

Abstract
The aim of this study was to test a hypothesis that ascorbate depletion could enhance carcinogenicity and acute toxicity of nickel. Homozygous L-gulono-<gamma>-lactone oxidase gene knock-out mice (Gulo-/- mice) unable to produce ascorbate and wild-type C57BL mice (WT mice) were injected intramuscularly with carcinogenic nickel subsulfide (Ni3S2), and observed for the development of injection site tumors for 57 weeks. Small pieces of one of the induced tumors were transplanted subcutaneously into separate groups of Gulo-/- and WT mice and the growth of these tumors was measured for up to 3 months. The two strains of mice differed significantly with regard to (1) Ni3S2 carcinogenesis: Gulo-/- mice were 40% more susceptible than WT mice; and (2) transplanted tumors development: Gulo-/- mice were more receptive to tumor growth than WT mice, but only in terms of a much shorter tumor latency; later in the exponential phase of growth, the growth rates were the same. And, with adequate ascorbate supplementation, the two strains were equally susceptible to acute toxicity of Ni3S2. Statistically significant effects of dietary ascorbate dosing levels were the following: (1) reduction in ascorbate supplementation increased acute toxicity of Ni3S2 in Gulo-/- mice; (2) ascorbate supplementation extended the latency of transplanted tumors in WT mice. In conclusion, the lack of endogenous ascorbate synthesis makes Gulo-/- mice more susceptible to Ni3S2 carcinogenesis. Dietary ascorbate tends to attenuate acute toxicity of Ni3S2 and to extend the latency of transplanted tumors. The latter effects may be of practical importance to humans and thus deserve further studies.
Keywords: Ascorbate, gulonolactone oxidase, Gulo-/- mice, nickel carcinogenesis
Introduction
The idea that ascorbic acid confers resistance to neoplasia was introduced by Linus Pauling in 1970-s following the findings of low reserves of ascorbate in cancer patients and beneficial effects of ascorbate supplementation on their survival (Cameron et al., 1979). It has been known that carcinogenic transition metals, including nickel, can catalyze ascorbate destruction (Buettner and Jurkiewicz, 1996; Kaczmarek et al., 2007). Would this effect contribute to the mechanisms of nickel-induced carcinogenesis? In our previous publications (Salnikow et al., 2004; Salnikow and Kasprzak, 2005), we hypothesized that ascorbate depletion could, indeed, be a critical step in nickel carcinogenesis. The depletion is important because of the key role ascorbate plays in maintaining the enzymatic activity of specific non-heme dioxygenases that are responsible for DNA repair, gene expression regulation, collagen assembly, and the inhibition of hypoxic stress (a hallmark of cancer) (Salnikow and Kasprzak, 2005; Arita and Costa, 2009). Ascorbate depletion should also weaken the antioxidant cellular guard against metal-induced oxidative DNA, lipid and protein damage that is thought to play a mechanistic role in carcinogenesis (Bal et al., 2010; Kasprzak, 2011). The aim of the present study was to test the above hypothesis on muscle carcinogenesis induced by nickel subsulfide (Ni3S2).
Unlike humans the laboratory rats and mice, on which most of the nickel (and other metals) carcinogenicity has been studied thus far, synthesize ascorbate endogenously. It was thus impossible to design a study in which their tissue ascorbate level was changed following metal exposure. In this study, to achieve our research goal, we decided to use mice with inactivated L-gulono-<gamma>-lactone oxidase gene (Gulo-/- mice), which make them unable to synthesize ascorbate. This allowed us to control dietary ascorbate uptake, as well as to evaluate changes in tissue ascorbate in nickel exposed animals. As a reference for comparison we used C57BL mice from which the Gulo-/- mice have been derived (Maeda et al., 2000).
Materials and Methods
Animals
This study was conducted in compliance with the Intramural Animal Care and Use (ACU) program of the National Institutes of Health. The homozygous L-gulono-γ-lactone oxidase knock-out mice (Gulo-/- mice) and wild-type C57BL/6 background mice (WT mice) were derived from heterozygous BL6.129P2-Gulotm1/Unc /Ucd mice by in-house breeding at animal facilities of the Laboratory Animal Science Program, SAIC - Frederick (NCI at Frederick, Frederick, MD). The heterozygous breeders were purchased from the University of California Center for Comparative Medicine (MMRRC) at Davis, CA. All WT mice were kept on an ascorbic acid-free diet, PicoLab Mouse Diet 20/5058 (Quality Lab Products, Inc., Elkridge, MD) and HCl-acidified drinking water (pH 4). The Gulo-/- mice were offered 330 mg/L of ascorbic acid (Sigma-Aldrich, St. Louis, MO)-containing water starting at weaning at 3 weeks of age (full supplementation; (Maeda et al., 2000; Telang et al., 2007). Some experimental groups were kept on reduced, 100 mg/L, ascorbic acid supplementation.
Genotyping was performed as described by Maeda et al. (Maeda et al., 2000), using three PCR primers: P2 (5’-CGCGCCTTAATTAAGGATCC-3’), P3 (5’-GTCGTGACAGAATGTCTTGC-3’), and P4 (5’-GCATCCCAGTGACTAAGGAT-3’). The amplification conditions were the following: initial DNA denaturation 94°C for 3 min; 35 cycles of 30 sec denaturing at 94°C, 30 sec. annealing at 58°C, 45 sec extension at 72°C and 8 min final extension at 72°C. All experiments were performed on 6-10 weeks old male mice.
Treatments
For the carcinogenicity tests, the mice, divided randomly into groups of 15 – 22 animals received single intramuscular (i.m.) injections of 2.0 or 2.5 mg/site of Ni3S2 powder, 8.3 μm geometric mean particle size (volume-based; determined by Elzone analyzer), suspended in 50% aqueous glycerol, 0.05 ml/site, into the thigh musculature of both hind limbs, using gauge 25 needle. The control mice received 0.05 ml/site of the injection vehicle alone. The Ni3S2 powder was a generous gift of Dr. A. Oller (NiPERA, Durham, NC).
The mice were examined weekly by palpation for tumor development, starting at week 24 post-injection. They were euthanized with CO2 when a local tumor reached approximately 1 cm in diameter, or at termination of the bioassay at week 57.
The effect of ascorbate on acute toxicity of Ni3S2-treated mice was assessed based on one-week mortality data from the two carcinogenicity experiments and an additional short-term test in which five groups of 18 mice were injected with 2.5 mg Ni3S2/site as in the carcinogenicity experiments and observed for one week. Kidneys of mice dying after nickel exposure were fixed in 10% formalin for microscopic examination.
To check the effect of ascorbic acid on transplanted tumor growth, two groups of six Gulo-/- plus two groups of five WT mice each, supplemented with different levels of ascorbic acid (as in the carcinogenesis experiment) were grafted subcutaneously on both flanks with 2 - 3 mm chunks of a muscle tumor induced with Ni3S2 in a Gulo-/- mouse kept on 330 mg/L of ascorbate in drinking water. The tumor to be transplanted, approx. 1 cm in size, free of necrosis, was collected at sacrifice and minced with scissors in normal saline. The transplants were inserted with fine forceps, randomly, under ether anesthesia, through 5 mm incisions, which were closed with metal clips (removed after 3 days). The growing tumors were measured in weekly intervals for up to 3 month post-transplantation.
To determine tissue levels of ascorbic acid in blood, kidneys, lungs, and muscles, groups of 12 – 14 Gulo-/- and WT mice were injected i.m. with 2.0 mg doses of Ni3S2 in the same way as described for the carcinogenicity bioassays. For analysis, surviving mice were sacrificed with CO2 and autopsied 1 or 7 days after the injection.
Ascorbic acid analysis
Tissue ascorbate levels were measured using Lykkesfeldt’s procedure (Lykkesfeldt, 2000) with HPLC separation and electrochemical detection as described elsewhere (Kaczmarek et al., 2007).
Statistical evaluation
The results of the carcinogenicity bioassay were assessed by testing the end point results (final survival populations) with Fisher’s exact test (Enderlein, 1987). The results of the Ni3S2 acute toxicity testing were evaluated statistically using a number of non-parametric analyses. The possibility of pooling the results of analogous groups from different experiments was examined using an ANOVA table with a Kruskal-Wallis non-parametric test (Hollander and Wolfe, 1999).
The transplanted tumors growth was evaluated considering four possible growth profiles. In profile 1, the tumors were considered to begin growing immediately after grafting in a linear progression. In profile 2, the tumors began to grow immediately in an exponential progression. In the third and forth profiles, an initial delay in the tumor growth (incubation period) followed by either a linear or exponential growth progression were considered. Different detailed descriptions of the transplanted tumor growth were unified by noting that there was a delay followed by a growth rate.
The results of the growth delay and growth rate parameters from non-linear regression analysis of each tumor were used to search for a linear relationship between the tumor growth and mouse flank. A correlation analysis showed the extent of the tumor growth dependence on individual mice.
An ANOVA analysis of the non-linear regression analysis results was used to search for significant differences in the rate and delay of tumor growth for the four mouse groups (Hogg and Ledolter, 1987).
A global nonlinear regression was used to look for differences in tumor growth delay among the four mouse groups (Seber and Wild, 1989). Significance of differences in tissue ascorbate levels was established using student’s t-test.
Results
Carcinogenesis
The survival data and incidence of tumors in the carcinogenesis bioassays are presented in Tables 1 and 2. All the injection site tumors developed only in one leg. They readily invaded the adjacent muscles and bone marrow. The tumors grew at uneven rates, reaching the terminal size of 1 cm in 4 to 8 weeks from the time of detection by palpation. The time of appearance of the first tumor (latent period) in each group varied from 30 to 40 weeks after injections, and the final tumor incidence range varied from 11 – 67% of mice that survived until the occurrence of the first tumor in this study (30 weeks). Control mice developed no tumors. Histologically, all the injection site tumors were fibrosarcomas. No other tumors, either primary or metastatic, were found.
Table 1.
Mortality and tumor incidence in mice injected with 2.0 mg Ni3S2/thigh muscle1
| Group No. | Treatment | Total No. of mice | Mice dead in 1 week4 | No. of mice at tumor risk (surviving > 30 wks.) | Mice with injection site tumors5 | Tumor-bearing mice euthanized at week6 | ||
|---|---|---|---|---|---|---|---|---|
| No. | % total | No. | % at risk | |||||
|
| ||||||||
| 1 | Gulo-/-, 1002 | 20 | 7 | 35.0 | 12 | 8 | 66.7 | 40, 43, 47, 49, 54, 54, 56, 56 |
|
| ||||||||
| 2 | Gulo-/-, 3302 | 20 | 9 | 45.0 | 11 | 4 | 36.4 | 40, 42, 48, 49 |
|
| ||||||||
| 3 | Gulo-/-, 3302/sham3 | 15 | 0 | 0 | 15 | 0 | 0 | No tumors |
|
| ||||||||
| 4 | WT | 21 | 5 | 23.8 | 16 | 4 | 25.0 | 34, 43, 48, 51 |
|
| ||||||||
| 5 | WT, 3302 | 19 | 10 | 52.6 | 9 | 1 | 11.1 | 38 |
Thigh musculature of both hind limbs; at week 0
Mice offered 100 or 330 mg ascorbic acid-containing drinking water, respectively, from weaning to life termination
Mice injected with the injection vehicle only
Renal failure
Tumor in one leg only. No tumors were found at other sites of the body
When tumor reached approx. 1 cm in diameter
Table 2.
Mortality and tumor incidence in mice injected with 2.5 mg Ni3S2/thigh muscle1
| Group No. | Treatment | Total No. of mice | Mice dead in 1 week4 | No. of mice at tumor risk (surviving > 30 wks.) | Mice with injection site tumors5 | Tumor-bearing mice euthanized at week6 | ||
|---|---|---|---|---|---|---|---|---|
| No. | % total | No. | % at risk | |||||
|
| ||||||||
| 6 | Gulo-/-, 1002 | 18 | 14 | 77.8 | 3 | 2 | 66.7 | 38, 42 |
|
| ||||||||
| 7 | Gulo-/-, 3302 | 18 | 9 | 50.0 | 9 | 5 | 55.6 | 45, 48, 48, 48, 54 |
|
| ||||||||
| 8 | Gulo-/-, 3302/sham3 | 17 | 2 | 11.8 | 15 | 0 | 0 | No tumors |
|
| ||||||||
| 9 | WT | 24 | 11 | 45.8 | 12 | 3 | 25.0 | 48, 54, 57 |
|
| ||||||||
| 10 | WT, 3302 | 24 | 9 | 37.5 | 15 | 3 | 20.0 | 42, 54, 57 |
Thigh musculature of both hind limbs; at week 0
Mice offered 100 or 330 mg ascorbic acid-containing drinking water, respectively, from weaning to life termination
Mice injected with the injection vehicle only
Renal failure
Tumor in one leg only. No tumors were found at other sites of the body
When tumor reached approx. 1 cm in diameter
The observed differences in the development of the injection site tumors appeared to depend on the treatments and/or mouse strain, but due to the high initial mortality among Ni3S2-treated mice (see below) the number of induced tumors was small and the real importance of these differences had to be confirmed by statistical means. The end point statistical analysis, using Fisher’s exact test, considered mice that survived past the acute stage of nickel toxicity, i.e., long enough to develop tumors. This analysis disclosed that the differences in tumor development between Gulo-/-, 100 mice and Gulo-/-, 330 mice and between WT and WT 330 mice in each experiment taken separately were not significant. Likewise non-significant were apparent differences between the remaining pairs of groups with obvious exceptions for pairs that included the sham-injected mice.
To overcome the statistical limitations of the small group sizes we next checked, using the log-rank test for survival, if those sizes could be increased by pooling similar groups given different Ni3S2 doses, i. e., groups 1 and 6, 2 and 7, 4 and 9, and 5 and 10, respectively (Tables 1 and 2). The results of this test clearly indicated that both Ni3S2 doses had the same effect on tumor development (p = 1) and thus the corresponding groups could be pooled together. The subsequent analysis of the pooled groups, too, showed no difference between the Gulo-/-, 100 and Gulo-/-, 330 (p = 1) and between the WT and WT, 330 mice (p = 0.19). Hence, the ascorbic acid supplementation regime had no significant effect on tumor yield in either mouse strain. This allowed for further pooling: of four Gulo-/- groups (1, 2, 6, and 7) and four WT (groups 4, 5, 9, 10) to compare strain response to Ni3S2. In this case, Fisher’s exact test indicated a significant difference (p = 0.005), with Gulo-/- mice being more susceptible than WT mice to Ni3S2 –induced carcinogenesis.
Acute toxicity
The mortality data are presented in Tables 1 - 3. They indicate that Ni3S2 at both doses tested was lethal to many animals in the first week after administration. Mice that survived the critical first week post-injection had a very low mortality afterwards and lived past the detection (palpation) of a local tumor or the termination of this experiment at wk. 57. Only 3 of them (in groups 1, 6, and 9) died before wk. 30, i.e. the time of appearance of the first tumor. At autopsy and histologic examination, the kidneys of mice dying in the first week post-injection were found to be enlarged with pitted and mottled surface. They often contained cysts and thickened Bowman capsules. Protein-containing droplets were visible in slightly increased mesangial matrix. Multiple foci of inflammation and necrosis were visible in renal tubular epithelium. There were also some necrotic foci in the glomeruli. Thus, renal failure was the most likely cause of death.
Table 3.
Short-term mortality in mice injected with 2.5 mg Ni3S2/thigh muscle1
| Treatment | Total No. of mice | No. of mice dead in 1 week4 | |
|---|---|---|---|
| No. | % total | ||
|
| |||
| Gulo-/-, 1002 | 18 | 13 | 72.2 |
|
| |||
| Gulo-/-, 3302 | 18 | 10 | 55.6 |
|
| |||
| Gulo-/-, 3302/sham3 | 18 | 0 | 0 |
|
| |||
| WT | 18 | 9 | 50.0 |
|
| |||
| WT, 3302 | 18 | 7 | 38.9 |
Thigh musculature of both hind limbs; at week 0
Mice offered 100 or 330 mg ascorbic acid-containing drinking water, respectively, from weaning to life termination
Mice injected with the injection vehicle only
Renal failure
The results were analyzed statistically for significantly differential mortality depending on Ni3S2, ascorbic acid doses, and mouse strain. In regard to the two Ni3S2 doses for the same ascorbate treatments, the analysis disclosed a significant difference in toxicity only in the Gulo-/-, 100 mice (groups 1 and 6; p < 0.02 by Kruskal-Wallis test). The relatively small increase in nickel dose from 2.0 to 2.5 mg Ni3S2/site caused over two-fold increase in mortality of these mice. In contrast, there was no significant nickel dose effect on the Gulo-/-, 330 mice (groups 2 and 7). Likewise, the mortality of WT mice given either 0 or 330 mg/L of ascorbate in drinking water (groups 4 and 9, and 5 and 10, respectively) was not significantly different at the two nickel doses.
To test the effect of ascorbic acid dosing on the acute toxicity of a given nickel dose (2.5 mg Ni3S2/site), the mortality data from Table 2 were pooled with the data from analogous treatment groups from the short-term toxicity experiment (Table 3) and analyzed using the Kruskal-Wallis test. This analysis showed a significant ascorbic acid dosing affect only for the Gulo-/-, 100 group versus Gulo-/-, 330 group (p < 0.009), but no such effect for the WT and WT, 330 groups. Since there were no significant differences in mortality among the WT groups in all three experiments, (Tables 1, 2, and 3), the corresponding groups could be pooled to increase the statistical base for analysis. However, this procedure, too, did not reveal any effect of the ascorbic acid supplementation on the one-week mortality of Ni3S2-injected WT mice.
Possible effect of strain difference on the acute toxicity of a given nickel dose at similar organismal ascorbic acid status (Maeda et al., 2000) could be assessed using Kruskal-Wallis test to compare the short-term mortality of WT mice (pooled groups 4, 9 from Tables 1 and 2, plus the WT group from Table 3) with that of Gulo-/-, 330 mice (pooled groups 2, 7 from Tables 1 and 2, plus the Gulo-/-, 330 group from Table 3). No significant difference was found (p = 0.26).
Tumor transplants growth
In the present study, the growth of the transplanted tumors was characterized by two parameters: incubation period (the length of time during which no growth was detected after the transplant) and growth rate (weekly size increase after the tumor started to grow). Single regression analysis of the tumor growth model was completed for each tumor in the study. Its results were then used to determine if there was a correlation between the tumor flank (right or left thigh) and the tumor growth rate or incubation period. No such correlations were found in any of the four groups injected with Ni3S2 (p = 0.5 based on a t-distribution). In addition, no significant correlation was found among flank and tumor growth for the pooled WT and pooled Gulo-/- mice (p = 0.3). This analysis also showed no evidence that the two tumors in a mouse had similar growth rates or incubation periods. Therefore, we could assume that the tumor growth results were independent observations.
The ANOVA tests revealed that tumor growth rates did not significantly vary between these four Ni3S2-treated mouse groups (similar slopes in Figure 1). In contrast, the incubation period did show significant variation between these four groups (p < 0.02). The extent of differences in this period was examined through a global regression.
Figure 1.
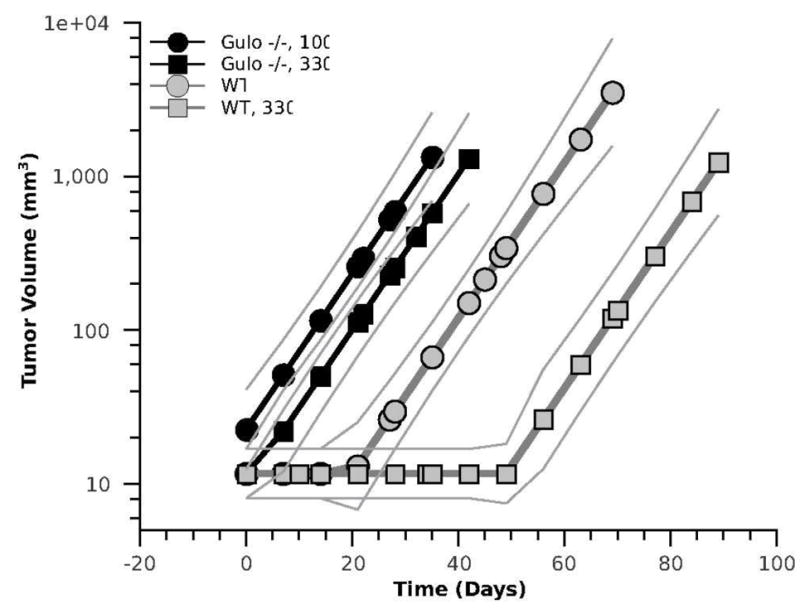
Regression analysis plots of tumor transplants growth (solid lines flanked by faint lines representing 95% confidence intervals). Tumors transplanted into Gulo-/- mice started to grow shortly after grafting with no significant difference between dietary ascorbate supplementation levels. Tumors transplanted into WT mice started to grow only after a 20-, or 50-day incubation period, depending on ascorbate supplementation.
A global fit of the tumor growth data was used to determine the significance of differences in the incubation periods between the four mouse groups (Wald, 1945). The results of this fit, shown in Figure 2, indicated that these differences were highly significant (p < 0.001) for the WT mice (WT versus WT, 330), but not for the Gulo-/- mice (Gulo-/-, 100 versus Gulo-/-, 330). The incubation period was also significantly longer in the WT mice than in the Gulo-/- mice.
Figure 2.
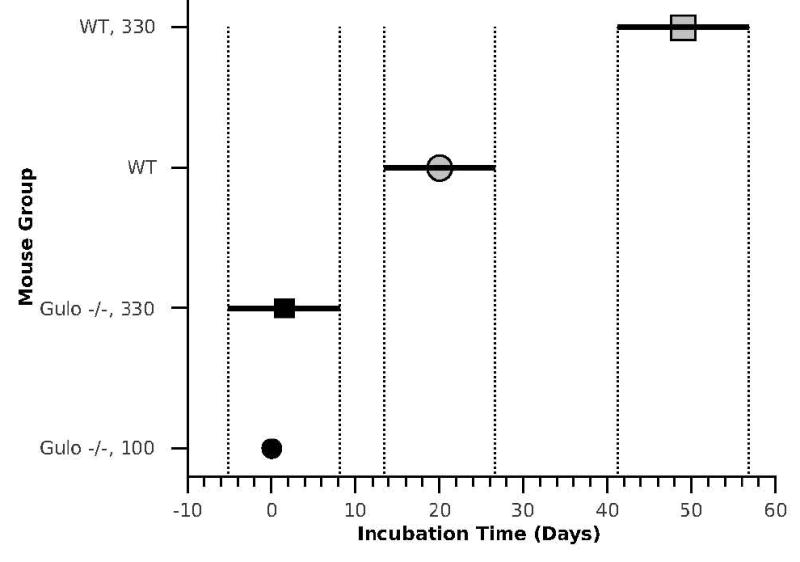
Incubation time of transplanted tumor growth as a function of mouse strain and ascorbic acid supplementation. The 95% confidence intervals for the growth delay values (vertical lines) are based on Tukey’s honest significance difference calculations (Hochberg and Tamhane, 1987). Unlike both groups of Gulo-/- mice, in which the tumor transplants started growing exponentially with no measurable delay, the WT mice show a significant delay in tumor growth after transplantation with ascorbate supplementation extending the delay even further.
Ascorbic acid analysis
The results of tissue ascorbate analysis are presented in Figure 3. As can be seen in this figure, the levels of ascorbate in the kidneys, lungs, muscles, and blood in Gulo-/-, 100 mice were markedly lower than in the corresponding tissues of WT mice. For the untreated mice, this difference was relatively greatest in the kidneys (4-fold, p < 0.002), followed by blood plasma (2.5-fold; p < 0.01), lung (2.2-fold; p < 0.01), and skeletal muscles (1.6-fold; p < 0.001). Nickel treatment significantly lowered ascorbate levels in the tissues of Gulo-/-, 100 mice (except for plasma) at day 1 after injection, which then tended to rebound (except in the muscles) at day 7. The effect of nickel on tissue ascorbate levels in WT mice, although apparently suppressive, did not reach statistically significant degree, except in muscles at day 7 post-injection (1.3-fold reduction vs. control; p < 0.01).
Figure 3.
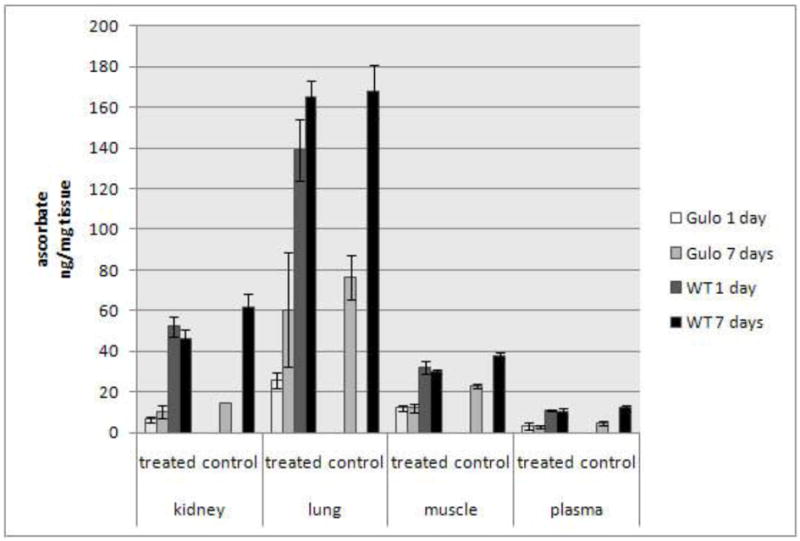
Tissue and blood plasma levels of ascorbate (ng/mg wet tissue +/- SE, n = 3 – 4) in WT and Gulo-/-, 100 mice at days 1 and 7 following i.m. injections of 0 (control; the same for days 1 and 7) or 2.0 mg Ni3S2/thigh (treated).
Discussion
In this study, we examined the role of ascorbate in the carcinogenicity and acute toxicity of Ni3S2 by comparing Ni3S2 effects in wild-type C57BL/6 mice (WT mice) with those in mice of the same strain with inactivated L-gulono-<gamma>-lactone oxidase gene (Gulo-/- mice). Inactivation of this gene eliminates the ability of mammals to endogenously synthesize ascorbate. The Ni3S2 dosing and treatment regimens were derived from our previous experiment in which the susceptibilities of three mouse strains (C57BL/6, (C57BL/6xC3H/He)F1, andC3H/He) to the acute toxicity and carcinogenicity of nickel were compared (Rodriguez et al., 1996). Among these strains, C57BL mice had the lowest susceptibility to Ni3S2-induced carcinogenesis and the highest susceptibility to the lethal effects of Ni3S2. The present results confirmed this pattern. They also indicate that the susceptibility to carcinogenesis may be increased by disabling endogenous ascorbate synthesis.
The dietary ascorbate supplementation levels had no significant influence on nickel carcinogenesis in either WT or Gulo-/- mice. It only affected nickel-induced acute toxicity in Gulo-/- mice: the mortality rates in Gulo-/-, 100 mice were significantly higher than those in Gulo-/-, 330 mice regardless of Ni3S2 dose. However, under the present experimental conditions, dietary ascorbate supplementation of WT mice did not improve their survival after Ni3S2 administration but rather tended to decrease it (although this trend did not reach statistical significance). The major cause of death following Ni3S2 injection was renal failure associated with a severe necrotizing/inflammatory response in the renal tubular epithelium. This concurs with previous reports (Waalkes et al., 1985; Rodriguez et al., 1996) and is most likely associated with metal-promoted oxidative damage (Misra et al., 1991). Ni3S2 in tissues undergoes oxidative dissolution to Ni(II), which is then distributed by blood throughout the body and finally excreted through the kidney (Sunderman and Selin, 1968; Kasprzak and Sunderman, 1977; Rodriguez et al., 1996), thus exposing the kidney to the entire dose of nickel. Ascorbate is generally considered as an antioxidant. Therefore, it might not be surprising that the relatively lower dosing of ascorbate made the Gulo-/-, 100 mice more sensitive to nickel toxicity than the Gulo-/, 330 or WT mice. However, this conclusion must be viewed with caution because, remarkably, giving the WT mice additional ascorbate did not attenuate Ni3S2 morbidity. These findings reveal the complex nature of ascorbate effects, especially in conjunction with nickel, one of several transition metals able to generate damaging reactive oxygen species with ascorbic acid (Kasprzak and Hernandez, 1989).
Ascorbate regulates many cell and tissue functions that may be critical for cancer development (Salnikow and Kasprzak, 2005). However, some of these functions may produce opposing effects. For example, ascorbate deficiency can activate angiogenic growth factor VEGF, which is necessary to promote tumor growth through neo-vascularization, and can also impair collagen fiber assembly that is important for blood vessel formation and tumor growth (Telang et al., 2007). In addition, ascorbate is important for immune response regulation, which may also affect tumor development (Naidu, 2003). Overall, because of the complexity of cellular/tissue functions of ascorbate, experimental studies on its effects on cancer have yielded equivocal results (Telang et al., 2007; Chen et al., 2008). Thus, tumor development will depend on a fine tuning of cellular ascorbate levels influenced not only by the ascorbate dosing but also by nickel-catalyzed ascorbate destruction (Kaczmarek et al., 2007).
As mentioned above, we found no significant differences in Ni3S2 carcinogenicity among the individual treatment groups to indicate possible effects of ascorbate dosing. This could be due to the inherent low susceptibility to nickel-induced carcinogenesis of C57BL mice combined with their high mortality, which made the number of mice at risk for tumor development in individual treatment groups, and the final number of tumors, too low to produce statistical differences, if any. Therefore we looked for possible effects of ascorbate on the growth of tumor transplants originating from a fibrosarcoma induced in a Gulo-/-, 300 mouse earlier in this study. The transplants could be investigated in abundance more amenable to statistical analysis. We found that in both groups of Gulo-/- mice, irrespectively of the dietary ascorbate supplementation level, the transplants started to grow into sizeable tumors immediately, whereas in WT mice the onset of tumor growth was delayed by approximately 20 days in WT to 50 days in WT, 330 mice. Hence, the tissue microenvironment of Gulo-/- mice was more receptive to tumor development than that of WT mice.
The mechanisms underlying this observation are not clear. One possibility is the lack of endogenous ascorbate synthesis and thus differences in ascorbate homeostasis between these two mouse strains. Ascorbate deficiency, which can develop only in Gulo-/- mice, could, through HIF-1α activation, promote secretion of the VEGF from the transplanted tumor and thus facilitate tumor vascularization and expansion. Preventing angiogenesis has been reported to induce dormancy in tumors growing in vivo (Gimbrone et al., 1972). Consequently, supplementation with ascorbate should inhibit tumor development, as we indeed observed in WT mice. However, whereas the low ascorbate levels in Gulo-/-, 100 mice at the time of tumor grafting (compare the control levels in Fig. 3) favored a prompt start of tumor growth, the levels of ascorbate in Gulo-/-, 330 mice should be similar to those in WT mice, in which the onset of growth was markedly delayed. Nonetheless, in both Gulo-/- treatment groups, the tumors grew with no delay. Thus, other factors specific for Gulo-/- mice must have contributed to supporting the tumor grafts in addition to low tissue ascorbate. Such factors could include impaired immunity, reported for Gulo-/- mice (Vissers and Wilkie, 2007). Host immunity could also contribute to the initial dormancy of the transplants in WT mice, but only if the transplanted tumor, originally induced in Gulo-/- mouse, was not fully immuno-compatible with the WT mouse. But this remains to be established. Likewise it remains unclear why the factors, which caused transplant dormancy, did not affect tumor growth rates at the exponential growth phase (the same slopes for all groups in Fig. 1). Answering these questions will require further investigations.
The initiation of nickel-induced carcinogenesis has been associated with oxidative DNA damage by reactive oxygen species (ROS) generated by this metal. Such damage, if not repaired, may lead to mutations and neoplastic transformation. ROS may also be involved in the promotion and progression steps of cancer development (Kasprzak et al., 2003; Bal et al., 2010; Kasprzak, 2011). Therefore, it is believed that antioxidants, including ascorbate, have the potential to prevent cancer. Accordingly, the low relative susceptibility of WT mice to nickel carcinogenesis has been correlated with their relatively high intrinsic antioxidant potential (Rodriguez et al., 1996). However, ascorbate is not the only factor influencing tumor development. There are many other factors, some clearly specie-, strain-, and tissue-specific (Sunderman, 1983; Kasprzak et al., 2003), and ascorbate’s interplay with them has yet to be explored.
As we found, Gulo-/- mice are only slightly more susceptible to muscle carcinogenesis by nickel than their already low susceptible parental wild-type C57BL mice. Therefore, despite their dependence on exogenous ascorbate, Gulo-/- mice may not be the best choice to test ascorbate effects on nickel carcinogenesis. Nickel is generally a stronger carcinogen in rats than in mice (Sunderman, 1983); and rats have been more frequently used in metal carcinogenicity experiments than mice. Hence, we speculate that the Wistar-derived mutants, ODS Shionogi rats, which also depend on dietary ascorbate, should be a better model than Gulo-/- mice. Guinea pigs, which, similarly to humans, are naturally dependent on dietary ascorbate, may also be a reasonable model; but there are no published data on guinea pig susceptibility to nickel-induced carcinogenesis. Also, because lung is the major carcinogenic target in humans, and ascorbate has been found to be depleted by nickel in lung tissue to a higher extent than in muscle (Fig. 3), a lung carcinogenesis model may be more appropriate for future experiments than the more common skeletal muscle model herein.
In conclusion, this study finds ascorbate as a potentially important part of the molecular mechanisms of nickel carcinogenesis and acute toxicity. Although the complexity of carcinogenesis and the cytotoxicity of the carcinogen might have obscured some ascorbic acid effects, our results reveal increased receptivities of the dietary ascorbate-dependent Gulo-/- mice to Ni3S2 carcinogenesis and transplanted tumors growth relative to those of WT mice. Ascorbate deficiency resulted in higher susceptibility of the Gulo-/- mice to the acute toxicity of nickel, and ascorbate supplementation resulted in extension of the incubation period of tumors transplanted into WT mice. The latter effect may inhibit the formation of cancer metastases. Thus, the possibility of attenuating harmful effects of nickel by ascorbate may be of practical importance to humans and this deserves further studies.
Research Highlights.
Destruction of ascorbate by heavy metals decreases antioxidant defense and suppresses function of numerous hydroxylases
We propose to use Gulo-/- mice, which are like humans unable to synthesize ascorbate to test the effect of carcinogenic nickel
The reduction in ascorbate levels increased acute toxicity induced by Ni3S2 in Gulo-/- mice
Gulo-/- mice were found more susceptible than wild-type mice to nickel-induced carcinogenesis
Acknowledgments
This paper is dedicated to the memory of Dr. F. William Sunderman, Jr. (1931 -2011), whose seminal discoveries in nickel toxicology and carcinogenesis have been inspirational to so many followers.
The authors are grateful to Lisa Riffle, Susan L. North, and Robert M. Bare for skillful technical assistance, and Dr. Anna Maciag for critical discussion of this paper. This publication has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- Gulo-/- mice
L-gulono-γ-lactone oxidase knock-out mice
- WT
wild type
- i.m.
intramuscular injections
Footnotes
Conflict of interest: The authors declare that there is no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arita A, Costa M. Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics. 2009;1:222–228. doi: 10.1039/b903049b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal W, Protas AM, Kasprzak KS, editors. Metal Ions in Toxicology: Effects, Interactions, Interdependencies. Royal Society of Chemistry Press; Cambridge, UK: 2010. [Google Scholar]
- Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145:532–541. [PubMed] [Google Scholar]
- Cameron E, Pauling L, Leibovitz B. Ascorbic acid and cancer: a review. Cancer Res. 1979;39:663–681. [PubMed] [Google Scholar]
- Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enderlein G. Cox, D. R.; Oakes, D.: Analysis of Survival Data Chapman and Hall, London – New York 1984, 201 S. Biometrical Journal. 1987;29:114–114. [Google Scholar]
- Gimbrone MA, Jr, Leapman SB, Cotran RS, Folkman J. Tumor dormancy in vivo by prevention of neovascularization. J Exp Med. 1972;136:261–276. doi: 10.1084/jem.136.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg Y, Tamhane AC. Multiple comparison procedures. John Wiley & Sons; New York, NY, USA: 1987. [Google Scholar]
- Hogg RV, Ledolter J. Applied Statistics for Engineers and Physical Scientists. Macmillan Publishing Company; New York: 1987. [Google Scholar]
- Hollander M, Wolfe DA. Nonparametric statistical methods. John Wiley & Sons Inc; New York: 1999. [Google Scholar]
- Kaczmarek M, Timofeeva OA, Karaczyn A, Malyguine A, Kasprzak KS, Salnikow K. The role of ascorbate in the modulation of HIF-1alpha protein and HIF-dependent transcription by chromium(VI) and nickel(II) Free Radic Biol Med. 2007;42:1246–1257. doi: 10.1016/j.freeradbiomed.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzak KS. Role of Oxidative Damage in Metal-Induced Carcinogenesis. Springer Verlag Dusseldorf; Germany: 2011. [Google Scholar]
- Kasprzak KS, Hernandez L. Enhancement of hydroxylation and deglycosylation of 2’-deoxyguanosine by carcinogenic nickel compounds. Cancer Res. 1989;49:5964–5968. [PubMed] [Google Scholar]
- Kasprzak KS, Sunderman FW., Jr Mechanisms of dissolution of nickel subsulfide in rat serum. Res Commun Chem Pathol Pharmacol. 1977;16:95–108. [PubMed] [Google Scholar]
- Kasprzak KS, Sunderman FW, Jr, Salnikow K. Nickel carcinogenesis. Mutat Res. 2003;533:67–97. doi: 10.1016/j.mrfmmm.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Lykkesfeldt J. Determination of ascorbic acid and dehydroascorbic acid in biological samples by high-performance liquid chromatography using subtraction methods: reliable reduction with tris[2-carboxyethyl]phosphine hydrochloride. Anal Biochem. 2000;282:89–93. doi: 10.1006/abio.2000.4592. [DOI] [PubMed] [Google Scholar]
- Maeda N, Hagihara H, Nakata Y, Hiller S, Wilder J, Reddick R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci U S A. 2000;97:841–846. doi: 10.1073/pnas.97.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra M, Rodriguez RE, North SL, Kasprzak KS. Nickel-induced renal lipid peroxidation in different strains of mice: concurrence with nickel effect on antioxidant defense systems. Toxicol Lett. 1991;58:121–133. doi: 10.1016/0378-4274(91)90166-4. [DOI] [PubMed] [Google Scholar]
- Naidu KA. Vitamin C in human health and disease is still a mystery? An overview. Nutr J. 2003;2:7. doi: 10.1186/1475-2891-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez RE, Misra M, Diwan BA, Riggs CW, Kasprzak KS. Relative susceptibilities of C57BL/6, (C57BL/6 x C3H/He)F1, and C3H/He mice to acute toxicity and carcinogenicity of nickel subsulfide. Toxicology. 1996;107:131–140. doi: 10.1016/0300-483x(95)03251-a. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279:40337–40344. doi: 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Kasprzak KS. Ascorbate depletion: a critical step in nickel carcinogenesis? Environ Health Perspect. 2005;113:577–584. doi: 10.1289/ehp.7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seber GA, Wild CJ. Nonlinear Regression. John Wiley & Sons; New York: 1989. [Google Scholar]
- Sunderman FW., Jr Organ and species specificity in nickel subsulfide carcinogenesis. Basic Life Sci. 1983;24:107–127. doi: 10.1007/978-1-4684-4400-1_6. [DOI] [PubMed] [Google Scholar]
- Sunderman FW, Jr, Selin CE. The metabolism of nickel-63 carbonyl. Toxicol Appl Pharmacol. 1968;12:207–218. doi: 10.1016/0041-008x(68)90033-1. [DOI] [PubMed] [Google Scholar]
- Telang S, Clem AL, Eaton JW, Chesney J. Depletion of ascorbic acid restricts angiogenesis and retards tumor growth in a mouse model. Neoplasia. 2007;9:47–56. doi: 10.1593/neo.06664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers MC, Wilkie RP. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1alpha. J Leukoc Biol. 2007;81:1236–1244. doi: 10.1189/jlb.0806541. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Kasprzak KS, Ohshima M, Poirier LA. Protective effects of zinc acetate toward the toxicity of nickelous acetate in rats. Toxicology. 1985;34:29–41. doi: 10.1016/0300-483x(85)90076-9. [DOI] [PubMed] [Google Scholar]
- Wald A. Sequential Tests of Statistical Hypotheses. Annals of Mathematical Statistics. 1945;16:117–186. [Google Scholar]