

Abstract
Identification of new molecular targets in heart failure could ultimately have a substantial positive impact on both the health and financial aspects of treating the large heart failure population. We originally identified reduced levels of the cell junction protein claudin-5 specifically in heart in the dystrophin/utrophin-deficient (Dmdmdx;Utrn−/−) mouse model of muscular dystrophy and cardiomyopathy, which demonstrates physiological hallmarks of heart failure. We then showed that at least 60% of cardiac explant samples from patients with heart failure resulting from diverse etiologies also have reduced claudin-5 levels. These claudin-5 reductions were independent of changes in other cell junction proteins previously linked to heart failure. The goal of this study was to determine whether sustaining claudin-5 levels is sufficient to prevent the onset of histological and functional indicators of heart failure. Here, we show the proof-of-concept rescue experiment in the Dmdmdx;Utrn−/− model, in which claudin-5 reductions were originally identified. Expression of claudin-5 4 weeks after a single administration of recombinant adeno-associated virus (rAAV) containing a claudin-5 expression cassette prevented the onset of physiological hallmarks of cardiomyopathy and improved histological signs of cardiac damage. This experiment demonstrates that claudin-5 may represent a novel treatment target for prevention of heart failure.
Introduction
Dystrophin is a protein that links the subsarcolemmal actin cytoskeleton with the extracellular matrix in striated muscle. Genetic mutations that lead to total loss of dystrophin result in the most severe form of X-linked muscular dystrophy, Duchenne muscular dystrophy (DMD). Mutations that lead to reduced levels or less functional dystrophin result in the milder Becker muscular dystrophy (BMD). The major, life-threatening clinical manifestation in DMD is progressive degeneration of skeletal muscles, particularly the diaphragm, leading to respiratory distress. However, 95% of DMD patients develop cardiomyopathy, and heart failure is the cause of death in at least 25% of patients.1,2 Studies from muscular dystrophy animal models demonstrate that treatments targeting skeletal muscle without improving the heart will lead to a higher incidence of cardiac deaths.3,4 In BMD, where skeletal muscle degeneration is less severe, heart failure is the ultimate cause of death in 90% of the patients.1,2 Therefore, treatment strategies that target the heart will be necessary to treat the entire spectrum of disease in DMD and BMD. Currently these patients lack early-stage, protective treatment options with heart failure standard-of-care drug treatment implemented after the heart is already severely compromised. Moreover, because the 5 million patients with heart failure in the United States display similar end-stage symptoms and have the same limited treatment options, strategies designed to treat muscular dystrophy-based cardiac dysfunction may also be applicable to most heart failure cases.
To identify novel molecular treatment targets for muscular dystrophy-based heart failure, we carried out messenger RNA microarray on cardiac tissue from two mouse models of muscular dystrophy: the dystrophin-deficient mouse (Dmdmdx, or mdx) and the dystrophin/utrophin-deficient mouse (Dmdmdx;Utrn−/−, or dko). Mdx mice, due to partial compensation from the dystrophin homolog, utrophin,5 have mild skeletal muscle pathology,6 mild cardiomyopathy,7 and lack physiological hallmarks of heart failure.8 However, dko mice show severe muscular dystrophy,6 severe cardiomyopathy,7 and functional hallmarks of heart failure.8 Messenger RNA levels of the cell junction protein claudin-5 were reduced threefold specifically in heart, but not skeletal muscle, of dko mice and claudin-5 protein was severely reduced.9,10 We then showed at least 60% of cardiac explant samples from patients with heart failure resulting from diverse etiologies also have reduced claudin-5 levels.11 Claudin-5 reductions were independent of changes in other cell junction proteins linked to heart failure11,12 and known secondary reductions in dystrophin.13,14
In the current study, we tested the hypothesis that sustaining claudin-5 levels in dko mice would prevent hallmarks of heart failure. After a single injection of a recombinant adeno-associated virus (rAAV) containing a claudin-5 expression cassette, we observed improvements in functional and histological markers of cardiomyopathy and heart failure in dko mice. These data support that claudin-5 represents a novel treatment target for cardiomyopathy in muscular dystrophy and possibly even for a majority of heart failure patients.
Results
Four-week-old dko mice, which do not yet display any histological signs of cardiomyopathy and do not yet have reduced cardiac claudin-5 levels, were injected with 1 × 1012 vector genomes of a recombinant AAV6 vector carrying a mouse claudin-5 cDNA expressed from a minimal cytomegalovirus (CMV) promoter (rAAV6-cldn5).15 In parallel, control dko mice were injected with an equivalent volume of phosphate-buffered saline (PBS). This AAV6-pseudotyped vector has previously been shown to have a high tropism for cardiac muscle and confers sustained expression of the delivered gene product within 2 weeks of a single intravenous injection.16,17 Six-week-old dko mice do not display histological indicators of cardiomyopathy (Figure 1a) and have higher levels of cardiac claudin-5 compared to 8- and 10-week-old dko hearts (Figure 1b). By 8 weeks of age, dko mice show initial signs of cardiac damage by histology (Figure 1a); concurrent reduction in levels of claudin-5 messenger RNA and protein9 (Figure 1b); altered localization of claudin-5 in cardiac tissue9; and physiological hallmarks of heart failure including severe contractile deficits, an overly negative force–frequency relationship, and blunted β-adrenergic response.8,18,19 However, these indicators of cardiac pathology precede whole-heart dysfunction (Figure 1c). Dko mice begin to succumb to their skeletal muscle disease after this time-point, making analysis of sufficient numbers of these mice at more advanced stages very challenging.5 By injecting 4-week-old dko mice with rAAV6-cldn5, we hypothesized that cardiac claudin-5 levels would be increased by 8 weeks-of-age compared to control dko mice, and that the rAAV6-cldn5 injected mice would have reduced histological and physiological indicators of cardiac damage.
Figure 1.

Progression of cardiomyopathy and loss of claudin-5 in dko hearts, with preserved whole heart function. (a) H&E stained sections from representative 6-, 8-, and 10-week-old dko hearts showing progressive fibrosis and cellular infiltrate. Bar = 50 µm. (b) Western blot showing claudin-5 protein levels in a 8-week-old wild-type C57BL/10 heart, and 6-, 8-, and 10-week-old dko hearts. α-Sarcomeric actin is shown as a control for cardiomyocyte content. Cardiac claudin-5 levels in 8-week-old dko mice (n = 6) are 24% lower than in age-matched wild-type mice (n = 2) and become further reduced to 42% of wild-type levels by 10 weeks-of-age (n = 5) (P < 0.05). (c) Representative end diastolic and end systolic cardiac MRI images of an 8-week-old dko mouse with normal ejection fraction (66.2 ± 3.0%). The circular and crescent areas of high contrast (bright white) indicate blood present in the left and right ventricles, respectively, at the level of papillary muscles at the end of diastole and systole. H&E, hematoxylin and eosin; MRI, magnetic resonance imaging.
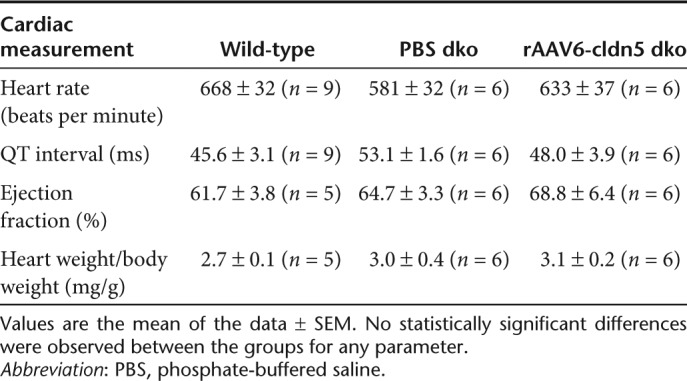
In conscious and unrestrained mice, heart rates did not differ significantly between rAAV6-cldn5–treated and PBS-treated dko mice, and C57 wild-type controls, and the QT interval was not prolonged by rAAV6-cldn5 treatment (Table 1). Cardiac magnetic resonance imaging (MRI) showed that in both dko groups (Table 1, Figure 1c), ejection fractions remained within the normal range for mice and were not different from C57 wild-type controls (Table 1). Analysis of variance analysis also revealed that the ratios of heart weight to body weight were not significantly different between the three groups (Table 1). These data demonstrate that rAAV6-cldn5 treatment did not alter whole-heart function.
Table 1. In vivo cardiac parameters.
Claudin-5 protein levels in hearts from all rAAV6-cldn5–injected dko mice were significantly higher (129 ± 17% compared to wild-type control levels) than in PBS-treated dko controls (28 ± 8% compared to wild-type) (P < 0.05) (Figure 2a). Although exogenous claudin-5 expression varied mildly, all rAAV6-cldn5–injected dko mice showed cardiac claudin-5 protein levels within twofold of that present in hearts from wild-type mice, supporting that claudin-5 in treated mice was present at near-physiological levels. Immunolocalization of claudin-5 on heart sections from rAAV6-cldn5–treated dko mice demonstrated that exogenous claudin-5 concentrates at the lateral membrane similar to claudin-5 localization in wild-type C57 controls9 (Figure 2b). PBS-treated dko hearts showed a very low level, nonspecific pattern of claudin-5 localization (Figure 2b).
Figure 2.

Claudin-5 levels and localization in rAAV6-cldn5 dko hearts. (a) Western blot showing levels of claudin-5 in hearts from representative 8-week-old wild-type, PBS-treated and rAAV6-cldn5–treated dko mice. α-Sarcomeric actin was used to normalize cardiomyocyte content for quantification. (b) Confocal micrographs of merged channels of anti-claudin-5 antibody staining (green) and DAPI nuclei staining (blue) show concentrated claudin-5 localization at the cardiomyocyte lateral membrane in wild-type and rAAV6-cldn5 treated dko hearts (arrowheads), but not in PBS-treated dko hearts.9 Bar = 20 µm. DAPI, 4′,6-diamidino-2-phenylindole; PBS, phosphate-buffered saline.
Cardiac muscle contractile force was assessed in isolated twitch-contracting cardiac trabeculae as described in detail previously8,20,21 (Figure 3a). We compared PBS- and rAAV6-cldn5–treated dko hearts to a group of 10 wild-type C57 controls. In two of the six PBS-treated dko mice, no suitably sized, non-damaged linear muscle preparations could be dissected. Baseline active developed force was significantly higher in rAAV6-cldn5–treated dko mice compared to PBS-treated dko mice (Figure 3b). rAAV6-cldn5 treatment restored levels to that of the C57 wild-type control group (Figure 3b). rAAV6-cldn5 also led to normal maximal and minimal rates of force development (Figure 3c). Contractile kinetics, including 90% relaxation time of twitch contractions, were not different between the three groups (Figure 3c,d).
Figure 3.

Contractile force in isolated cardiac trabeculae from wild-type, rAAV6-cldn5 and PBS-treated dko mice. (a) Schematic showing the parameters of twitch contractions for which measurements are shown in b–d. (b) rAAV6-cldn5 dko mice show increases in baseline cardiac contractile force at 4 Hz stimulation frequency and optimal preload and (c) increases in maximal and minimal rates of force development compared to PBS-treated dko mice. These parameters are not different between rAAV6-cldn5 dko mice and wild-type C57 (WT) controls. (d) Twitch timing kinetics and their balance were not different between both dko groups and WT controls. (e) The enhanced force development conferred by rAAV6-cldn5 treatment was maintained at WT levels during the physiological cardiac regulatory mechanism of length-dependent behavior. (f) Frequency-dependent behavior that spanned the entire in vivo range for the mouse was also improved by rAAV6-cldn5 treatment. (g) The blunted β-adrenergic response normally characterizing dko hearts was enhanced in preparations from rAAV6-cldn5–treated dko mice. Temperature 37 °C, n = 1–3 muscles/mouse, n = 4–10 mice/group. Error bars indicate SEM. *P < 0.05 between the PBS-treated dko and the other two groups (rAAV6-cldn5–treated dko and WT). **P ≤ 0.05 between rAAV6-cldn5–treated dko and C57 WT mice. PBS, phosphate-buffered saline; RT90%, 90% relaxation time.
Additional comparisons of active contractile force were made at various levels of preload, at different stimulation frequencies spanning the in vivo range, and at different levels of the β-adrenergic agonist isoproterenol, and all demonstrated improved cardiac contractile physiology in the rAAV6-cldn5–treated mice. rAAV6-cldn5 treatment elevated active developed force on average to 197% of PBS-treated mice, ranging from 172% at optimal preload length at 4 Hz, not different from wild-type levels, (Figure 3e) to 217% at 14 Hz (Figure 3f). In addition to the already higher baseline force, the increase in developed force in response to isoproterenol that is blunted in dko mice, and is a strong physiological indicator of heart failure,18 was significantly improved by rAAV6-cldn5 treatment (9.8 ± 3.3 mN/mm2 in rAAV6-cldn5–treated dko mice versus 7.7 ± 3.1 mN/mm2 in PBS-treated mice) (Figure 3g). The improvement with rAAV6-cldn5 treatment in response to isoproterenol challenge, however, demonstrated only a partial recovery compared to wild-type mice (28.3 ± 4.9 mN/mm2).
At 8 weeks-of-age dko mice display the initial signs of histopathological cardiac damage that rapidly transforms into large amounts of fibrotic scarring by 10 weeks-of-age7 (Figure 1a). As cardiac enzymes such as creatine kinase leak out of damaged cardiomyocytes, and serve clinically as the initial diagnostic tool of cardiac damage, serum proteins also leak in and serve as an excellent marker of damaged cardiomyocytes.22,23,24 We therefore stained heart cryosections from PBS- and rAAV6-cldn5–treated dko mice (at least two 8 µm sections per mouse at greater than 150 µm apart) with a fluorescently conjugated anti-mouse IgG antibody to assess intracellular localization of serum proteins. All sections from the six PBS-treated mice showed localization of serum IgG within cardiomyocytes (Figure 4a). Extracellular IgG localization can also be observed in regions of more advanced damage where cardiomyocytes are being replaced by fibrotic scarring (Figure 4b). Five of seven rAAV6-cldn5–treated mice had no detectable cardiac damage (Figure 4c). Two of seven rAAV6-cldn5–treated mice showed evidence of cardiac damage by intracellular IgG localization in only one of the spatially separated sections. Confocal microscopy imaging of co-stained PBS-treated (Figure 4d) and rAAV6-cldn5–treated (Figure 4e) dko heart sections showed that only some cardiomyocytes in the occasional small areas of IgG positive damage in rAAV6-cldn5–treated dko hearts were not expressing recombinant claudin-5.
Figure 4.

Histological analysis of rAAV6-cldn5 dko hearts. (a) Localization of endogenous serum IgG within cardiomyocytes (top) is present in PBS-treated dko mice, where H&E staining confirms cardiomyocyte presence (bottom). Mononuclear cells, as detected by the increased concentration of DAPI-stained nuclei (2nd row), colocalize with damaged cardiomyocytes in the merged image (3rd row). (b) Fibrotic damage can be observed in H&E stained sections of other PBS-treated hearts; IgG is present both intracellularly, and extracellularly where cardiomyocytes are absent. (c) rAAV6-cldn5 prevents histological signs of damage in nearly all observed sections of the treated dko mice, evidenced by absence of concentrated IgG staining (top), concentrated nuclei (2nd and 3rd rows), and morphological damage (bottom). Bar = 50 µm. Confocal microscopy images of heart sections co-immunostained for IgG (red), claudin-5 (green), and nuclei (DAPI-blue) comparing frequent regions of damage present in (d) PBS-treated dko mice with the occasional area of cardiac damage observed in a subset of (e) rAAV6-cldn5–treated dko mice. Bar = 20 µm. DAPI, 4′,6-diamidino-2-phenylindole; H&E, hematoxylin and eosin; PBS, phosphate-buffered saline.
Discussion
This experiment provides initial proof-of-principle that treatment with rAAV6-cldn5 in the dko model of severe muscular dystrophy, the original model where reduced cardiac claudin-5 levels were first identified, is able to prevent physiological and histological indicators of cardiac pathology. This single treatment of rAAV6-cldn5 resulted in an improvement of dko cardiac muscle force from 57 to 100% of wild-type baseline force. This improvement falls within the normal range of several different strains of control mice analyzed by the same investigators.20 In addition, even under stress conditions of increased stimulation frequency and β-adrenergic agonist (isoproterenol) challenge, rAAV6-cldn5 treatment recovered half to two-thirds of wild-type levels. When comparing the results here with our previous study, where we assessed specific active contractile force development in wild-type, mdx, and dko cardiac muscles,8 even the partially rescued force-frequency and β-adrenergic responses are at least similar to the milder mdx cardiomyopathic model. The cardiac muscle relaxation data suggest that sustained claudin-5 expression does not impair cardiac relaxation and potentially improves the trend towards diastolic dysfunction normally present in dko mice.
The histological indicators of cardiomyopathy were substantially improved in rAAV6-cldn5–treated dko hearts compared to control dko hearts, but we observed a minimal amount of cardiac damage in a subset of the treatment group. Together, the partial recovery of some parameters of cardiac muscle contractile function and the observation that there were minimal areas of damage in rAAV6-cldn5–treated mice may be explained by all cardiomyocytes in the area of damage requiring a threshold level of claudin-5 required for maximal protection. Another possibility is that recombinant claudin-5 needs to be present earlier to completely restore wild-type function. The cardiac muscles isolated from some of the rAAV6-cldn5–treated dko mice may also have included partially damaged but still contracting muscle preparations. It is possible that optimizing the treatment regimen could eliminate cardiac damage and further improve all physiological responses of isolated muscles to wild-type levels.
At this time, we can only speculate about the role claudin-5 plays in the heart. Interestingly, in hearts of both wild-type mice9 and rAAV6-cldn5–treated dko mice, claudin-5 localizes to the lateral membrane of cardiomyocytes near the extracellular matrix (ECM). Claudin-5, previously identified as a cell-to-cell tight junction protein in other cell types,25 does not localize to the cell-to-cell junction (intercalated disc) in the heart. It is possible that cardiac claudin-5 instead makes a cardiomyocyte–ECM junction. The ECM helps support the structural integrity of the heart and cardiomyocyte–ECM interactions are disrupted in the ventricular remodelling observed in the progression to heart failure.26 The loss of cardiac claudin-5 in the dko model of cardiomyopathy and at least 60% of human heart failure cases supports the possibility that claudin-5 may contribute to the cardiomyocyte–ECM interactions required for structural integrity.
Here, we present evidence that claudin-5 levels are normal before initial cardiac damage, but begin to be reduced concurrent with the first physiological and histological indicators of heart failure that precede whole-heart dysfunction. These data support that claudin-5 reductions occur as a very early step in disease progression. Cardiac damage and physiological deficits also occur before reduced ejection fraction in DMD and BMD patients.27,28 Therefore, the timing of endogenous claudin-5 reductions and its ability to prevent initial signs of dystrophic cardiomyopathy, support claudin-5 as a potentially useful molecular target for muscular dystrophy-based cardiomyopathy. Additional experiments will be required to optimize the treatment strategy, determine whether the improvements present in the rAAV6-cldn5–treated mice result from a prevention or only a delay in disease progression, and whether claudin-5 has the ability to halt or reverse heart failure progression at more advanced stages in dystrophin-related cardiomyopathy.29 Testing claudin-5–targeted treatment strategies in additional models may reveal general applicability. However, the reduction of claudin-5 in a majority of human heart failure samples resulting from diverse etiologies already supports a role for involvement of claudin-5 in the general heart failure population. Further optimization could make claudin-5–based treatment a viable therapeutic approach for a majority of heart failure patients.
Materials and Methods
rAAV6-cldn5 vector. The single exon encoding the claudin-5 protein coding sequence was amplified from cDNA prepared from C57BL/10 mouse hearts and inserted with an upstream Kozak consensus sequence into the NotI site of pAAV:CMV-MCS-SV40pA, and used to prepare recombinant rAAV6-cldn5 as described.15
Treatment and physiological analyses. Animal protocols were approved by the Institutional Animal Care and Use Committee at The Ohio State University. Dmdmdx;Utrn−/− (dko) mice on a C57BL/10 background were produced from matings between Dmdmdx; Utrn+/− parents.7 rAAV6-cldn5 (1 × 1012 viral genomes) or an equal volume (167 µl) of PBS was injected once into male and female 4-week-old dko mice via the tail vein (n = 7 rAAV6-cldn5–treated, n = 6 PBS-treated). Small groups of aged-matched mice were injected on four separate days over the course of 6 weeks. In each group, there were both rAAV6-cldn5– and PBS-injected dko mice so analysis of control and experimental mice were interspersed. One day before 8 weeks-of-age, dko mice were weighed and underwent cardiac MRI. Previously unpublished whole-heart function data from 20-week-old C57BL/10 wild-type mice that underwent both cardiac MRI and electrocardiograms on subsequent days during the same time-frame and carried out by the same investigators as for the two dko groups, are included as a wild-type comparison in Table 1. For PBS and rAAV6-cldn5–treated dko mice, electrocardiograms were recorded the day following MRI, then mice were euthanized for in vitro cardiac muscle contraction analysis8 by experimenters blinded to the treatment groups. Isolated cardiac trabeculae were assessed by in vitro physiological measurements at 37 °C, 4 Hz stimulation frequency, optimal length, and 2.0 mmol/l bathing (Ca2+) as described.20 A previously unpublished data set of in vitro cardiac physiological measurements from 8-week-old C57 mice done by the same investigators with an almost identical protocol was included as a wild-type comparison for PBS and rAAV6-cldn5–treated mice in Figure 3. After removal of cardiac trabeculae from dko mice, remaining heart tissue was divided; half flash frozen for subsequent protein isolation and half embedded in optimal cutting temperature medium and frozen on liquid-nitrogen cooled isopentane for subsequent histological analyses. From two PBS controls and one rAAV6-cldn5 dko, heart tissue was fixed in 1% paraformaldehyde before freezing for improved confocal microscopy analysis of claudin-5 immunolocalization. Hearts from uninjected 6-, 8-, and 10-week-old dko mice (n = 6, 6, 5, respectively) and 8-week-old wild-type isogenic C57BL/10 mice (n = 2) were harvested and flash frozen or frozen in optimal cutting temperature medium.
Immunoblotting and histological analyses. Claudin-5 levels were assessed by western blot with a mouse monoclonal antibody (Invitrogen 35-2500, 1:100 dilution; Invitrogen, Camarillo, CA) and normalized to cardiomyocyte-specific α-sarcomeric actin (Sigma A2172, 1:5,000; Sigma-Aldrich, St Louis, MO) to control for cardiomyocyte content.9,11 Quantification of bands were performed by densitometric measurement using NIH Image J (NIH, Bethesda, MD). Specifically, the same size box was drawn around each protein band and pixel-densitometry histograms were generated using the Image J algorithm for each selected area (analyze ≥ gels ≥ plot lanes). The area under the peak of the histogram represented the band intensity. The claudin-5 band intensity was divided by the actin band intensity to normalize claudin-5 levels for cardiomyocyte content. The data are reported as a percentage of the mean of wild-type C57 control levels.
For immunolocalization of claudin-5, 8 µm thick cryosections were incubated with a rabbit polyclonal antibody against claudin-5 (Abcam ab15106, 1:100; Abcam, Cambridge, MA), then with AlexaFluor 555-anti-rabbit IgG (Invitrogen Molecular Probes A21429, 1:100; Invitrogen Molecular Probes, Eugene, OR) similar to previously described methods.9 IgG immunostaining was performed using a CY3-anti-mouse IgG antibody (Jackson Immunoresearch 115-165-146, 1:100; Jackson Immunoresearch, West Grove, PA) or, for costaining with claudin-5 followed by confocal microscopy, an AlexaFluor 488-anti-mouse IgG antibody (Invitrogen A11029, 1:100; Invitrogen) as described.7 Epifluorescence images were taken on a Nikon Eclipse 800 microscope (Nikon, Melville, NY). Confocal images were taken using an Olympus FV1000 Filter Confocal microscope (Olympus, Center Valley, PA). Hematoxylin and eosin staining was performed by standard methods.
Statistics. Analysis of variance followed by a post-hoc t-test using Bonferroni adjustment, where applicable, was used to determine statistically significant differences.
Acknowledgments
We would like to thank Jeffrey S Chamberlain for providing rAAV6-cldn5 and reading the manuscript, Eric Finn for preparing rAAV6-cldn5, the OSU Small Animal Imaging Core, the Campus Microscopy and Imaging Facility, and Cameron Black for technical assistance. This work was funded by the Muscular Dystrophy Association (to J.A.R.-F.); the American Heart Association (to P.M.L.J.); NIH T32 HL098039 and a UNCF-Merck Science Initiatives postdoctoral fellowship (supporting D.A.D.). The authors declared no conflict of interest.
References
- Kaspar RW, Allen HD., and, Montanaro F. Current understanding and management of dilated cardiomyopathy in Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract. 2009;21:241–249. doi: 10.1111/j.1745-7599.2009.00404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans MC, Pinto YM, Merkies IS, de Die-Smulders CE, Crijns HJ., and, Faber CG. Hereditary muscular dystrophies and the heart. Neuromuscul Disord. 2010;20:479–492. doi: 10.1016/j.nmd.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Duan D. Challenges and opportunities in dystrophin-deficient cardiomyopathy gene therapy. Hum Mol Genet. 2006;15 Spec No 2:R253–R261. doi: 10.1093/hmg/ddl180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D, Yasuda S, Li S, Chamberlain JS., and, Metzger JM. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol Ther. 2008;16:832–835. doi: 10.1038/mt.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafael JA, Tinsley JM, Potter AC, Deconinck AE., and, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L.et al. (1997Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy Cell 90717–727. [DOI] [PubMed] [Google Scholar]
- Hainsey TA, Senapati S, Kuhn DE., and, Rafael JA. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul Disord. 2003;13:294–302. doi: 10.1016/s0960-8966(02)00286-9. [DOI] [PubMed] [Google Scholar]
- Janssen PM, Hiranandani N, Mays TA., and, Rafael-Fortney JA. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2005;289:H2373–H2378. doi: 10.1152/ajpheart.00448.2005. [DOI] [PubMed] [Google Scholar]
- Sanford JL, Edwards JD, Mays TA, Gong B, Merriam AP., and, Rafael-Fortney JA. Claudin-5 localizes to the lateral membranes of cardiomyocytes and is altered in utrophin/dystrophin-deficient cardiomyopathic mice. J Mol Cell Cardiol. 2005;38:323–332. doi: 10.1016/j.yjmcc.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Baker PE, Kearney JA, Gong B, Merriam AP, Kuhn DE, Porter JD.et al. (2006Analysis of gene expression differences between utrophin/dystrophin-deficient vs mdx skeletal muscles reveals a specific upregulation of slow muscle genes in limb muscles Neurogenetics 781–91. [DOI] [PubMed] [Google Scholar]
- Mays TA, Binkley PF, Lesinski A, Doshi AA, Quaile MP, Margulies KB.et al. (2008Claudin-5 levels are reduced in human end-stage cardiomyopathy J Mol Cell Cardiol 4581–87. [DOI] [PubMed] [Google Scholar]
- Dupont E, Matsushita T, Kaba RA, Vozzi C, Coppen SR, Khan N.et al. (2001Altered connexin expression in human congestive heart failure J Mol Cell Cardiol 33359–371. [DOI] [PubMed] [Google Scholar]
- Müller AL., and, Dhalla NS. Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail Rev. 2012;17:395–409. doi: 10.1007/s10741-011-9269-8. [DOI] [PubMed] [Google Scholar]
- Vatta M, Stetson SJ, Perez-Verdia A, Entman ML, Noon GP, Torre-Amione G.et al. (2002Molecular remodelling of dystrophin in patients with end-stage cardiomyopathies and reversal in patients on assistance-device therapy Lancet 359936–941. [DOI] [PubMed] [Google Scholar]
- Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L.et al. (2006rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice Nat Med 12787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG.et al. (2004Systemic delivery of genes to striated muscles using adeno-associated viral vectors Nat Med 10828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS., and, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K.et al. (1982Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts N Engl J Med 307205–211. [DOI] [PubMed] [Google Scholar]
- Au CG, Butler TL, Sherwood MC, Egan JR, North KN., and, Winlaw DS. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int J Exp Pathol. 2011;92:57–65. doi: 10.1111/j.1365-2613.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen PM. Kinetics of cardiac muscle contraction and relaxation are linked and determined by properties of the cardiac sarcomere. Am J Physiol Heart Circ Physiol. 2010;299:H1092–H1099. doi: 10.1152/ajpheart.00417.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull LB, Leppo MK, Marbán E., and, Janssen PM. Physiological determinants of contractile force generation and calcium handling in mouse myocardium. J Mol Cell Cardiol. 2002;34:1367–1376. doi: 10.1006/jmcc.2002.2065. [DOI] [PubMed] [Google Scholar]
- Straub V, Rafael JA, Chamberlain JS., and, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabaeva Z, Meekhof KE., and, Michele DE. Sarcolemma instability during mechanical activity in Largemyd cardiac myocytes with loss of dystroglycan extracellular matrix receptor function. Hum Mol Genet. 2011;20:3346–3355. doi: 10.1093/hmg/ddr240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michele DE, Kabaeva Z, Davis SL, Weiss RM., and, Campbell KP. Dystroglycan matrix receptor function in cardiac myocytes is important for limiting activity-induced myocardial damage. Circ Res. 2009;105:984–993. doi: 10.1161/CIRCRESAHA.109.199489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Sasaki H, Furuse M., and, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147:185–194. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedak PW, Verma S, Weisel RD., and, Li RK. Cardiac remodeling and failure: from molecules to man (Part I) Cardiovasc Pathol. 2005;14:1–11. doi: 10.1016/j.carpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Puchalski MD, Williams RV, Askovich B, Sower CT, Hor KH, Su JT.et al. (2009Late gadolinium enhancement: precursor to cardiomyopathy in Duchenne muscular dystrophy Int J Cardiovasc Imaging 2557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaert D, Richards K, Rafael-Fortney JA., and, Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging. 2011;4:67–76. doi: 10.1161/CIRCIMAGING.110.960740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Long C., and, Duan D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102:121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]