

Abstract
Experimental probes of the acidity of weak carbon acids have been developed and used to determine the carbon acid pKas of glycine, glycine derivatives and iminium ion adducts of glycine to the carbonyl group, including 5′-deoxypyridoxal (DPL). The high reactivity of the DPL-stabilized glycyl carbanion towards nucleophilic addition to both DPL and the glycine-DPL iminium ion favors the formation of Claisen condensation products at enzyme active sites. The formation of the iminium ion between glycine and DPL is accompanied by a 12-unit decrease in the pKa of 29 for glycine. The complicated effects of formation of glycine iminium ions to DPL and other aromatic and aliphatic aldehydes and ketones on carbon acid pKa are discussed. These data provide insight into the contribution of the individual pyridine ring substituents to the catalytic efficiency of DPL It is suggested that the 5′-phosphodianion group of PLP may play an important role in enzymatic catalysis of carbon deprotonation by providing up to 12 kcal/mol of binding energy that is utilized to stabilize the transition state for the enzymatic reaction.
Keywords: pyridoxal, proton transfer, carbon acid, carbanion, catalysis
1. Introduction
Pyridoxal 5′-phosphate serves as a cofactor for an extraordinarily large number of enzyme-catalyzed reactions of amino acids [1, 2]. The primary catalytic role of PLP is to reduce the activation barrier for conversion of α–amino acids to a zwitterionic carbanion [3]. The primary functional roles of the protein catalyst are; (a) to provide additional catalytic assistance to carbanion formation [4], (b) to ensure cleavage of the proper bond at the reactant amino acid [5] and, (c) to direct the reaction of the carbanion intermediate to the desired product.
Our interest in defining and rationalizing substituent effects on carbanion stability in water [6–9] and in understanding the mechanism for catalysis of these reactions by enzymes [10, 11] prompted an examination of the carbon acidity of the simple amino acid glycine and derivatives of glycine. This led to the determination of pKa for deprotonation of the α–amino carbon of glycine [12], of glycyl peptides [13] and of several simple derivatives of glycine [14–16]. There have been extensive studies to characterize covalent catalysis by PLP in enzymatic [1], in model reactions[17–22], and in chemical syntheses that employ PLP analogues as reagents [23]. By comparison, there is relatively little quantitative data on the carbon acid pKa(s) of the key iminium ion intermediate of these reactions. For example, it was stated as late as 2004 that there is “no available experimental information on the energies of the carbanionic intermediate for the uncatalyzed or PLP catalyzed reactions (or on their existence)” [24]. We therefore extended our work and have evaluated the effect of the PLP cofactor and related electrophilic catalysts on the carbon acidity of glycine [25], and examined the relationship between the structure of PLP and its reactivity as a catalyst of deprotonation of glycine [14, 26–28] and alanine [29]. We summarize here the results of these studies, and their relevance to the mechanism of PLP-catalyzed reactions in water and at enzyme active sites.
2.1. Methods for the Determination of Carbon Acid pKas
The thermodynamic barrier to deprotonation of weak carbon acids and carbon acid pKas might be calculated directly from the equilibrium constant for ionization of the carbon acid in water, but the concentration of the carbanion product is often too low to detect for strongly unfavorable proton transfer reactions. These barriers can also be determined by kinetic methods, because ΔG° (Figure 1) for thermodynamically unfavorable deprotonation of a carbon acid is equal to (eq 1 for Scheme 1), the difference between the kinetic activation barrier to formation of the carbanion and the activation barrier to carbanion protonation in the reverse direction.
Figure 1.

Profiles that show the changes in free energy for deprotonation of weak carbon acids (eq 1) to form carbanion products. Note that the barrier to thermodynamically unfavorable proton transfer (ΔG†HO) is equal to the sum of the thermodynamic barriers to proton transfer (ΔG°) and the barrier to downhill protonation of the carbanion in the reverse direction (ΔG†HOH). (A) Strongly thermodynamically unfavorable proton transfer reaction, for which there is a limiting small barrier to protonation of the carbanion by water. (B) Thermodynamically unfavorable formation of a strongly resonance stabilized carbanion, for which there is a substantial kinetic barrier to protonation of the carbanion by water [61].
Scheme 1.
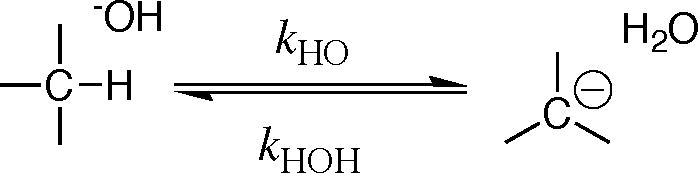
| (1) |
| (2) |
| (3) |
| (4) |
Kinetic methods to determine carbon acid pKas have been summarized in a recent review [11]. Briefly, carbon acid pKascan be determined from the ratio of rate constants for formation and reaction of the carbanion (e.g., eq 2 and 3), provided it is possible to estimate the value of the rate constant for fast carbanion protonation. For example, the kinetic barrier to carbanion formation is the dominant term (ΔGHO† ≫ ΔGHOH†, eq 1) for unfavorable ionization of weak carbon acids, such as for deprotonation of the amino acid glycine to form the highly unstable glycyl carbanion (Figure 1A). In this case, the small barrier for protonation of the unstable carbanion by water can be estimated from the limiting rate constant of kr = 1011 s−1 for rate-determining rotation of water into a reactive conformation [8]. There is a much larger barrier ΔGHOH† (Figure 1B) to protonation of glycyl carbanions stabilized by formation of a Schiffs base adduct to carbonyl compounds, including 5′-deoxypyridoxal (Figure 1B). In these cases the pKa for the carbon acid has been estimated from the rate constant kHO for carbon deprotonation, using the linear free energy relationship (eq 4) between the rate constant kHO and pKCH for the carbon acid [12, 25].
The essential experimental parameter used to estimate these carbon acid pKas (eqs 3 and 4) is the second order rate constant kHO (M−1s−1) for deprotonation of the carbon acid by hydroxide ion. In our hands, the fastest and most convenient method to determine kDO is to monitor the DO−-catalyzed deuterium exchange reaction of an acidic methyl or methylene group in DOD (Scheme 2). This reaction can be conveniently monitored using 1H NMR, because the incorporation of deuterium results in a decrease in the area of the singlet for an X-CH3 or an X-CH2-R group (Scheme 2), and the appearance of a 0.01 – 0.02 ppm upfield shifted triplet for the X-CH2D or X-CHD-R groups [6, 7]. Values of kHO for carbon acid deprotonation in HOH are then estimated from kDO for reactions in DOD, using representative values of the solvent deuterium isotope effects (kHO/kDO) for carbon acid deprotonation [6–8, 12, 13].
Scheme 2.

3. Novel reactions between pyridoxal and glycine
1H NMR analysis of the reaction of glycine with the pyridoxal analog DPL in D2O at pD 7.0 reveals the first-order disappearance of DPL to give a mixture at chemical equilibrium that contains 3% DPL and 97% of the diastereomeric products shown in Scheme 3, but no detectable (< 1%) incorporation of deuterium from D2O into glycine or transamination to give 5′-deoxypyridoxamine [27]. It was therefore not possible to determine the effect of DPL on the carbon acidity of glycine by monitoring the pyridoxal-catalyzed deuterium exchange reaction of glycine. The Claisen-type addition of glycine to pyridoxal shown in Scheme 3 was reported many years ago in a study on the role of metal cations in PLP-catalyzed reactions [30]. These same Claisen-type adducts were also reported to form as a product of the reaction of aminomalonate with DPL, presumably by decarboxylation of the DPL-aminomalonate iminium ion to form the DPL-stabilized glycyl carbanion (Scheme 3), which then reacts with the carbonyl group of a second molecule of DPL [31].
Scheme 3.

The rate-determining step for the Claisen-type condensation reaction between glycine and pyridoxal at pH 2 – 7 is deprotonation of the glycine iminium ion (kp = kHO[HO] + kB[B], Scheme 3) to form the pyridoxal stabilized glycyl carbanion. This is because the rate of addition of the carbanion to DPL (kadd[DPL]o, [DPL]o =10 mM, Scheme 3) is much faster than the rate of carbanion protonation by the water and by buffer catalysts (k−p = kHOH + kBH[BH]). Under these reaction conditions, the observed first-order rate constant for the reaction of DPL is equal to the rate constant for deprotonation of H-DPL=Gly: kobsd = kHO[HO] + kB[B], (Scheme 3). An examination of the reaction kinetics between pH 2 and 7 has provided a thorough description of the kinetic acidity of the different ionic forms of H-DPL=Gly [25]. There is a large increase, with increasing pH, in the equilibrium constant for addition of glycine to DPL to form the iminium ion H-DPL=Gly. This favors formation at high pH of an additional novel product by addition of the DPL-stabilized glycyl carbanion to the α–pyridyl carbon of H-DPL=Gly [28].
Enzymes such as serine palmitoyl transferase [32] and 5-aminolevulinate synthase [33] catalyze Claisen-type addition reactions that involve addition of PLP-stabilized α-amino carbanions to thioesters. For example, the key step for the 5-aminolevulinate synthase-catalyzed reaction is addition of the PLP-stabilized glycyl carbanion to succinyl-CoA to form a β–keto acid (Scheme 4), which then undergoes decarboxylation to 5-aminolevulinate. Our results show that while the glycyl carbanion is strongly stabilized by interactions with the PLP cofactor, it maintains a high kinetic reactivity toward addition to the carbonyl group. Therefore, the key condensation step in these enzyme-catalyzed Claisen reactions probably requires no assistance by the enzyme beyond orientation of the carbanion and thioester at the active site.
Scheme 4.

4. The Burden Borne by Pyridoxal in Catalysis of Deprotonation of Glycine
An examination of the changes in carbon acid pKa for a broad range glycyl derivatives and glycine-iminium ions provides considerable insight into the contribution of the different structural elements of the PLP cofactor to the enhanced carbon acidity of H-DPL=Gly.
Chart 1 shows that addition of the –NH3+ group to acetate anion causes a 4.5 unit decrease in the carbon acid pKa from 33.5 [9] to 29 [12]. The conversion of glycine to its acetone iminium ion 1-H results in a seven unit decrease in the carbon acid pKa from 29 to 22 (Chart 1) [34]. This relatively large effect on carbon acid pKa represents the sum of two smaller effects:
Chart 1.

An enhancement of intramolecular electrostatic stabilization of the enolate anion by interaction with the cationic nitrogen when the amino protons are replaced by an organic fragment to give the iminium ion [3, 12]. This results in a 2 unit larger acidifying effect of the α-NMe3+ group at betaine methyl ester (pKCH = 27) than of the α-NH3+ group at glycine methyl ester (pKCH = 29) [12].
Stabilization of the enolate by inductive and resonance interactions with the -N+=C(CH3)2 substituent. Similar resonance and inductive interactions between an enolate carbon and a vinyl (-C=C-) substituent result in a 3 unit lower pKa of 15.2 for the C-2 proton of 3-cyclohexenone [35] compared with the pKa of 18.1 for cyclohexanone [36]. Part of this change in pKa must be due to an inductive effect of the vinyl group, because a similar vinyl substitution causes a 1.0 unit decrease in the pKa = 10.7 for CH3CH2CH2NH3+ to 9.7 for H2C=CHCH2NH3+ [37], for a reaction where there is no resonance interaction between the substituent and the basic site.
The carbon acid pKa of 27 for the neutral benzaldehyde imine 2 (Chart 2) is similar to that for betaine (Chart 1) and the pKa of ≈ 23 for the protonated iminium ion 2-H (Chart 2) is similar to that for the acetone iminium ion 1-H (Chart 1). These comparisons show that there is no significant stabilization of negative charge at the enolate of 2-H by delocalization of negative charge onto the phenyl ring substituent. The results suggest that the inductive, and not the resonance effect of the –N+=C(R1)(R2) substituent at 1-H and 2-H, is largely responsible for the reduction in the carbon acid pKss for these compounds compared with the pKa of betaine.
Chart 2.

The 2-O− substituent at the salicylaldehyde iminium ion 3-Hcauses only a small increase in the carbon acid pKa of the benzaldehyde iminium ion 2-H. This reflects the destabilizing interaction between the oxyanion ring substituent and the carbanionic glycyl carbon. By contrast, there is an 8 kcal/mole larger thermodynamic driving force for formation of the iminium ion 3-H compared to 2-H. This reflects the stabilization of 3-H by the resonance effect of the 2-O− substituent, and the additional stabilization of 3-H by an intramolecular hydrogen bond between the 2-O− substituent and the iminium nitrogen [26]. This large effect of the 2-O− substituent on iminium ion stability dominates over the smaller effect on carbon acidity (Chart 2), so that salicylaldehyde is more effective than benzaldehyde as a catalyst of deprotonation of glycine [34]. The ring oxyanion at DPL should provide a similar catalytic advantage to deprotonation of glycine through the H-DPL=Gly iminium ion intermediate [26].
The α-benzyl carboxylate group at benzaldehyde causes a large increase in the reactivity of the carbonyl group as an electrophilic catalyst [14, 34], which reflects the decrease in the carbon acid pK ≈ 23 for 2-H to pKa =14 for 4-H. This large substituent effect on carbon acidity is consistent with a significant delocalization of negative charge from the enolate carbon to both the α-amino acid carboxylate (Scheme 5, resonance form A) and to the α-benzylic carboxylate (resonance form C). The results suggest that the carboxylate group at enzymatic pyruvoyl prosthetic groups is essential for effective catalysis of reactions that proceed through α-amino carbanion reaction intermediates [38–41]. Despite the strong carbon acidity of 4-H, DPL is more effective than phenylglyoxalate as a catalyst of deprotonation of glycine [14], because of the very favourable equilibrium constant for the addition of glycine to DPL to form the iminium ion H-DPL=Gly that is stabilized by an intramolecular hydrogen bond between the iminium nitrogen and the 2-O− ring substituent [25, 26].
Scheme 5.

A very low carbon acid pKa of 6 was estimated for the most highly protonated form of the PLP cofactor H2-DPL=Gly-H. This would favor enzymatic catalysis of deprotonation at the α-imino acid carbon of H2-DPL=Gly-H. However, X-ray crystal structures of pyridoxal enzymes show the cofactor bound in the form analogous to H-DPL=Gly [42–44], or in the case of alanine racemase, in the form analogous to DPL=Gly (basic pyridine nitrogen [45]). Despite the relatively weak carbon acidity of H-DPL=Gly (pKa ≈ 17) and the even weaker carbon acidity of DPL=Gly (not determined) there are several reasons why enzyme-catalyzed reactions are unlikely to proceed by deprotonation of strongest carbon acid H2-DPL=Gly-H. First, the equilibrium constant for formation of H2-DPL=Gly-H from glycine and DPL is less favorable than for formation of H-DPL=Gly [25]. Second, there is a substantial thermodynamic barrier at pH 7 to formation of H2-DPL=Gly-H from H-DPL=Gly because of the relatively weak basicity of the phenoxide oxygen (pKa = 3.0 [46]) and the carboxylate oxygen (pKa = 2.1 [46]). Third, the minimal ionic sites at the fully protonated cofactor H2-DPL=Gly-H compared with H-DPL=Gly provide few targets for recognition by enzyme catalysts.
The low carbon acid pKa of ≈ 18 for 5-H (Chart 3) [47] shows that the α-pyridyl group provides stabilization of negative charge by delocalization onto the pyridinium ring (Scheme 6A). The large electron-demand of the pyridinium ring favors delocalization of charge from the α-amino to the α-pyridyl carbon at the DPL-stabilized glycyl carbanion, as required for cofactor catalysis of transamination reactions by a 1,3 hydrogen shift (Scheme 6B). The large effects of the pyridinium ion at H-DPL=Gly (pKa = 17, Chart 3) and the –CO2− substituent at 4-H (pKa = 14, Chart 2) on the acidity of the glycyl carbon suggest that a strongly resonance electron withdrawing substituent at the iminium carbon is required to drive delocalization of negative charge across the extended π-systems, as shown in Scheme 5 and 6B.
Chart 3.

Scheme 6.

The acidity of the guanidinium amino acid side chain next to the pyridine ring nitrogen of PLP at the active site of alanine racemase (Arg-219 [45]) is apparently much weaker than for the carboxylic acid side chains next to this nitrogen at the active sites of D-amino acid transaminase (Glu-177 [48, 49]) and alanine glyoxylate aminotransferase (Asp 180, [50]). These observations suggest that the pyridine nitrogen is protonated during PLP enzyme-catalyzed transaminase reactions, where there is requirement for delocalization of charge onto the α-pyridyl carbon (Scheme 6B), but not during PLP enzyme-catalyzed racemization. This is because holding the pyridine nitrogen in the basic form should minimize delocalization of charge onto the α-pyridyl carbon, and protonation of this carbon to give the unwanted product of transamination of alanine [45, 51, 52].
5. The Burden Borne by Pyridoxal Enzymes
The highly zwitterionic imine H-DPL=Gly undergoes nonenzymatic deprotonation by phosphate dianion (pKa = 6.5) at 25 °C, with a second order rate constant kB = 1.7×10−3 M−1s−1 [25] that is substantially smaller than the value of kcat/Kd = 32 M−1 s−1 for the tryptophan synthase-catalyzed deuterium exchange reaction of the pro-2R hydrogen of glycine at pD 7.0 [53], and of kcat/Km = 4 × 105 M−1 s−1 for alanine racemase (Bacillus stearothermophilus) catalyzed racemization of D-alanine at 25 °C [52]. A comparison of the second-order rate constants for the nonenzymatic and alanine racemase-catalyzed reactions shows that the presence of the protein catalysts causes a 2 × 108-fold rate increase in the apparent second-order rate constant for the nonenzymatic reaction [25]. This is similar to the true rate acceleration for carbon-acid deprotonation, because proton transfer is partly rate determining for the alanine racemase catalyzed reaction [24, 45, 52]. It corresponds to a ca 11-kcal/mol stabilization of the transition state for deprotonation of the α-imino carbon. A larger 19 kcal/mole transition state stabilization was estimated for proline racemase, which does not require the assistance of the PLP cofactor [54]. This comparison shows that the PLP cofactor relieves about one-half of the catalytic burden borne by proline racemase. The cofactor therefore plays a critical, but not indispensible role in catalysis of the racemization of amino acids.
6. Phosphodianion binding energy and catalysis
The interactions between the spectator phosphodianion group of PLP and enzyme catalysts are certainly utilized to stabilize the enzyme-cofactor Michaelis complex. One of the most important differences between enzyme and small molecule catalysts is that enzymes often make efficient use of the interactions between the protein and nonreacting substrate parts in the stabilization of the transition state for the enzymatic reaction at a distant site [55]. In other cases the phosphodianion groups of D-glyceraldehyde 3-phosphate, orotidine 5′-monophosphate and dihydroxyacetone phosphate each provide a ca. 12 kcal/mol stabilization of the respective transition states for proton transfer catalyzed by triosephosphate isomerase (TIM) [56], decarboxylation catalyzed by orotidine 5′-monophosphate decarboxylase [57], and hydride transfer catalyzed by glycerol 3-phosphate dehydrogenase [58]. Part of this binding energy is expressed at the ground state Michaelis complex, but in all three cases more than 50% of the intrinsic binding energy of the substrate phosphodianion group is expressed specifically at the transition state for the catalyzed reaction [56–59]. There is little additional experimental data relevant to the importance of the utilization of phosphodianion interactions in the stabilization of other enzymatic transition states. However, it would be surprising if our recent work has uncovered the full set of reactions where these interactions make an important contribution to the enzymatic rate acceleration.
The PLP-dependent enzymes alanine racemase, ornithine decarboxylase and diaminopimelate decarboxylase are members of the PLP-binding barrel superfamily [4, 60]. This superfamily exhibits a highly conserved phosphodianion binding motif that resembles the phosphate binding motif of the TIM superfamily and the ribulose-phosphate binding barrel superfamily, of which orotidine 5′-monophosphate decarboxylase is a member [60]. It is interesting to speculate that these phosphodianion interactions do not simply serve the pedestrian function of stabilizing the Michaelis complex between the cofactor and the protein catalyst, but that they may be specifically expressed at the transition state for deprotonation of the α-imino carbon, and contribute to the protein stabilization of the reaction transition state.
Acknowledgments
We acknowledge the NIH (GM39754), the Ministerio de Educación y Ciencia and the European Regional Development Fund (ERDF) (Grant CTQ2004-06594) for their generous support of the work from our laboratories described in this review.
References
- 1.Toney MD. The chemistry of PLP-dependent enzymes. Wiley Encyclopedia of Chemical Biology. 2009;3:731–735. [Google Scholar]
- 2.Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch Biochem Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 3.Richard JP, Amyes TL. On the importance of being zwitterionic: enzymic catalysis of decarboxylation and deprotonation of cationic carbon. Bioorg Chem. 2004;32:354–366. doi: 10.1016/j.bioorg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Richard JP, Amyes TL, Crugeiras J, Rios A. Pyridoxal 5′-phosphate: electrophilic catalyst extraordinaire. Curr Opin Chem Biol. 2009;13:475–483. doi: 10.1016/j.cbpa.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc Nat Acad Sci. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amyes TL, Richard JP. Generation and Stability of a Simple Thiol Ester Enolate in Aqueous Solution. J Am Chem Soc. 1992;114:10297–10302. [Google Scholar]
- 7.Amyes TL, Richard JP. Determination of the pKa of ethyl acetate: Brønsted correlation for deprotonation of a simple oxygen ester In aqueous solution. J Am Chem Soc. 1996;118:3129–3141. [Google Scholar]
- 8.Richard JP, Williams G, Gao J. Experimental and computational determination of the effect of the cyano group on carbon acidity in water. J Am Chem Soc. 1999;121:715–726. [Google Scholar]
- 9.Richard JP, Williams G, O’Donoghue AC, Amyes TL. Formation and stability of enolates of acetamide and acetate ion: An Eigen plot for proton transfer at α-carbonyl carbon. J Am Chem Soc. 2002;124:2957–2968. doi: 10.1021/ja0125321. [DOI] [PubMed] [Google Scholar]
- 10.Richard JP, Amyes TL. Proton transfer at carbon. Curr Opin Chem Biol. 2001;5:626–633. doi: 10.1016/s1367-5931(01)00258-7. [DOI] [PubMed] [Google Scholar]
- 11.Amyes TL, Richard JP. Proton Transfer to and from carbon in model systems. In: Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Hydrogen-Transfer Reactions, Volume 3, Biological Aspects I-II. Vol. 3. Wiley-VCH; Weinheim: 2007. pp. 949–973. [Google Scholar]
- 12.Rios A, Amyes TL, Richard JP. Formation and stability of organic zwitterions in aqueous solution: enolates of the amino acid glycine and its derivatives. J Am Chem Soc. 2000;122:9373–9385. [Google Scholar]
- 13.Rios A, Richard JP, Amyes TL. Formation and stability of peptide enolates in aqueous solution. J Am Chem Soc. 2002;124:8251–8259. doi: 10.1021/ja026267a. [DOI] [PubMed] [Google Scholar]
- 14.Crugeiras J, Rios A, Riveiros E, Amyes TL, Richard JP. Glycine enolates: The effect of formation of iminium ions to simple ketones on α-amino carbon acidity and a comparison with pyridoxal iminium ions. J Am Chem Soc. 2008;130:2041–2050. doi: 10.1021/ja078006c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rios A, Crugeiras J, Amyes TL, Richard JP. Glycine enolates: The large effect of iminium ion formation on α-amino carbon acidity. J Am Chem Soc. 2001;123:7949–7950. doi: 10.1021/ja016250c. [DOI] [PubMed] [Google Scholar]
- 16.Rios A, Richard JP. Biological enolates: Generation and stability of the enolate of N-protonated glycine methyl ester in water. J Am Chem Soc. 1997;119:8375–8376. [Google Scholar]
- 17.Auld DS, Bruice TC. Catalytic reactions involving azomethines. IX. General base catalysis of the transamination of 3-hydroxypridine-4-carboxaldehyde by alanine. J Am Chem Soc. 1967;89:2098–2106. [Google Scholar]
- 18.Auld DS, Bruice TC. Catalytic reactions involving azomethines. VIII. Water and alanine catalysis of the transamination of 3-hydroxy-pyridine-4-carboxaldehyde by alanine. J Am Chem Soc. 1967;89:2090–2097. [Google Scholar]
- 19.Dixon JE, Bruice TC. Comparison of the rate constants for general base catatlyzed prototropy and racemization of the aldimine species formed from 3-hydroxypyridine-4-carboxaldehyde and alanine. Biochemistry. 1973;12:4762–4766. doi: 10.1021/bi00747a031. [DOI] [PubMed] [Google Scholar]
- 20.Chruma JJ, Liu L, Zhou W, Breslow R. Hydrophobic and electronic factors in the design of dialkylglycine decarboxylase mimics. Biochem Med Chem Lett. 2005;13:5873–5883. doi: 10.1016/j.bmc.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 21.Koh JT, Delaude L, Breslow R. Geometric control of a pyridoxal-catalyzed aldol condensation. J Am Chem Soc. 1994;116:11234–11240. [Google Scholar]
- 22.Weiner W, Winkler J, Zimmerman SC, Czarnik AW, Breslow R. Mimics of tryptophan synthetase and of biochemical dehydroalanine formation. J Am Chem Soc. 1985;107:4093–4094. [Google Scholar]
- 23.Felten AE, Zhu G, Aron ZD. Simplifying pyridoxal: Practical methods for amino acid dynamic kinetic resolution. Organic Letters. 12:1916–1919. doi: 10.1021/ol100319b. [DOI] [PubMed] [Google Scholar]
- 24.Spies MA, Woodward JJ, Watnik MR, Toney MD. Alanine racemase free energy profiles from global analyses of progress curves. J Am Chem Soc. 2004;126:7464–7475. doi: 10.1021/ja049579h. [DOI] [PubMed] [Google Scholar]
- 25.Toth K, Richard JP. Covalent catalysis by pyridoxal: Evaluation of the effect of the cofactor on the carbon acidity of glycine. J Am Chem Soc. 2007;129:3013–3021. doi: 10.1021/ja0679228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crugeiras J, Rios A, Riveiros E, Richard JP. Substituent effects on the thermodynamic stability of imines formed from glycine and aromatic aldehydes: Implications for the catalytic activity of pyridoxal-5′-phosphate. J Am Chem Soc. 2009;131:15815–15824. doi: 10.1021/ja906230n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toth K, Amyes TL, Richard JP, Malthouse JPG, NíBeilliu ME. Claisen-type addition of glycine to pyridoxal in water. J Am Chem Soc. 2004;126:10538–10539. doi: 10.1021/ja047501v. [DOI] [PubMed] [Google Scholar]
- 28.Toth K, Gaskell LM, Richard JP. Claisen-type addition of glycine to a pyridoxal iminium ion in water. J Org Chem. 2006;71:7094–7096. doi: 10.1021/jo061090e. [DOI] [PubMed] [Google Scholar]
- 29.Go MK, Richard JP. Alanine-dependent reactions of 5′-deoxypyridoxal in water. Bioorg Chem. 2008;36:295–298. doi: 10.1016/j.bioorg.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metzler DE, Longenecker JB, Snell EE. The reversible catalytic cleavage of hydroxyamino acids by pyridoxal and metal salts. J Am Chem Soc. 1954;76:639–644. [Google Scholar]
- 31.Thanassi JW. Aminomalonic acid. Spontaneous decarboxylation and reaction with 5′-deoxypyridoxal. Biochemistry. 1970;9:525–532. doi: 10.1021/bi00805a011. [DOI] [PubMed] [Google Scholar]
- 32.Ikushiro H, Fujii S, Shiraiwa Y, Hayashi H. Acceleration of the substrate C-α deprotonation by an analogue of the second substrate palmitoyl-CoA in serine palmitoyltransferase. J Biol Chem. 2008;283:7542–7553. doi: 10.1074/jbc.M706874200. [DOI] [PubMed] [Google Scholar]
- 33.Hunter GA, Zhang J, Ferreira GC. Transient kinetic studies support refinements to the chemical and kinetic mechanisms of aminolevulinate synthase. J Biol Chem. 2007;282:23025–23035. doi: 10.1074/jbc.M609330200. [DOI] [PubMed] [Google Scholar]
- 34.Crugeiras J, Rios A, Riveiros E, Amyes TL, Richard JP. 2010 In preparation for publication. [Google Scholar]
- 35.Dzingeleski GD, Blotny G, Pollack RM. Kinetics of the acetate ion catalyzed ketonization of 1,3-cyclohexadienol: equilibrium constants for the enolization of 2- and 3-cyclohexenone. J Org Chem. 1990;55:1019–1023. [Google Scholar]
- 36.Keeffe JR, Kresge AJ. Kinetics and mechanism of enolization and ketonization. In: Rappoport Z, editor. The Chemistry of Enols. John Wiley and Sons; Chichester: 1990. p. 460. [Google Scholar]
- 37.Jencks WP, Regenstein J. Ionization constants of acids and bases. In: Fasman GD, editor. Handbook of Biochemistry and Molecular Biology (Physical and Chemical Data) Vol. 1. CRC Press; Cleveland, OH: 1976. pp. 305–351. [Google Scholar]
- 38.Tolbert WD, Zhang Y, Cottet SE, Bennett EM, Ekstrom JL, Pegg AE, Ealick SE. Mechanism of human S-adenosylmethionine decarboxylase proenzyme processing as revealed by the structure of the S68A mutant. Biochemistry. 2003;42:2386–2395. doi: 10.1021/bi0268854. [DOI] [PubMed] [Google Scholar]
- 39.Graham DE, Xu H, White RH. Methanococcus jannaschii uses a pyruvoyl-dependent arginine decarboxylase in polyamine biosynthesis. J Biol Chem. 2002;277:23500–23507. doi: 10.1074/jbc.M203467200. [DOI] [PubMed] [Google Scholar]
- 40.Bach RD, Canepa C. Theoretical model for pyruvoyl-dependent enzymic decarboxylation of a-amino acids. J Am Chem Soc. 1997;119:11725–11733. [Google Scholar]
- 41.Gallagher T, Rozwarski DA, Ernst SR, Hackert ML. Refined structure of the pyruvoyl-dependent histidine decarboxylase from Lactobacillus. J Mol Biol. 1993;230:516–528. doi: 10.1006/jmbi.1993.1168. [DOI] [PubMed] [Google Scholar]
- 42.Toney MD, Hohenester E, Cowan SW, Jansonius JN. Dialkylglycine decarboxylase structure: bifunctional active site and alkali metal sites. Science. 1993;261:756–759. doi: 10.1126/science.8342040. [DOI] [PubMed] [Google Scholar]
- 43.Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN. Crystal structure of aspartate aminotransferase. J Mol Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 44.Almo SL, Smith DL, Danishefsky AT, Ringe D. Strucure of E. Coli aspartate transaminase. Prot Eng. 1994;7:405–412. doi: 10.1093/protein/7.3.405. [DOI] [PubMed] [Google Scholar]
- 45.Shaw JP, Petsko GA, Ringe D. Determination of the structure of alanine racemase from Bacillus stearothermophilus at 1.9-Å resolution. Biochemistry. 1997;36:1329–1342. doi: 10.1021/bi961856c. [DOI] [PubMed] [Google Scholar]
- 46.Vazquez MA, Echevarria G, Munoz F, Donoso J, Garcia Blanco F. Kinetic study of the Schiff-base formation between glycine and pyridoxal 5′-phosphate (PLP), pyridoxal (PL), and 5′-deoxypyridoxal (DPL) J Chem Soc, Perkin Trans. 1989;2:1617–1622. [Google Scholar]
- 47.Crugeiras J, Rios A, Amyes TL, Richard JP. Carbon acidity of the α-pyridinium carbon of a pyridoxamine analog. Org Biomol Chem. 2005;3:2145–2149. doi: 10.1039/b504399a. [DOI] [PubMed] [Google Scholar]
- 48.Sugio S, Petsko GA, Manning JM, Soda K, Ringe D. Crystal structure of a D-amino acid aminotransferase: How the protein controls stereoselectivity. Biochemistry. 1995;34:9661–9669. doi: 10.1021/bi00030a002. [DOI] [PubMed] [Google Scholar]
- 49.Van Ophem PW, Peisach D, Erickson SD, Soda K, Ringe D, Manning JM. Effects of the E177K mutation in D-amino acid transaminase. Studies on an essential coenzyme anchoring group that contributes to stereochemical fidelity. Biochemistry. 1999;38:1323–1331. doi: 10.1021/bi982414z. [DOI] [PubMed] [Google Scholar]
- 50.Han Q, Robinson H, Gao Y, Vogelaar N, Wilson S, Rizzi M, Li J. Crystal structures of Aedes aegypti alanine glyoxylate aminotransferase. J Biol Chem. 2006;281:37175–37182. doi: 10.1074/jbc.M607032200. [DOI] [PubMed] [Google Scholar]
- 51.Morollo AA, Petsko GA, Ringe D. Structure of a Michaelis complex analog: Propionate binds in the substrate carboxylate site of alanine racemase. Biochemistry. 1999;38:3293–3301. doi: 10.1021/bi9822729. [DOI] [PubMed] [Google Scholar]
- 52.Sun S, Toney MD. Evidence for a two-base mechanism involving tyrosine-265 from arginine-219 mutants of alanine racemase. Biochemistry. 1999;38:4058–4065. doi: 10.1021/bi982924t. [DOI] [PubMed] [Google Scholar]
- 53.Milne JJ, Malthouse JPG. The effect of different amino acid side chains on the stereospecificity and catalytic efficiency of the tryptophan synthase-catalyzed exchange of the a-protons of amino acids. Biochem J. 1996;314:787–791. doi: 10.1042/bj3140787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams G, Maziarz EP, Amyes TL, Wood TD, Richard JP. Formation and stability of the enolates of N-protonated proline methyl ester and proline zwitterion in aqueous solution: A nonenzymatic model for the first step in the racemization of proline catalyzed by proline racemase. Biochemistry. 2003;42:8354–8361. doi: 10.1021/bi0345992. [DOI] [PubMed] [Google Scholar]
- 55.Jencks WP. Binding Energy, Specificity and Enzymic Catalysis: The Circe Effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 56.Amyes TL, Richard JP. Enzymatic catalysis of proton transfer at carbon: Activation of triosephosphate isomerase by phosphite dianion. Biochemistry. 2007;46:5841–5854. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amyes TL, Richard JP, Tait JJ. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 58.Tsang W-Y, Amyes TL, Richard JP. A substrate in pieces: Allosteric activation of glycerol 3-phosphate dehydrogenase (NAD+) by phosphite dianion. Biochemistry. 2008;47:4575–4582. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morrow JR, Amyes TL, Richard JP. Phosphate binding energy and catalysis by small and large molecules. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Denesyuk A. Functional attributes of the phosphate group binding cup of pyridoxal phosphate-dependent enzymes. J Mol Biol. 2002;316:155–172. doi: 10.1006/jmbi.2001.5310. [DOI] [PubMed] [Google Scholar]
- 61.Richard JP, Amyes TL, Toteva MM. Formation and stability of carbocations and carbanions in water and intrinsic barriers to their reactions. Acc Chem Res. 2001;34:981–988. doi: 10.1021/ar0000556. [DOI] [PubMed] [Google Scholar]