

Abstract
Increasing amounts of data suggest that inflammatory responses have an important role in the pathophysiology of depression. Depressed patients have been found to have higher levels of proinflammatory cytokines, acute phase proteins, chemokines and cellular adhesion molecules. In addition, therapeutic administration of the cytokine interferon-α leads to depression in up to 50% of patients. Moreover, proinflammatory cytokines have been found to interact with many of the pathophysiological domains that characterize depression, including neurotransmitter metabolism, neuroendocrine function, synaptic plasticity and behavior. Stress, which can precipitate depression, can also promote inflammatory responses through effects on sympathetic and parasympathetic nervous system pathways. Finally, depression might be a behavioral byproduct of early adaptive advantages conferred by genes that promote inflammation. These findings suggest that targeting proinflammatory cytokines and their signaling pathways might represent a novel strategy to treat depression.
So much is lost from the lives of people who suffer with major depression that it was perhaps natural for initial discoveries regarding mood disorders and immunity to focus on yet another loss – in this case, diminished functioning of the humoral and cellular arms of the acquired immune response [1]. Nevertheless, data amassed over the past 15 years have led to a dramatic paradigm shift in which the early focus on immunosuppression has been subsumed within, and supplanted by, an increasing recognition that depressive disorders might be best characterized as conditions of immune activation, especially hyperactivity of innate immune inflammatory responses. This profound change in our view of depression and immunity has not occurred in isolation but rather is part of a larger scientific movement built around an increasing appreciation that inflammatory processes are central to the pathogenesis of several modern maladies, including cardiovascular disease, diabetes and cancer [2–4]. Indeed, much of the recent work linking depression with inflammation has been prompted by the search for potential shared etiological mechanisms that might explain the striking co-morbidity between these medical illnesses and major depression [5].
Evidence for increased inflammation in depression
Patients with major depression who are otherwise medically healthy have been repeatedly observed to have activated inflammatory pathways, as manifested by increased proinflammatory cytokines, increased acute-phase proteins and increased expression of chemokines and adhesion molecules [6–22]. Increased serum and/or plasma concentrations of interleukin (IL)-6 and/or C-reactive protein have been most frequently observed[6–15,17,19,20], although elevations in IL-1-β and tumor necrosis factor (TNF)-α have also been described, both in the peripheral blood circulation and in the central nervous system (CNS; in particular, in the cerebrospinal fluid)[10,12,15–17,21,23–26]. In addition to C-reactive protein, other acute-phase proteins found to be elevated include α-1-acid glycoprotein, α-1-antichymotrypsin and haptoglobin [11,15,22]. Increased levels of chemokines and adhesion molecules, including human macrophage chemoattractant protein-1 (MCP-1), soluble intracellular adhesion molecule-1 (sICAM-1) and E-selectin, have also been described [18]. Although most studies have compared inflammatory markers in depressed versus nondepressed subjects, several have reported positive correlations between plasma concentrations of various inflammatory mediators and depressive symptom severity [6,7,26]. Finally, a nascent literature suggests that functional allelic variants of the genes for IL-1β and TNF-α increase the risk for depression and are associated with reduced responsiveness to antidepressant therapy [27,28].
The association between depression and inflammation is apparent across the adult lifespan [8,10,11,18] and is evident even in the context of mild depressive symptoms that do not meet criteria for major depression [29]. Indeed, even single depression-related symptoms – such as fatigue, insomnia and anger and/or hostility – have been associated with evidence of inflammatory activation in otherwise healthy individuals [29–32]. Inflammation has generally been measured at a single time point; however, a recent study found that abnormal IL-6 production is apparent across the circadian cycle in patients with major depression [6]. In addition to findings in medically healthy subjects, depression and depressive symptoms (including fatigue and cognitive dysfunction) have been associated with inflammatory markers in several medical illnesses, including cardiovascular disease [33,34], cancer [9,35,36] and postviral infection [25,30].
Questions and controversies: does association imply causality?
Although several studies support the idea that inflammatory processes contribute to the pathogenesis of major depression, other studies have failed to find an association between the two [37,38] and, in some cases, associations have been attenuated or obviated when potential mediating or moderating factors (e.g. body mass index, gender or personality) have been included in the analyses[8,13,14,39]. Some positive studies have failed to find a correlation between inflammation and depressive severity[12,25] or have found disparate and occasionally opposite correlations for different proinflammatory mediators[7,12,16,26,34]. These inconsistencies indicate that strong pronouncements about the role of the immune system in depression might be premature and suggest that inflammation contributes to some, but not all, cases of depression. Consistent with this, a wide range of inflammatory activity is typically observed within any given sample of depressed subjects, and associations between inflammation and groups of depressed subjects are often accounted for by a subset of individuals at the upper range of cytokine production and/or release [11]. Moreover, individual symptoms, especially sleep disturbance [40], have been reported to contribute disproportionately to the association between depression and inflammation. These points of controversy suggest avenues of future research, including clarification of factors most closely associated with immune activation.
Pathophysiological mechanisms: immunological pathways to psychopathology
A rich neuropsychiatric literature attests to fundamental mechanisms that conspire to cause the syndrome of major depression. At a neurobiological level, alterations in neurotransmitter function involving serotonin, norepinephrine and dopamine are well known to induce depression and are primary targets for currently available psychopharmacological (antidepressant) treatments [41]. Abnormalities in neuropeptide function are also believed to contribute. Indeed, hypersecretion of the neuropeptide hormone corticotrophin-releasing hormone (CRH) has been reliably demonstrated in depressed patients [42]. CRH is the primary regulator of the hormonal response to stress [activation of the hypothalamic–pituitary–adrenal (HPA) axis and the sympathetic nervous system]. Hypersecretion of CRH is considered to be the crucial biological substrate of the well-known link between psychosocial stress and depression [42]. In addition, the well-characterized hyperactivity of the HPA axis and sympathetic nervous system in depression is believed to be secondary to CRH hyperactivity, in part because of impaired negative regulation of CRH by glucocorticoids [43]. Altered CRH regulation by glucocorticoids is thought to be a result of alterations in the functioning of the glucocorticoid receptor [44]. Changes in regional brain activity have also been documented in depression and involve abnormal metabolic activity in the prefrontal cortex and altered dopamine metabolism in the basal ganglia (caudate) [45,46]. There has also been increasing interest in the role of neural plasticity in depression, with special emphasis on factors that influence neuronal growth and survival [47].
At the interface between immunology and behavior
Given the primary factors that are associated with the pathophysiology of depression, it is intriguing that peripherally released inflammatory cytokines can access the brain and influence all of the relevant pathophysiological domains (Figure 1). Because of the large size of cytokines and their resultant inability to readily penetrate the blood–brain barrier, attention has been paid to routes by which cytokines access the brain, including (i) entry through leaky regions in the blood–brain barrier, such as the circumventricular organs; (ii) binding to cytokinespecific transport molecules expressed on brain endothelium and (iii) activation of vagal afferent fibers which transmit cytokine signals to specific brain nuclei, such as the nucleus of the solitary tract, which then serves as a relay station to other brain nuclei, including the paraventricular nucleus in the hypothalamus [48] (Figure 1c).
Figure 1.

Stress–immune interactions and depression. (a) Activation of NF-κB through Toll-like receptors (TLR) during immune challenge leads to an inflammatory response including (b) the release of the proinflammatory cytokines TNF-α, IL-1 and IL-6. (c) These cytokines, in turn, access the brain via leaky regions in the blood–brain barrier, active transport molecules and afferent nerve fibers (e.g. sensory vagus), which relay information through the nucleus tractus solitarius (NTS) [48]. (d) Once in the brain, cytokine signals participate in pathways (indicated in orange) known to be involved in the development of depression, including: (i) altered metabolism of relevant neurotransmitters such as serotonin (5HT) and dopamine (DA) [50,51]; (ii) activation of CRH in the paraventricular nucleus (PVN) and the subsequent production and/or release of ACTH and glucocorticoids (cortisol) [52,53] and (iii) disruption of synaptic plasticity through alterations in relevant growth factors [e.g. brain-derived neurotrophic factor (BDNF)] [59,60]. (e) Exposure to environmental stressors promotes activation of inflammatory signaling (NF-κB) through increased outflow of proinflammatory sympathetic nervous system responses [release of norepinephrine (NE), which binds to α (αAR) and β (βAR) adrenoceptors] (orange). (f) Stressors also induce withdrawal of inhibitory motor vagal input [release of acetylcholine (ACh), which binds to the α7 subunit of the nicotinic acetylcholine receptor (α7nAChR)] (blue) [73,77]. (g) Activation of the mitogen activated protein kinase pathways, including p38 and Jun amino-terminal kinase (JNK), inhibit the function of glucocorticoid receptors (GR), thereby releasing NF-κB from negative regulation by glucocorticoids released as a result of the HPA axis in response to stress (blue) [55,56].
Once in the brain, there is a CNS cytokine network that is made up of cells (neurons and glial elements) that not only produce cytokines and express cytokine receptors, but also amplify cytokine signals, which in turn can have profound effects on neurotransmitter and CRH function, as well as on behavior [43,49]. For example, studies in laboratory animals provide compelling evidence that inflammatory cytokines induce a syndrome of sickness behavior that has many overlapping features with major depression, including anhedonia (an inabililty to experience pleasure), anorexia, impaired sleep and reduced locomotor activity [49]. Consistent with the pathophysiology of depression, these cytokine-induced behavioral changes are associated with alterations in the metabolism of serotonin, norepinephrine and dopamine in brain regions essential to the regulation of emotion, including the limbic system (amygdala, hippocampus and nucleus accumbens), as well as the regulation of psychomotor function and reward, including the basal ganglia [50,51].
In addition to effects on neurotransmitter metabolism, inflammatory cytokines have profound stimulatory effects on HPA axis hormones as well as CRH (mRNA and protein), in both the hypothalamus and the amygdala, a brain region that has an important role in fear and anxiety[52–54]. These effects are, in large part, mediated by a rich network of cytokines and their receptors within HPA axis tissues which facilitate the integration of cytokine signals [53]. Downstream cytokine signal transduction pathways, including mitogen-activated protein kinases (MAPKs) and nuclear factor κB (NF-κB), also disrupt glucocorticoid receptor signaling [55,56], and thus might contribute to altered glucocorticoid-mediated feedback regulation of both CRH and further proinflammatory cytokine release. In addition, activation of p38 MAPKs might contribute to alterations in neurotransmitter function through effects on the serotonin transporter [57].
Induction of NF-κB in the brain following peripheral cytokine (IL-1) exposure has been shown to mediate many of the behavioral effects of IL-1, including social withdrawal and decreased food intake [58]. NF-κB induction in the brain also might contribute to alterations in neuronal growth and survival, especially through the induction of nitric oxide (e.g. via inducible nitric oxide synthase, iNOS) and, ultimately, oxidative stress, which has been shown to alter promoter function for several genes central to synaptic plasticity [59,60]. Finally, humans who are administered cytokines exogenously for infectious diseases and cancer show altered function in brain regions relevant to the development of depressive symptoms [54].
Interferon-α: a clinical model of cytokine-induced depression
To study the effects of innate immune cytokines on behavior in humans, several investigators have seized upon the profound clinical observation that a high percentage of patients who are administered the cytokine interferon (IFN)-α for the treatment of infectious diseases or cancer develop a behavioral syndrome that is strikingly similar to major depression (Figure 2). IFN-α is a potent inducer of proinflammatory cytokines, including IL-6 and, to lesser extent, IL-1 β and TNF-α [54]. Substantiating the fact that the IFN-α-induced behavioral syndrome is indeed depression as encountered in other venues (e.g. mental health clinics) is that IFN-α-induced depression: (i) is responsive to treatment with standard antidepressant therapy [61]; (ii) is associated with alterations in serotonin metabolism, in part related to increased activity of the metabolic enzyme indoleamine 2,3, dioxygenase, which degrades tryptophan to kynurenine, which is then metabolized to quinolinic acid [62]; and (iii) is associated with alterations in CRH function as manifested by an exaggerated adrenocorticotropic hormone (ACTH) and cortisol response to the first administration of IFN-α in patients who ultimately develop depression [63].
Figure 2.

IFN-α: modeling of cytokine-induced depression. Therapeutic administration of IFN-α is associated with depression in 30–50% of patients, depending on the dose [61,62]. IFN-α-induced depression is associated with pathophysiological changes that overlap with those found in medically healthy depressed patients, including activation of neuroendocrine (HPA axis) pathways (a), alterations in neurotransmitter metabolism (b), responsiveness to antidepressant treatment (c) and alterations in brain circuitry relevant to information processing (d). (a) The initial injection of IFN-α to patients with malignant melanoma is associated with a marked induction of ACTH and cortisol, which was significantly higher in patients who eventually developed depression (blue) than in those who never became depressed during IFN-α treatment (red). a, significantly different from 0 hours; P<0.01; b, significant difference between groups, P<0.01. (b) The relationship between the severity of depressive symptoms [as measured by the Montgomery–Asberg Depression Rating Scale (MADRS)] and changes in plasma tryptophan (TRP) concentrations during IFN-α therapy for cancer. TRP is the primary amino acid precursor of serotonin, a major regulator of limbic brain circuitry that subserves emotion. Decreases in TRP were significantly correlated (R=−0.50, P<0.05) with increases in depression severity scores during IFN-α treatment. (c) Patients who received the serotonin reuptake inhibitor paroxetine, a commonly used antidepressant, before and during IFN-α therapy for malignant melanoma (red triangles), were significantly more likely to remain free of depression during IFN-α administration than a placebo-treated control group (black squares). (d) Significantly greater activation (yellow and orange) of the dorsal ACC, as measured by functional magnetic resonance imaging (fMRI) during a task of visuospatial attention was found in IFN-α-treated patients compared with controls. (e) A significant linear relationship was found between activation of the ACC and the number of task-related errors in IFN-α-treated patients (red stars) but not in control subjects (black triangles). Increased ACC activation in response to relatively low task error rates has been associated with cognitive styles that predispose to anxiety and depression, suggesting that IFN-α-induced changes in ACC function might represent a cognitive pathway to psychopathology [65]. Reproduced, with permission, from (a) Ref. [63], (b) Ref. [62], (c) Ref. [61] and (d,e) Ref. [65].
In addition, using positron emission tomography, IFN-α has been shown to alter metabolic activity in the basal ganglia [64], possibly representing disruption of basal ganglia circuitry involving dopamine and subserving hedonic tone (i.e. the capacity to experience pleasure and reward), as well as psychomotor speed. IFN-α has also been found to alter fundamental information processing in the dorsal part of the anterior cingulate cortex (ACC), as revealed by functional magnetic resonance imaging. In comparison with control subjects, IFN-α-treated patients exhibited increased ACC activity in response to a task with a low error rate, possibly representing an increased sensitivity to negative events or conflict [65]. Similar alterations have been demonstrated in populations at risk for mood and anxiety disorders, including subjects with high-trait anxiety, neuroticism and obsessive compulsive disorder. Taken together, these data demonstrate that cytokine (IFN-α) administration to humans replicates multiple pathologies central to depression, thereby providing support for the notion that endogenous cytokines that mediate innate immune responses can contribute to the state of depression.
Stress and immunity: a crucial link in the cytokine–depression chain
Inflammation is the sine qua non of pathology, so it is understandable that proinflammatory cytokines might contribute to depression in the context of medical illness, thus potentially accounting for the five–tenfold greater prevalence of depression in individuals with a wide range of medical disorders [5]. Nevertheless, it is not as readily apparent what might drive increased inflammation in patients with major depression who are presumably physically healthy. One possibility is the impact of stress on the immune response (Figures 1 and 3).
Figure 3.
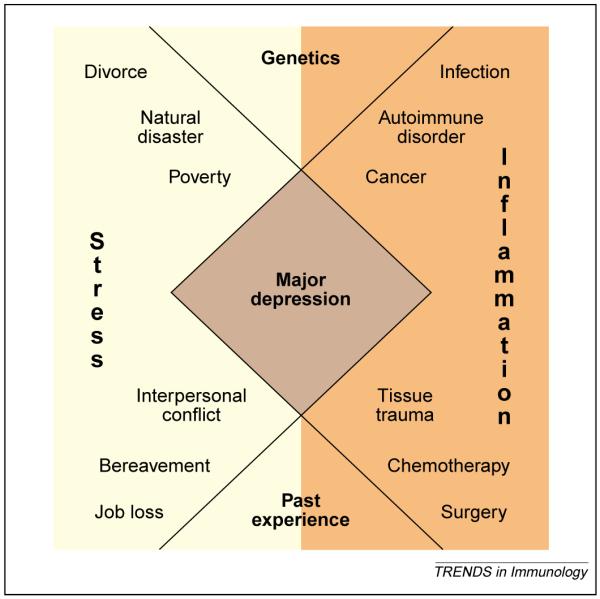
Acute and chronic immune and inflammatory processes, combined with relevant contributions from immunogenetics (such as polymorphisms in cytokine genes) and past immune experiences (such as prior infections and vaccination history) (orange) interact with acute and chronic stressors combined with relevant contributions from psychiatric genetics (such as polymorphisms in neurotransmitter transporter genes) and past emotional experiences (such as adversity in early life) (yellow) to promote the syndrome of major depression (brown). A diagnosis of major depression is based on the presence of five of the following symptoms: depressed mood, anhedonia, fatigue, guilt and/or worthlessness, suicidal ideation, impaired concentration and/or memory, psychomotor retardation and/or agitation and disturbances of sleep or appetite. Symptoms must persist for at least two weeks and cause significant functional impairment [91].
Psychological stress is a common risk factor for the development of major depression in every culture examined, and most initial episodes of major depression are preceded by an identifiable stressor [66]. Consistent with the notion that stress might provide a link between depression and inflammation, increasing data indicate that psychological stress activates proinflammatory cytokines and their signaling pathways in the periphery and CNS. In laboratory animals, a variety of psychological stressors (e.g. restraint, open-field exposure or social isolation) increase concentrations of proinflammatory cytokines, including IL-1β and TNF-α, in brain regions involved in emotional regulation, as well as in the periphery [60,67]. Moreover, behavioral changes induced in rodents by social isolation can be reversed by intracerebroventricular administration of the soluble IL-1 receptor antagonist (IL-1ra) [68]. Proinflammatory cytokine activation also appears to mediate other stress-related biochemical changes in the brain. For example, TNF-α is required for the production of nitric oxide within the CNS following immobilization stress [60], and IL-1-b has a key role in the inhibition of the expression of brain-derived neural growth factor in the hippocampus of rats following social isolation [69]. Consistent with these findings is the fact that stress-induced neuronal cell loss in rodents is linked to increased concentrations of TNF-α and NF-κB [60].
In humans, acute stress (e.g. laboratory stressor) and chronic stress (including lack of social support) have both been associated with increased production and/or release of proinflammatory cytokines and decreases in anti-inflammatory cytokines such as IL-10 [70–72]. Recent work indicates that psychosocial stress also activates NF-κB in peripheral blood mononuclear cells of healthy volunteers [73].
Interestingly, stress-induced activation of proinflammatory cytokines might provide some insight into the decreases in acquired immune responses found in both stress and depression. For example, stress-induced decreases in the IgM and IgG antibody responses to immunization with keyhole limpet hemocyanin were found to be attenuated significantly if rats were pretreated with IL-1ra [74]. Combined with data suggesting that TNF-α can significantly disrupt T-cell signaling, these findings suggest that overactivation of innate immune responses following stress and during depression might come at the expense of decreased cellular and humoral acquired immune responses [43].
Stress system activation might promote cytokine production through several mechanisms (Figure 1). Despite suppressing certain immune processes, activation of the sympathetic nervous system has been linked in several studies to proinflammatory activation in the periphery, which might, in turn, influence inflammatory processes in the CNS [43]. For example, stress-induced activation of NF-κB in peripheral blood mononuclear cells appears to be dependent on norepinephrine and can be abrogated by α1-adrenoceptor blockade [73]. Similarly, β-adrenoceptor stimulation has been shown to increase gene expression and protein production of TNF-α, IL-1β and IL-6 in myocardial cells [75], and chronic b-blockade reduces plasma levels of IL-6 in concert with symptomatic improvement in patients with congestive heart failure [76]. Vagal withdrawal in response to stress might also promote inflammation, given the evidence that vagal activity inhibits NFkB activation (and the release of TNF-α from macrophages) via cholinergic signaling through the α-7 subunit of the nicotinic acetylcholine receptor [77]. Indeed, decreased vagal tone, as manifested by reduced heart rate variability, has been associated with increased inflammatory markers in women with coronary artery disease [78]. Stress and depression have both been associated with reduced heart rate variability [79]. Finally, chronic stress promotes the development of glucocorticoid resistance, which has been associated with increased cytokine production and which might also release the sympathetic nervous system from inhibitory control, further promoting inflammatory activation [43].
Evolutionary perspectives
The relationship between depression and activation of innate immune responses might have its roots in the evolutionary advantages of a behavioral repertoire that enables diversion of crucial energy resources to the metabolic demands of fever during times of pathogen exposure. In addition, pathophysiological changes central to depression might protect inflammatory responses from negative regulation by neuroendocrine (glucocorticoid) responses [48,80] (Box 1). Thus, depression might have been a necessary behavioral accoutrement to early, endogenous antibiotic strategies that have been rendered unnecessary in modern times.
Box 1. Evolutionary imperatives for the depression–inflammation link.
The discovery that depression shares an underlying physiology with inflammation and stress system activation provides a novel perspective on why vulnerability genes for major depression might have been retained in the human gene pool despite the negative impact of depression on morbidity and mortality. Although current adaptive theories focus on the potential benefits of depressive symptoms themselves (i.e. serving as signals to increase aid from, or decrease competition with, others), viewing depression as a requisite behavioral counterpart of immune activation suggests that the high prevalence of mood disorders came about not as a result of providing hidden social benefits but because these disorders are pleiotropically linked to genes that directly, or indirectly, promote enhanced inflammatory responses and, hence, survival in times of stress. Despite posing a risk for depression, such allelic variants might have simultaneously improved overall fitness by increasing resistance to pathogen assault before the advent of modern sanitation and antibiotic therapy [48]. Consistent with this hypothesis is the observation that many symptoms of depression (e.g. psychomotor retardation, fatigue and anhedonia) promote the conservation of essential energy resources that are adaptive in the context of subserving the metabolic demands of endogenous antibiotic strategies such as fever [48].
The benefits of heightened inflammatory activity in response to infection might also help to explain a common neuroendocrine abnormality found in major depression. In hierarchical social species (including humans), psychosocial stress in males is frequently accounted for by dominance struggles in which the risk of wounding, especially in subordinates, is high. This presents individuals with the paradox of needing HPA axis activation (and the release of glucocorticoids) to provide energy for the stressful encounter, while avoiding the increased danger of wound infection presented by glucocorticoid-induced immunosuppression. Studies using a social disruption paradigm in rodents suggest that this impasse is resolved through the induction of glucocorticoid resistance, which correlates with assumption of a subordinate behavioral profile following defeat and with the number of wounds received in fighting with aggressive intruder mice [80]. Interestingly, depression in humans is highly associated with both social defeat (especially in males) and with endocrine and immune tissue resistance to glucocorticoids.
These considerations suggest that, rather than harboring hidden adaptive features, major depression is a disease fueled in the modern world by an evolutionary mismatch between current conditions and our ancestral environment. This also suggests a surprising hypothesis: to the degree that inflammatory processes are rendered less essential by cultural inventions (i.e. improved medicines or laws against physical violence), genes that promote major depression should decrease in frequency as the survival benefits of immune activation become outweighed by disadvantages, including an increased risk of major depression in the context of psychological stress.
Treatment implications
Because paradigm shifts within medicine are judged (finally) by their clinical utility, it is encouraging that important treatment implications have already emerged from our nascent understanding of the role of inflammation in the pathogenesis of major depression. Indeed, data increasingly suggest that inflammatory processes contribute to the therapeutic effects of the currently available antidepressants and could provide targets for novel pharmacological and nonpharmacological treatment strategies.
Consistent with their ability to ameliorate cytokine-induced depression [61], antidepressants have been shown in animals and humans to inhibit the production and/or release of proinflammatory cytokines and to stimulate the production of anti-inflammatory cytokines (such as IL-10) [81]. These effects might contribute to therapeutic efficacy, given recent data that antidepressant-like effects of desipramine in the forced swim test (an animal model frequently used to evaluate antidepressant efficacy) depend on the ability of the drug to inhibit CNS production of TNF-α [82]. In humans, a variety of antidepressant strategies (including medication, electro-convulsive shock therapy and psychotherapy) appear to attenuate inflammatory activity in concert with improvements in depressive symptoms, suggesting that reductions in inflammation might contribute to treatment response [16,17,19,23,24,83].
An inflammatory perspective on depression also provides novel insights into inadequacies in our current treatment options. For example, patients with a history of nonresponse to antidepressants have been found to exhibit increased plasma concentrations of IL-6 and acute-phase proteins when compared with treatment-responsive patients [20,22]. Similarly, patients with evidence of increased inflammatory activity before treatment have been reported to be less responsive to antidepressants, lithium or sleep deprivation (a potent short-term mood elevator) [17,21,22,84]. Moreover, work by our group and by others suggests that in the context of cytokine exposure, antidepressants might only address selected symptom domains (e.g. depressed mood and anxiety), while leaving several symptoms relatively unaffected (e.g. fatigue or psychomotor slowing) [85,86]. Interestingly, this pattern of symptom response has also been observed in the antidepressant treatment of medically healthy patients with depression [87].
These observations suggest that inhibition of proinflammatory cytokine signaling represents a viable strategy for the treatment of depression, especially in patients with evidence of increased inflammatory activity before therapy who might be less likely to respond to conventional agents. In support of this idea, studies in laboratory animals demonstrate that cytokine-induced sickness syndromes (which overlap symptomatically with depression) can be ameliorated or reversed by administering specific cytokine antagonists (e.g. IL-1ra) or anti-inflammatory cytokines (e.g. IL-10) directly into the brain[68,88]. Moreover, cytokine antagonists appear to have antidepressant-like effects, even in the absence of an immune challenge. For example, in rodents intracerebro-ventricular administration of IL-1ra prevents memory deficits following the psychological stress of social isolation [68], and antibodies to TNF-α demonstrate anti-depressant effects on the forced swim test when administered via the intracerebroventricular route [82]. In humans, antagonists of TNF-α (i.e. etanercept, infliximab) have been reported to reduce depressive symptoms in the context of treating a variety of autoimmune conditions [89,90]. Interestingly, in these patients, improvements in mood often appear to precede improvements in the underlying disease state.
Conclusion
A great amount of compelling data suggest that inflammatory innate immune responses might contribute to the development of depression, in part through complex interactions with stress-responsive pathways involving the neuroendocrine and autonomic nervous systems. The role of the immune system in the pathophysiology of depression probably derives from evolutionary imperatives, and presents intriguing and unique therapeutic opportunities to expand the treatment targets for this disabling and common disorder.
References
- 1.Herbert TB, Cohen S. Depression and immunity: a meta-analytic review. Psychol. Bull. 1993;113:472–486. doi: 10.1037/0033-2909.113.3.472. [DOI] [PubMed] [Google Scholar]
- 2.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J. Clin. Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willerson JT, Ridker PM. Inflammation as a cardiovascular risk factor. Circulation. 2004;109(21, Suppl 1):II2–10. doi: 10.1161/01.CIR.0000129535.04194.38. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, et al. Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol. 2005;26:318–325. doi: 10.1016/j.it.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Evans DL, et al. Mood disorders in the medically ill: scientific review and recommendations. Biol. Psychiatry. 2005;58:175–189. doi: 10.1016/j.biopsych.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Alesci S, et al. Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J. Clin. Endocrinol. Metab. 2005;90:2522–2530. doi: 10.1210/jc.2004-1667. [DOI] [PubMed] [Google Scholar]
- 7.Miller GE, et al. Clinical depression and inflammatory risk markers for coronary heart disease. Am. J. Cardiol. 2002;90:1279–1283. doi: 10.1016/s0002-9149(02)02863-1. [DOI] [PubMed] [Google Scholar]
- 8.Bouhuys AL, et al. Potential psychosocial mechanisms linking depression to immune function in elderly subjects. Psychiatry Res. 2004;127:237–245. doi: 10.1016/j.psychres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Musselman DL, et al. Higher than normal plasma interleukin-6 concentrations in cancer patients with depression: preliminary findings. Am. J. Psychiatry. 2001;158:1252–1257. doi: 10.1176/appi.ajp.158.8.1252. [DOI] [PubMed] [Google Scholar]
- 10.Kahl KG, et al. Bone mineral density, markers of bone turnover, and cytokines in young women with borderline personality disorder with and without comorbid major depressive disorder. Am. J. Psychiatry. 2005;162:168–174. doi: 10.1176/appi.ajp.162.1.168. [DOI] [PubMed] [Google Scholar]
- 11.Tiemeier H, et al. Inflammatory proteins and depression in the elderly. Epidemiology. 2003;14:103–107. doi: 10.1097/00001648-200301000-00025. [DOI] [PubMed] [Google Scholar]
- 12.Schlatter J, et al. Monocytic parameters in patients with dysthymia versus major depression. J. Affect. Disord. 2004;78:243–247. doi: 10.1016/S0165-0327(02)00316-6. [DOI] [PubMed] [Google Scholar]
- 13.Ford DE, Erlinger TP. Depression and C-reactive protein in US adults. Arch. Intern. Med. 2004;164:1010–1014. doi: 10.1001/archinte.164.9.1010. [DOI] [PubMed] [Google Scholar]
- 14.Danner M, et al. Association between depression and elevated C-reactive protein. Psychosom. Med. 2003;65:347–356. doi: 10.1097/01.psy.0000041542.29808.01. [DOI] [PubMed] [Google Scholar]
- 15.Maes M. Major depression and activation of the inflammatory response system. Adv. Exp. Med. Biol. 1999;461:25–46. doi: 10.1007/978-0-585-37970-8_2. [DOI] [PubMed] [Google Scholar]
- 16.Levine J, et al. Cerebrospinal cytokine levels in patients with acute depression. Neuropsychobiology. 1999;40:171–176. doi: 10.1159/000026615. [DOI] [PubMed] [Google Scholar]
- 17.Lanquillon S, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22:370–379. doi: 10.1016/S0893-133X(99)00134-7. [DOI] [PubMed] [Google Scholar]
- 18.Rajagopalan S, et al. Abnormal brachial artery flow-mediated vasodilation in young adults with major depression. Am. J. Cardiol. 2001;88:196–198. doi: 10.1016/s0002-9149(01)01623-x. [DOI] [PubMed] [Google Scholar]
- 19.Sluzewska A, et al. Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine. Ann. N. Y. Acad. Sci. 1995;762:474–476. doi: 10.1111/j.1749-6632.1995.tb32372.x. [DOI] [PubMed] [Google Scholar]
- 20.Maes M, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–858. doi: 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- 21.Mikova O, et al. Increased serum tumor necrosis factor alpha concentrations in major depression and multiple sclerosis. Eur. Neuropsychopharmacol. 2001;11:203–208. doi: 10.1016/s0924-977x(01)00081-5. [DOI] [PubMed] [Google Scholar]
- 22.Sluzewska A, et al. Changes in acute-phase proteins during lithium potentiation of antidepressants in refractory depression. Neuropsychobiology. 1997;35:123–127. doi: 10.1159/000119332. [DOI] [PubMed] [Google Scholar]
- 23.Tuglu C, et al. Increased serum tumor necrosis factor-alpha levels and treatment response in major depressive disorder. Psychopharmacology (Berl.) 2003;170:429–433. doi: 10.1007/s00213-003-1566-z. [DOI] [PubMed] [Google Scholar]
- 24.Hestad KA, et al. Raised plasma levels of tumor necrosis factor alpha in patients with depression. J. ECT. 2003;19:183–188. doi: 10.1097/00124509-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Owen BM, et al. Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr. Scand. 2001;103:226–228. doi: 10.1034/j.1600-0447.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- 26.Thomas AJ, et al. Increase in interleukin-1beta in late-life depression. Am. J. Psychiatry. 2005;162:175–177. doi: 10.1176/appi.ajp.162.1.175. [DOI] [PubMed] [Google Scholar]
- 27.Yu YW, et al. Association study of the interleukin-1 beta (C-511T) genetic polymorphism with major depressive disorder, associated symptomatology, and antidepressant response. Neuropsychopharmacology. 2003;28:1182–1185. doi: 10.1038/sj.npp.1300172. [DOI] [PubMed] [Google Scholar]
- 28.Jun TY, et al. Possible association between -G308A tumour necrosis factor-alpha gene polymorphism and major depressive disorder in the Korean population. Psychiatr. Genet. 2003;13:179–181. doi: 10.1097/00041444-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Suarez EC, et al. Enhanced expression of cytokines and chemokines by blood monocytes to in vitro lipopolysaccharide stimulation are associated with hostility and severity of depressive symptoms in healthy women. Psychoneuroendocrinology. 2004;29:1119–1128. doi: 10.1016/j.psyneuen.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 30.van der Ven A, et al. Herpes viruses, cytokines, and altered hemostasis in vital exhaustion. Psychosom. Med. 2003;65:194–200. doi: 10.1097/01.psy.0000058378.50240.80. [DOI] [PubMed] [Google Scholar]
- 31.Irwin M, et al. Nocturnal proinflammatory cytokine-associated sleep disturbances in abstinent African American alcoholics. Brain Behav. Immun. 2004;18:349–360. doi: 10.1016/j.bbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Suarez EC, et al. The relation of aggression, hostility, and anger to lipopolysaccharide-stimulated tumor necrosis factor (TNF)-alpha by blood monocytes from normal men. Brain Behav. Immun. 2002;16:675–684. doi: 10.1016/s0889-1591(02)00019-3. [DOI] [PubMed] [Google Scholar]
- 33.Lesperance F, et al. The association between major depression and levels of soluble intercellular adhesion molecule 1, interleukin-6, and C-reactive protein in patients with recent acute coronary syndromes. Am. J. Psychiatry. 2004;161:271–277. doi: 10.1176/appi.ajp.161.2.271. [DOI] [PubMed] [Google Scholar]
- 34.Miller GE, et al. Relation of depressive symptoms to C-reactive protein and pathogen burden (cytomegalovirus, herpes simplex virus, Epstein-Barr virus) in patients with earlier acute coronary syndromes. Am. J. Cardiol. 2005;95:317–321. doi: 10.1016/j.amjcard.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 35.Bower JE, et al. Fatigue and proinflammatory cytokine activity in breast cancer survivors. Psychosom. Med. 2002;64:604–611. doi: 10.1097/00006842-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 36.Meyers CA, et al. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer. 2005;104:788–793. doi: 10.1002/cncr.21234. [DOI] [PubMed] [Google Scholar]
- 37.Haack M, et al. Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: effects of confounding factors and diagnosis. J. Psychiatr. Res. 1999;33:407–418. doi: 10.1016/s0022-3956(99)00021-7. [DOI] [PubMed] [Google Scholar]
- 38.Steptoe A, et al. Lack of association between depressive symptoms and markers of immune and vascular inflammation in middle-aged men and women. Psychol. Med. 2003;33:667–674. doi: 10.1017/s0033291702007250. [DOI] [PubMed] [Google Scholar]
- 39.Miller GE, et al. Pathways linking depression, adiposity, and inflammatory markers in healthy young adults. Brain Behav. Immun. 2003;17:276–285. doi: 10.1016/s0889-1591(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 40.Motivala SJ, et al. Inflammatory markers and sleep disturbance in major depression. Psychosom. Med. 2005;67:187–194. doi: 10.1097/01.psy.0000149259.72488.09. [DOI] [PubMed] [Google Scholar]
- 41.Szabo ST, et al. Neurotransmitters, receptors, signal transduction, and second messengers in psychiatric disorders. In: Schatzberg AF, Nemeroff CB, editors. Textbook of Psychopharmacology. American Psychiatric Publishing; 2004. pp. 3–52. [Google Scholar]
- 42.Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol. Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- 43.Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am. J. Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 44.Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol. Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- 45.Martinot M, et al. Decreased presynaptic dopamine function in the left caudate of depressed patients with affective flattening and psychomotor retardation. Am. J. Psychiatry. 2001;158:314–316. doi: 10.1176/appi.ajp.158.2.314. [DOI] [PubMed] [Google Scholar]
- 46.Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr. Opin. Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- 47.Duman RS. Depression: a case of neuronal life and death? Biol. Psychiatry. 2004;56:140–145. doi: 10.1016/j.biopsych.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 48.Maier SF, Watkins LR. Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol. Rev. 1998;105:83–107. doi: 10.1037/0033-295x.105.1.83. [DOI] [PubMed] [Google Scholar]
- 49.Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur. J. Pharmacol. 2004;500:399–411. doi: 10.1016/j.ejphar.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 50.Gao HM, et al. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J. Neurochem. 2002;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- 51.Dunn AJ, et al. Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv. Exp. Med. Biol. 1999;461:117–127. doi: 10.1007/978-0-585-37970-8_8. [DOI] [PubMed] [Google Scholar]
- 52.Besedovsky HO, del Rey A. Immune-neuro-endocrine interactions: facts and hypotheses. Endocr. Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 53.Silverman MN, et al. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol. Psychiatry. 2004;56:819–824. doi: 10.1016/j.biopsych.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, et al. Interleukin-1 alpha-induced activation of p38 mitogen-activated kinase inhibits glucocorticoid receptor function. Mol. Psychiatry. 2004;9:65–75. doi: 10.1038/sj.mp.4001339. [DOI] [PubMed] [Google Scholar]
- 56.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr. Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 57.Zhu CB, et al. p38 MAPK activation elevates serotonin transport activity via a trafficking-independent, protein phosphatase 2A-dependent process. J. Biol. Chem. 2005;280:15649–15658. doi: 10.1074/jbc.M410858200. [DOI] [PubMed] [Google Scholar]
- 58.Nadjar A, et al. Inactivation of the cerebral NFkappaB pathway inhibits interleukin-1beta-induced sickness behavior and c-Fos expression in various brain nuclei. Neuropsychopharmacology. doi: 10.1038/sj.npp.1300755. (in press) [DOI] [PubMed] [Google Scholar]
- 59.Lu T, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 60.Madrigal JL, et al. The increase in TNF-αlpha levels is implicated in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology. 2002;26:155–163. doi: 10.1016/S0893-133X(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 61.Musselman DL, et al. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N. Engl. J. Med. 2001;344:961–966. doi: 10.1056/NEJM200103293441303. [DOI] [PubMed] [Google Scholar]
- 62.Capuron L, et al. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol. Psychiatry. 2002;7:468–473. doi: 10.1038/sj.mp.4000995. [DOI] [PubMed] [Google Scholar]
- 63.Capuron L, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am. J. Psychiatry. 2003;160:1342–1345. doi: 10.1176/appi.ajp.160.7.1342. [DOI] [PubMed] [Google Scholar]
- 64.Juengling FD, et al. Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmacology (Berl.) 2000;152:383–389. doi: 10.1007/s002130000549. [DOI] [PubMed] [Google Scholar]
- 65.Capuron L, et al. Anterior cingulate activation and error processing during interferon-alpha treatment. Biol. Psychiatry. 2005;58:190–196. doi: 10.1016/j.biopsych.2005.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kendler KS, et al. Stressful life events and previous episodes in the etiology of major depression in women: an evaluation of the ‘kindling’ hypothesis. Am. J. Psychiatry. 2000;157:1243–1251. doi: 10.1176/appi.ajp.157.8.1243. [DOI] [PubMed] [Google Scholar]
- 67.O’Connor KA, et al. Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res. 2003;991:123–132. doi: 10.1016/j.brainres.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 68.Pugh CR, et al. Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav. Brain Res. 1999;106:109–118. doi: 10.1016/s0166-4328(99)00098-4. [DOI] [PubMed] [Google Scholar]
- 69.Barrientos RM, et al. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience. 2003;121:847–853. doi: 10.1016/s0306-4522(03)00564-5. [DOI] [PubMed] [Google Scholar]
- 70.Goebel MU, et al. Interleukin-6 and tumor necrosis factor-alpha production after acute psychological stress, exercise, and infused isoproterenol: differential effects and pathways. Psychosom. Med. 2000;62:591–598. doi: 10.1097/00006842-200007000-00019. [DOI] [PubMed] [Google Scholar]
- 71.Deinzer R, et al. Acute stress effects on local Il-1beta responses to pathogens in a human in vivo model. Brain Behav. Immun. 2004;18:458–467. doi: 10.1016/j.bbi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 72.Maes M, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine. 1998;10:313–318. doi: 10.1006/cyto.1997.0290. [DOI] [PubMed] [Google Scholar]
- 73.Bierhaus A, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moraska A, et al. Elevated IL-1beta contributes to antibody suppression produced by stress. J. Appl. Physiol. 2002;93:207–215. doi: 10.1152/japplphysiol.01151.2001. [DOI] [PubMed] [Google Scholar]
- 75.Murray DR, et al. Chronic beta-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation. 2000;101:2338–2341. doi: 10.1161/01.cir.101.20.2338. [DOI] [PubMed] [Google Scholar]
- 76.Mayer B, et al. Functional improvement in heart failure patients treated with beta-blockers is associated with a decline of cytokine levels. Int. J. Cardiol. 2005;103:182–186. doi: 10.1016/j.ijcard.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 77.Pavlov VA, Tracey KJ. The cholinergic anti-inflammatory pathway. Brain Behav. Immun. 2005;19:493–499. doi: 10.1016/j.bbi.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 78.Janszky I, et al. Inflammatory markers and heart rate variability in women with coronary heart disease. J. Intern. Med. 2004;256:421–428. doi: 10.1111/j.1365-2796.2004.01403.x. [DOI] [PubMed] [Google Scholar]
- 79.Kim CK, et al. Depressive symptoms and heart rate variability in postmenopausal women. Arch. Intern. Med. 2005;165:1239–1244. doi: 10.1001/archinte.165.11.1239. [DOI] [PubMed] [Google Scholar]
- 80.Avitsur R, et al. Social stress induces glucocorticoid resistance in subordinate animals. Horm. Behav. 2001;39:247–257. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- 81.Kenis G, Maes M. Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol. 2002;5:401–412. doi: 10.1017/S1461145702003164. [DOI] [PubMed] [Google Scholar]
- 82.Reynolds JL, et al. Brain-derived tumor necrosis factor-alpha and its involvement in noradrenergic neuron functioning involved in the mechanism of action of an antidepressant. J. Pharmacol. Exp. Ther. 2004;310:1216–1225. doi: 10.1124/jpet.104.067835. [DOI] [PubMed] [Google Scholar]
- 83.Frommberger UH, et al. Interleukin-6-(IL-6) plasma levels in depression and schizophrenia: comparison between the acute state and after remission. Eur. Arch. Psychiatry Clin. Neurosci. 1997;247:228–233. doi: 10.1007/BF02900219. [DOI] [PubMed] [Google Scholar]
- 84.Benedetti F, et al. Interleukine-6 serum levels correlate with response to antidepressant sleep deprivation and sleep phase advance. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2002;26:1167–1170. doi: 10.1016/s0278-5846(02)00255-5. [DOI] [PubMed] [Google Scholar]
- 85.Capuron L, et al. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002;26:643–652. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- 86.Morrow GR, et al. Differential effects of paroxetine on fatigue and depression: a randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J. Clin. Oncol. 2003;21:4635–4641. doi: 10.1200/JCO.2003.04.070. [DOI] [PubMed] [Google Scholar]
- 87.Greco T, et al. The outcome of physical symptoms with treatment of depression. J. Gen. Intern. Med. 2004;19:813–818. doi: 10.1111/j.1525-1497.2004.30531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695:279–282. doi: 10.1016/0006-8993(95)00930-o. [DOI] [PubMed] [Google Scholar]
- 89.Lichtenstein GR, et al. Infliximab improves quality of life in patients with Crohn’s disease. Inflamm. Bowel Dis. 2002;8:237–243. doi: 10.1097/00054725-200207000-00001. [DOI] [PubMed] [Google Scholar]
- 90.Mathias SD, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin. Therap. 2000;22:128–139. doi: 10.1016/s0149-2918(00)87984-9. [DOI] [PubMed] [Google Scholar]
- 91.APA . Diagnostic and Statistical Manual of Mental Disorders. 4th edn American Psychiatric Association; 2000. [Google Scholar]