmetry is the absolute value of length differences between sides. Measurement error is estimated as the difference between paired measurements on one side of a fish. (D) Overall skeletal asymmetry of 7.5-dpf fish, calculated as A=(|Sy shortl-r| + |Sy-Ch fusionl-r| + |Me-Pq fusionl-r|) / 3N. (E-G′) Second arch joint region of right (E-G) and left (E′-G′) individuals imaged live at 7.5 dpf; anterior to left, dorsal up. Compared with WT fish (E,E′), fras1 mutant phenotypes are often asymmetric (F-G′). For instance, a fish presenting a short, but unfused, symplectic cartilage on one side (F) also shows an extended, but fused, symplectic cartilage on the opposite side (F′). Conversely, the ‘short’ and ‘fused’ symplectic phenotypes (G) can be found on one side of a fish that presents only subtle defects (G′) on the opposite side. Error bars show mean ± 1.96 times the s.e. Scale bar: 100 μm.
In fras1 mutants, asymmetry between left and right symplectic lengths is twice that of their WT counterparts (Fig. 7B). Although fras1 mutants show a possible mild right side bias to the symplectic length defects, symplectic lengths do not differ significantly between sides (supplementary material Fig. S4). Instead, asymmetry in symplectic lengths appears to fluctuate randomly between the left and right sides of embryos (Fig. 7C). Such ‘fluctuating asymmetry’ is often interpreted as being due, at least in part, to the presence of random processes perturbing development (e.g. Dongen, 2006) (see Discussion). The degree of fluctuating asymmetry in the symplectic length of fras1 mutants is at least twice that of WT siblings (Fig. 7C). When defects in all skeletal elements are considered, fras1 mutants are much more asymmetric than WT siblings, but do not show an antisymmetric (biased towards unilateral) pattern (Fig. 7D). Furthermore, individual fras1 mutants can present any combination of skeletal defects (Fig. 7E-G′), and the presence of any one overt cartilage defect is a very poor indicator of any other defect (not shown). We hypothesize that the consistent failure of late-p1 formation allows stochastic processes to prominently influence the degree of cartilage defects.
DISCUSSION
We have identified a fras1-dependent endodermal outpocket, termed late-p1 (Fig. 8). Late-p1 differs from the more dorsal and early forming portion of endodermal pouch 1 that was previously assumed to constitute the entirety of pouch 1 (Kimmel et al., 2001). It is possible that these previous studies failed to notice late-p1 because it develops after the classical ‘pharyngula’ period of embryogenesis (Kimmel et al., 1995). However, four lines of evidence indicate that late-p1 is a portion of the first pharyngeal pouch. First, late-p1 forms via lateral protrusion of endoderm (Fig. 8), similar to other pouches. Second, late-p1 is the most anterior pouch in the 72-hpf pharynx, similar to human pouch 1 (Hamilton et al., 1947). Third, late-p1 separates first arch-derived cartilages from second arch-derived cartilages (Fig. 8B), also similar to human pouch 1 (Arey, 1966). Although the second arch-derived symplectic cartilage extends dorsal to late-p1, this intrusion is likely to be related to the long-studied function of the symplectic as a first arch-supporting cartilage (De Beer, 1937). Fourth, an illustration of salamander pouch 1 (Lehman, 1987) shows striking similarity to zebrafish late-p1, further linking the morphology of this pouch across vertebrate phylogeny. Hence, we infer that zebrafish late-p1 is indeed a late-forming portion of pouch 1 that has simply gone unnoticed in previous studies. A more complete description of Eustachian tube morphology (pouch 1 derived) and middle ear skeleton morphology (derived from the first two arches) in Fraser syndrome patients might yield fruitful insights as they often have symptoms in both the ear canal and middle ear (e.g. Gattuso et al., 1987) and conductive hearing loss (Smyth and Scambler, 2005). If this homology can be taken as a guide, then endodermal pouching defects might underlie some ear defects in Fraser patients.
Fig. 8.

fras1-dependent late-p1 formation stabilizes zebrafish skeletal development. (A-C) The movement of late-p1 is primarily across a plane (sagittal, A) orthogonal to the primary plane (transverse, B,C) of cartilage morphogenesis. These processes are intertwined in three dimensions. (B) In the second arch, late-p1 formation brings endoderm through space previously occupied by symplectic precursor cells, and into a region that separates symplectic from ceratohyal. (C) In the first arch, late-p1 formation brings endodermal signaling cues (red-orange gradient) closer to the first arch joint precursors, and might pull Meckel's and palatoquadrate cartilages apart from one another. Late-p1 formation fails in fras1 mutants, potentially reducing the forces that separate first arch cartilages, and leaving the first arch cartilage joint precursors distant from endodermal signaling cues. Red arrowheads indicate late-p1 movements. Arrows indicate skeletal movements between time points: Sy growth (black), Sy fusion (brown) and Me-Pq separation (purple). Ch, ceratohyal; Me, Meckel's; Pq, palatoquadrate; St, stomadeum; Sy, symplectic.
fras1 is expressed in the basal lamina of endoderm and ectoderm surrounding pharyngeal arches. Because late-p1 (endodermal) shape is disrupted in fras1 mutants, but ectoderm shape is normal, our model focuses on endodermal fras1 functions (Fig. 8). We find that endodermal fras1 influences both skeletal and epithelial morphogenesis (Fig. 8A). For instance, endodermal fras1 is able to rescue facial development in fish that are otherwise mutant for fras1. However, WT fish mosaically carrying fras1 mutant endoderm show subtler defects than non-mosaic fras1 mutants, perhaps owing to rescue by a remnant of WT endodermal cells (see Carney et al., 2010), or partial anterior late-p1 formation due to anterior connections between late-p1 and the ectodermally derived stomadeum (Fig. 8B,C). Alternatively, the fras1→WT mosaic data could be explained by a model wherein fras1-dependent signals are sent from facial ectoderm to pharyngeal skeleton. Nonetheless, it is notable that some mosaic fish lacking fras1 only in the endoderm show facial defects, supporting a crucial role for endodermal fras1.
Fras1 protein might generate late-p1 by increasing adhesion between pharyngeal endoderm and underlying arch mesenchyme (Fig. 8B). For example, interaction between Fras1 and the mesenchymally expressed protein Frem1 is vital to Fras1 localization and function (Kiyozumi et al., 2006). Previous studies in mammalian cell culture have suggested Itga8 as another mesenchymally expressed partner of the Fraser complex (Kiyozumi et al., 2005); indeed, mammalian Itga8 [eMAGE (Richardson et al., 2010)] and zebrafish itga8 (J.C.T., unpublished observations) are expressed in pharyngeal arch mesenchyme. Many other studies have linked endodermal pouching to skeletal morphology (reviewed by Knight and Schilling, 2006). For instance, loss of early-p1 in zebrafish itga5 mutants reduces second arch mesenchymal adhesion, resulting in hyomandibular cartilage defects (Crump et al., 2004b). Furthermore, variable loss of posterior pouches in fgf3 mutants results in variable posterior cartilage fusions (Albertson and Yelick, 2005), analogous to the variable anterior arch cartilage fusions seen in fras1 mutants after late-p1 loss. Hence, we propose that fras1 acts in endoderm to autonomously sculpt late-p1 and to non-autonomously sculpt nearby skeletal elements.
Late-p1 might form a physical guide for skeletal development (Fig. 8). WT late-p1 morphogenesis brings endoderm through the symplectic-forming field (Fig. 8A,B), potentially exposing these cells to signals and physical forces that help drive symplectic extension (Fig. 8A, black arrows). WT late-p1 separates symplectic and ceratohyal cartilages (Fig. 8B); loss of late-p1 in fras1 mutants removes this barrier, facilitating the possibility of fusion later in development (Fig. 8A,B, brown arrows). We cannot explain Me-Pq fusions with an endodermal barrier model because these cartilage elements are not separated by endoderm, even in WT fish (Fig. 8C). Instead, late-p1 might tug on first arch cartilages to pull them apart (Fig. 8A,C, purple arrows), consistent with the physical models proposed for Sy-Ch fusion. Alternatively, late-p1 formation might bring signaling cues that activate joint formation (Fig. 8C, red-orange gradient) closer to the presumptive first arch joint.
The cartilage fusions in fras1 mutants highlight a more general feature of skeletal fusions: skeletal elements that possess similar identities tend to fuse to one another when normal patterning is disrupted. For example, symplectic and dorsal ceratohyal cartilages are both derived from the intermediate domain of the second arch (Coffin Talbot et al., 2010) and hence have very similar cell identities, allowing their fusion in fras1 mutants. By contrast, the first arch-derived palatoquadrate cartilage does not fuse to the second arch-derived symplectic cartilages in either WT or fras1 mutant embryos, even though they are not separated by epithelia, suggesting that WT symplectic cartilages are specified not to fuse with the palatoquadrate cartilage. When A-P identity is lost, as is seen in moz (kat6a – Zebrafish Information Network) mutants (Crump et al., 2006) and hoxa2-overexpressing embryos (Hunter and Prince, 2002), second arch cartilages are often found fused to first arch cartilages. Similarly, when arch identity is reduced along the dorsal-ventral axis, as is seen with edn1 (Walker et al., 2006) and Bmp (Alexander et al., 2011) pathway knockdowns, cartilages often fuse together along the dorsal-ventral axis. These findings suggest that nearby skeletal elements have a tendency to fuse together, unless molecularly instructed to do otherwise.
We propose that fras1 functions in anterior medial endoderm to sculpt late-p1 and thereby stabilize skeletal morphogenesis. The skeletal elements that require fras1 are intimately associated with late-p1, both in their time of developmental onset and in their spatial locations. In further support of this model, we find a strong correlation between the presence of late-p1 defects and the presence of cartilage defects. All fras1 mutants show severe late-p1 defects by 72 hpf, and almost all fras1 mutants present at least one skeletal defect. However, among fras1 mutants, the degree of endoderm defect does not predict symplectic length. We interpret this to mean that a critical threshold of late-p1 formation is required to buffer skeletal variation, but this threshold is not achieved in any fras1 mutant. Numerous studies have suggested that loss of developmental buffering might underlie stochastic phenotypic variation (reviewed by Dongen, 2006; Graham et al., 2010; Polak, 2003). In our model, the loss of fras1-dependent developmental stabilization provided by late-p1 allows inherent developmental instability (i.e. stochastic influences) to sometimes result in overt skeletal defects.
Developmental instability may also influence symptomatic expressivity in human Fraser syndrome (Cavalcanti et al., 2007). It is likely that genetic variation also influences some of the observed phenotypic variation in patients with Fraser syndrome (Slavotinek and Tifft, 2002). Indeed, mouse Frem1 mutants can show different phenotypic expressivity on different genetic backgrounds (Smyth et al., 2004). However, given that we expect low genetic and environmental variation between the left and right sides of individual fish heads, we interpret the increased variation between the two sides of individual fras1 mutant fish as evidence for a stochastic model, consistent with previous interpretations of fluctuating asymmetry (e.g. Polak, 2003). Further evidence for stochasticity arises when multiple axes are examined (Graham et al., 2010); we find facial defects to be asymmetric between the left versus right sides, arch one versus two (not shown), and early versus late developmental periods. Conversely, if genetic factors other than fras1 are the sole cause of phenotypic variation in fras1 mutants, we would expect to find different levels of phenotypic variation on different genetic backgrounds. This expectation is not met; zebrafish fras1 mutants identified in different genetic backgrounds appear very similar to one another in terms of both the type of facial defect and extent of facial variation. Similar to zebrafish fras1 mutants, human Fraser syndrome patients also show profound variation between siblings (Cavalcanti et al., 2007; Prasun et al., 2007), asymmetry within individual patients (Cavalcanti et al., 2007), and similar variation across distinct families (van Haelst et al., 2008). These observations of Fraser syndrome patients, combined with our observations of genetically similar fras1 mutant zebrafish raised in tightly controlled environments, indicate that stochastic processes influence Fraser syndrome variation.
Supplementary Material
Acknowledgements
We thank John Dowd and the University of Oregon Fish Facility for the care and maintenance of fish; Poh Kheng Loi and the University of Oregon Histology Laboratory for tissue sectioning; Joseph Okray for help in preparing pertinent background material; and Chi-Bin Chien's laboratory for providing sox9a:EGFP transgenics prior to publication.
Footnotes
Funding
This work was supported by the National Institutes of Health [grants DE13834 to C.B.K.; DE020076 to J.H.P.; HD22486 to J.H.P, C.B.K. and J.C.T.; T32 HD007348 to J.CT.; GM07413 to M.B.W.] and the German Research Foundation [Collaborative Research Centre SFB 829 (M.H.)]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.074906/-/DC1
References
- Alazami A. M., Shaheen R., Alzahrani F., Snape K., Saggar A., Brinkmann B., Bavi P., Al-Gazali L. I., Alkuraya F. S. (2009). FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am. J. Hum. Genet. 85, 414-418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson R. C., Yelick P. C. (2005). Roles for fgf8 signaling in left-right patterning of the visceral organs and craniofacial skeleton. Dev. Biol. 283, 310-321 [DOI] [PubMed] [Google Scholar]
- Alexander C., Zuniga E., Blitz I. L., Wada N., Le Pabic P., Javidan Y., Zhang T., Cho K. W., Crump J. G., Schilling T. F. (2011). Combinatorial roles for BMPs and Endothelin 1 in patterning the dorsal-ventral axis of the craniofacial skeleton. Development 138, 5135-5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arey L. B. (1966). Developmental Anatomy: a Textbook and Laboratory Manual of Embryology. Philadelphia and London: W. B. Saunders; [Google Scholar]
- Carney T. J., Feitosa N. M., Sonntag C., Slanchev K., Kluger J., Kiyozumi D., Gebauer J. M., Coffin Talbot J., Kimmel C. B., Sekiguchi K., et al. (2010). Genetic analysis of fin development in zebrafish identifies furin and hemicentin1 as potential novel fraser syndrome disease genes. PLoS Genet. 6, e1000907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcanti D. P., Matejas V., Luquetti D., Mello M. F., Zenker M. (2007). Fraser and Ablepharon macrostomia phenotypes: concurrence in one family and association with mutated FRAS1. Am. J. Med. Genet. A 143, 241-247 [DOI] [PubMed] [Google Scholar]
- Chiotaki R., Petrou P., Giakoumaki E., Pavlakis E., Sitaru C., Chalepakis G. (2007). Spatiotemporal distribution of Fras1/Frem proteins during mouse embryonic development. Gene Expr. Patterns 7, 381-388 [DOI] [PubMed] [Google Scholar]
- Coffin Talbot J., Johnson S. L., Kimmel C. B. (2010). hand2 and Dlx genes specify dorsal, intermediate and ventral domains within zebrafish pharyngeal arches. Development 137, 2507-2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump J. G., Maves L., Lawson N. D., Weinstein B. M., Kimmel C. B. (2004a). An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development 131, 5703-5716 [DOI] [PubMed] [Google Scholar]
- Crump J. G., Swartz M. E., Kimmel C. B. (2004b). An integrin-dependent role of pouch endoderm in hyoid cartilage development. PLoS Biol. 2, E244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump J. G., Swartz M. E., Eberhart J. K., Kimmel C. B. (2006). Moz-dependent Hox expression controls segment-specific fate maps of skeletal precursors in the face. Development 133, 2661-2669 [DOI] [PubMed] [Google Scholar]
- De Beer G. R. (1937). The Development of the Vertebrate Skull. Oxford, UK: Oxford University Press; [Google Scholar]
- Dongen S. V. (2006). Fluctuating asymmetry and developmental instability in evolutionary biology: past, present and future. J. Evol. Biol. 19, 1727-1743 [DOI] [PubMed] [Google Scholar]
- Fraser G. R. (1962). Our genetical ‘load’. A review of some aspects of genetical variation. Ann. Hum. Genet. 25, 387-415 [Google Scholar]
- Gattuso J., Patton M. A., Baraitser M. (1987). The clinical spectrum of the Fraser syndrome: report of three new cases and review. J. Med. Genet. 24, 549-555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier P., Naranjo-Golborne C., Taylor M. S., Jackson I. J., Smyth I. (2008). Expression of the fras1/frem gene family during zebrafish development and fin morphogenesis. Dev. Dyn. 237, 3295-3304 [DOI] [PubMed] [Google Scholar]
- Graham A., Okabe M., Quinlan R. (2005). The role of the endoderm in the development and evolution of the pharyngeal arches. J. Anat. 207, 479-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham J. H., Raz S., Hel-Or H., Nevo E. (2010). Fluctuating asymmetry: methods, theory, and applications. Symmetry 2, 466-540 [Google Scholar]
- Hamilton W. J., Boyd J. D., Mossman H. W. (1947). Human Embryology (Prenatal Development of Form and Function). Baltimore, MD: Williams and Wilkins; [Google Scholar]
- Hunter M. P., Prince V. E. (2002). Zebrafish hox paralogue group 2 genes function redundantly as selector genes to pattern the second pharyngeal arch. Dev. Biol. 247, 367-389 [DOI] [PubMed] [Google Scholar]
- Jadeja S., Smyth I., Pitera J. E., Taylor M. S., van Haelst M., Bentley E., McGregor L., Hopkins J., Chalepakis G., Philip N., et al. (2005). Identification of a new gene mutated in Fraser syndrome and mouse myelencephalic blebs. Nat. Genet. 37, 520-525 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253-310 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Miller C. T., Keynes R. J. (2001). Neural crest patterning and the evolution of the jaw. J. Anat. 199, 105-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby B. B., Takada N., Latimer A. J., Shin J., Carney T. J., Kelsh R. N., Appel B. (2006). In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci. 9, 1506-1511 [DOI] [PubMed] [Google Scholar]
- Kiyozumi D., Osada A., Sugimoto N., Weber C. N., Ono Y., Imai T., Okada A., Sekiguchi K. (2005). Identification of a novel cell-adhesive protein spatiotemporally expressed in the basement membrane of mouse developing hair follicle. Exp. Cell Res. 306, 9-23 [DOI] [PubMed] [Google Scholar]
- Kiyozumi D., Sugimoto N., Sekiguchi K. (2006). Breakdown of the reciprocal stabilization of QBRICK/Frem1, Fras1, and Frem2 at the basement membrane provokes Fraser syndrome-like defects. Proc. Natl. Acad. Sci. USA 103, 11981-11986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R. D., Schilling T. F. (2006). Cranial neural crest and development of the head skeleton. Adv. Exp. Med. Biol. 589, 120-133 [DOI] [PubMed] [Google Scholar]
- Lehman H. E. (1987). Chordate Development: a Practical Textbook with Atlases and Techniques for Experimental and Descriptive Embryology. Winston-Salem, NC: Hunter Textbooks; [Google Scholar]
- McGregor L., Makela V., Darling S. M., Vrontou S., Chalepakis G., Roberts C., Smart N., Rutland P., Prescott N., Hopkins J., et al. (2003). Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat. Genet. 34, 203-208 [DOI] [PubMed] [Google Scholar]
- Palmer R. A., Strobeck C. (2003). Fluctuating asymmetry analyses revisited. In Developmental Instability: Causes and Consequences (ed. Polak M.), pp. 279-319 Oxford, UK: Oxford University Press; [Google Scholar]
- Petrou P., Chiotaki R., Dalezios Y., Chalepakis G. (2007). Overlapping and divergent localization of Frem1 and Fras1 and its functional implications during mouse embryonic development. Exp. Cell Res. 313, 910-920 [DOI] [PubMed] [Google Scholar]
- Polak M. (ed.) (2003). Developmental Instability: Causes and Consequences. Oxford, UK: Oxford University Press; [Google Scholar]
- Prasun P., Pradhan M., Goel H. (2007). Intrafamilial variability in Fraser syndrome. Prenat. Diagn. 27, 778-782 [DOI] [PubMed] [Google Scholar]
- Richardson L., Venkataraman S., Stevenson P., Yang Y., Burton N., Rao J., Fisher M., Baldock R. A., Davidson D. R., Christiansen J. H. (2010). EMAGE mouse embryo spatial gene expression database: 2010 update. Nucleic Acids Res. 38, D703-D709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Mari A., Yan Y. L., Bremiller R. A., Wilson C., Canestro C., Postlethwait J. H. (2005). Characterization and expression pattern of zebrafish Anti-Mullerian hormone (Amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development. Gene Expr. Patterns 5, 655-667 [DOI] [PubMed] [Google Scholar]
- Shafeghati Y., Kniepert A., Vakili G., Zenker M. (2008). Fraser syndrome due to homozygosity for a splice site mutation of FREM2. Am. J. Med. Genet. A 146A, 529-531 [DOI] [PubMed] [Google Scholar]
- Slavotinek A., Li C., Sherr E. H., Chudley A. E. (2006). Mutation analysis of the FRAS1 gene demonstrates new mutations in a propositus with Fraser syndrome. Am. J. Med. Genet. A 140, 1909-1914 [DOI] [PubMed] [Google Scholar]
- Slavotinek A. M., Tifft C. J. (2002). Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. J. Med. Genet. 39, 623-633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek A. M., Baranzini S. E., Schanze D., Labelle-Dumais C., Short K. M., Chao R., Yahyavi M., Bijlsma E. K., Chu C., Musone S., et al. (2011). Manitoba-oculo-tricho-anal (MOTA) syndrome is caused by mutations in FREM1. J. Med. Genet. 48, 375-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth I., Scambler P. (2005). The genetics of Fraser syndrome and the blebs mouse mutants. Hum. Mol. Genet. 14 Spec. No. 2, R269-R274 [DOI] [PubMed] [Google Scholar]
- Smyth I., Du X., Taylor M. S., Justice M. J., Beutler B., Jackson I. J. (2004). The extracellular matrix gene Frem1 is essential for the normal adhesion of the embryonic epidermis. Proc. Natl. Acad. Sci. USA 101, 13560-13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallafuss A., Bally-Cuif L. (2003). Tracing of her5 progeny in zebrafish transgenics reveals the dynamics of midbrain-hindbrain neurogenesis and maintenance. Development 130, 4307-4323 [DOI] [PubMed] [Google Scholar]
- Thomas I. T., Frias J. L., Felix V., Sanchez de Leon L., Hernandez R. A., Jones M. C. (1986). Isolated and syndromic cryptophthalmos. Am. J. Med. Genet. 25, 85-98 [DOI] [PubMed] [Google Scholar]
- van Haelst M. M., Scambler P. J., Hennekam R. C. (2007). Fraser syndrome: a clinical study of 59 cases and evaluation of diagnostic criteria. Am. J. Med. Genet. A 143, 3194-3203 [DOI] [PubMed] [Google Scholar]
- van Haelst M. M., Maiburg M., Baujat G., Jadeja S., Monti E., Bland E., Pearce K., Hennekam R. C., Scambler P. J. (2008). Molecular study of 33 families with Fraser syndrome new data and mutation review. Am. J. Med. Genet. A 146, 2252-2257 [DOI] [PubMed] [Google Scholar]
- Vrontou S., Petrou P., Meyer B. I., Galanopoulos V. K., Imai K., Yanagi M., Chowdhury K., Scambler P. J., Chalepakis G. (2003). Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat. Genet. 34, 209-214 [DOI] [PubMed] [Google Scholar]
- Walker M. B., Kimmel C. B. (2007). A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem. 82, 23-28 [DOI] [PubMed] [Google Scholar]
- Walker M. B., Miller C. T., Coffin Talbot J., Stock D. W., Kimmel C. B. (2006). Zebrafish furin mutants reveal intricacies in regulating Endothelin1 signaling in craniofacial patterning. Dev. Biol. 295, 194-205 [DOI] [PubMed] [Google Scholar]
- Walker M. B., Miller C. T., Swartz M. E., Eberhart J. K., Kimmel C. B. (2007). phospholipase C, beta 3 is required for Endothelin1 regulation of pharyngeal arch patterning in zebrafish. Dev. Biol. 304, 194-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}