

Abstract
Various substituted arylcuprates undergo stereocontrolled additions to L-serine-derived γamino- and γ-carbamato-α,β-enoates with high syn-selectivities. The stereochemical outcome of these reactions is fully consistent with the reductive elimination-based model proposed previously. This method is well suited for the preparation of a broad range of biologically active amaryllidaceae constituents and their aromatic analogues.
1. Introduction
Numerous natural products found in the plants of the amaryllidaceae family have been the focus of intense research effort. Their diverse biological activities and, therefore, medicinal utility attract the attention of biochemists and pharmacologists.1 Meanwhile, the complex structures and limited natural abundance of many of these plant metabolites continue to fuel the interest of synthetic chemists.2
According to a recent comprehensive review, over 100 small molecule amaryllidaceae constituents that belong to pancratistatin, lycorine or lycorenine structural types have been isolated (Figure 1).3 Many of these exhibit anticancer,4 antiviral,5 antiparasitic,6 and anti-inflammatory7 activities amongst others. Pancratistatin has been in preclinical evaluation as an anticancer agent for many years, with limited availability being a major hurdle for its advancement to clinical trials. Significantly, a number of recent reports revealed that this agent displays much better activity/toxicity ratios than the currently used anticancer drugs etoposide and paclitaxel.8 This discovery is expected to further stimulate the development of efficient synthetic pathways to this class of structurally complex natural products.
Figure 1.

Selected amaryllidaceae constituents.
The major synthetic challenge stems from the dense stereochemistry of the cyclitol ring, in which the creation of the benzylic stereocenter C10b can be particularly arduous due to the necessity of stereoselective installation of an aromatic group.9 A close inspection of the structures in Figure 1 reveals consistent stereochemical relationships. Thus, the aromatic moiety is nearly always cis to the adjacent hydroxyl substituent (C, O) and trans to the nitrogen-bearing stereocenter on the other side (C, N). It follows that a flexible synthetic methodology that allows the introduction of an aromatic moiety with stereocontrol exerted by either oxygen- or nitrogen-containing adjacent stereocenters would be generally applicable to the synthesis of compounds that belong to these structural types. Furthermore, if the methodology is not sensitive to the identity and positioning of the substituents on the aromatic ring, a library of analogues of these natural products could be prepared for structure-activity studies.
We have recently reported the utilization of a highly, anti-selective arylcuprate conjugate addition to a γ-alkoxy-α,β-enoate as one such methodology.10 The anti-stereochemical relationship of the oxygen-bearing and benzylic stereocenters in the addition products corresponds to their cis positioning in the target cyclic structures (Figure 2). A complementary strategy would use a syn-selective arylcuprate conjugate addition to α,β-enoates with stereocontrol exerted by a γ-nitrogen-containing stereocenter. Since the feasibility of this stereochemical divergence had been reported previously,11c we explored this alternative approach and report our results herein.
Figure 2.

Correlation of stereochemical relationships in open-chain and cyclic structures.
2. Results and discussion
Syn-selective conjugate additions of organocuprates have been reported for both γ-amino- and γ-carbamato-α,β-enoates and they have been utilized in the syntheses of various medicinally relevant complex targets.11 Therefore, to evaluate this methodology for the introduction of multisubstituted aromatic rings, particularly those possessing the multiple alkoxy groups required in the synthesis of amaryllidaceae constituents, we prepared γ-amino- and γ-carbamato-enoates 1 and 2 (Scheme 1). While the synthesis of enoate 2 from L-serine had previously been described in the literature,12 the key to the successful straightforward preparation of enoate 1 was a one-pot Swern oxidation – Wittig olefination method that resulted in exclusive formation of the E-enoate.13
Scheme 1.

Gratifyingly, the reactions of arylcuprates derived from aromatic Grignard reagents with enoate 1 gave addition products 3a–f as single diastereomers (Scheme 2). The NMR analyses of crude and purified reaction mixtures revealed the presence of the β-epimeric compounds in only trace quantities. However, the reactions were sluggish and provided only modest yields of 3a-f, while the addition product 3g could not be detected at all. The steric congestion arising from the bulky t-butyldiphenylsilyl protection clearly plays a role in this unsatisfactory outcome.
Scheme 2.

In contrast, the addition products 4a-g, resulting form the reactions of enoate 2, were formed in excellent yields. However, the rotational isomerism associated with the carbamate moiety led to NMR peak broadening and this prevented any immediate conclusions regarding the diastereomeric purity of 4a-g. Therefore, each addition product was reacted with methanolic HCl to remove the Boc and isopropylidene protecting groups to give ammonium salts 5a-g. These underwent facile lactam formation upon treatment with MeONa in MeOH to give 6a-g in good overall yields. The NMR analyses of 6a-g clearly showed the presence of a single diastereomer in each case.
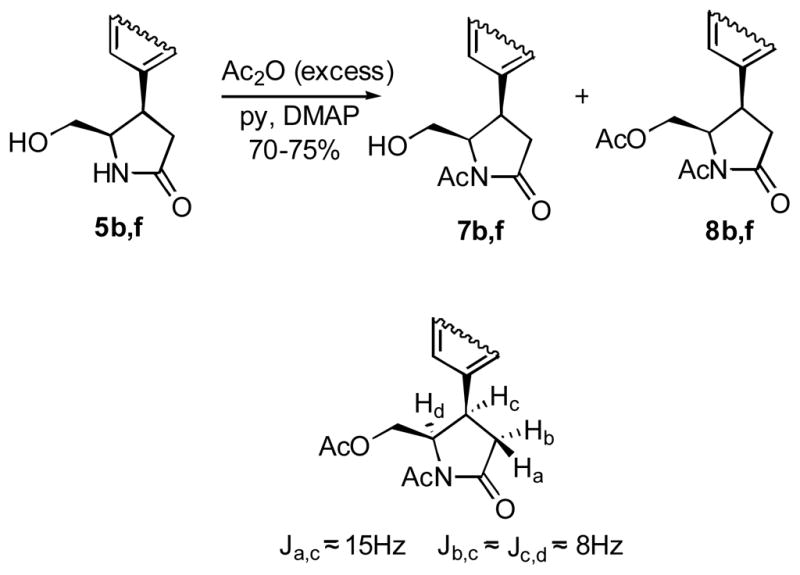
To facilitate the NMR-based stereochemistry assignment, we attempted to acetylate the primary hydroxyl group in lactams 5b and 5f (Scheme 3). Unexpectedly, the N-acetylated products 7b and 7f were isolated instead of the expected esters when the starting lactams were treated with equimolar amounts of acetic anhydride. Evidently, the intramolecular oxygen to nitrogen acetyl transfer leads to the formation of the thermodynamically more stable imides. However, treatment of 5b and f with excess of Ac2O gives diacetylated lactams 8b and 8f, whose 1H NMR contain well-resolved lactam proton signals allowing the unequivocal assignment of the cis stereochemical relationship between Hc and Hd.
Scheme 3.
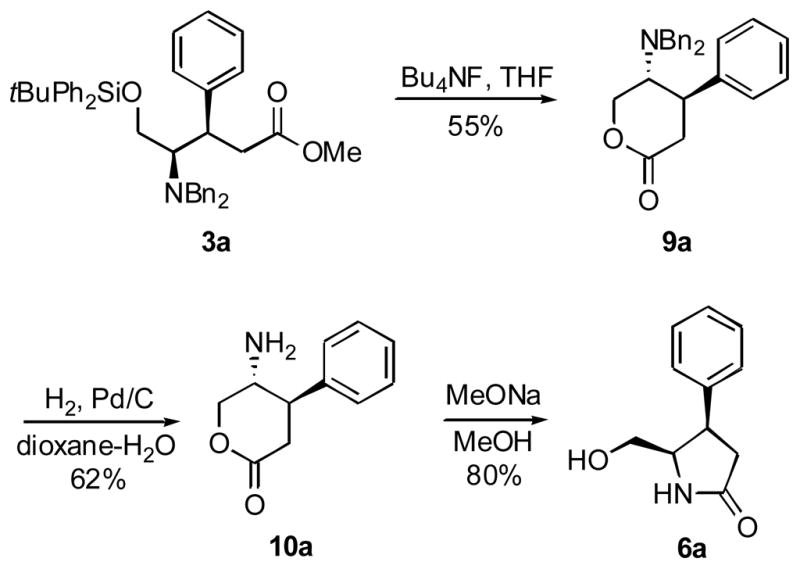
To confirm the syn-stereochemistry in addition products 3a-f by chemical correlation, we attempted to convert 3a to 6a by way of N-debenzylation, lactamization and O-desilylation. To this end, 3a was treated with hydrogen over various palladium catalysts and high pressures. Surprisingly, ester 3a is highly resistant to hydrogenolysis, possibly due to the steric bulk of the silyl protection and, consequently, an impaired contact with the catalyst surface. Reversing the order of the deprotections by performing the desilylation first, gave lactone 9a (Scheme 4). This compound underwent facile double debenzylation when stirred in a mixture of dioxane-water under a hydrogen balloon. Finally, lactone to lactam isomerization 10a to 6a was brought about with sodium methoxide in methanol. This transformation conceivably proceeds through the intermediacy of the ring-opened methyl ester.
Scheme 4.
3. Conclusions
Highly stereoselective processes with the participation of acyclic systems are not commonplace in organic synthesis. Therefore, we hope that our previously reported highly anti-selective arylcuprate additions to γ-alkoxy-α,β-enoates10 and the present syn-selective additions to γ-amino- and γ-carbamato-α,β-enoates will find utility in the introduction of multisubstituted aromatic groups into complex acyclic structures with high stereocontrol. Work in our laboratory is currently underway to apply this chemistry to the development of practical synthetic pathways to biologically active amaryllidaceae constituents to facilitate their advancement to clinical trials. Furthermore, the independence of the stereochemical outcome of the substitution pattern on the aromatic ring makes these processes particularly promising for the synthesis of aromatic analogues of these natural products.14
Recently, we proposed a stereochemical model to predict the outcomes of organocuprate addition reactions to γ-alkoxy-α,β-enoates, based on reductive elimination as a rate- and stereochemistry-determining step.10b We suggested that the model could be of general utility encompassing reactions of other α,β-enoates containing a γ-stereocenter. The results of the present investigation with γ-amino- and γ-carbamato–α,β-enoates are fully consistent with this model that predicts syn-selectivities for these processes (Figure 3).
Figure 3.

Reductive elimination and modified Felkin-Anh stereochemical models.
In contrast, the modified Felkin-Anh model11c,15 leads to the prediction of anti-isomers as the favored addition products. Furthermore, the reductive elimination model explains the low reactivity of γ-amino–α,β-enoates with a sterically demanding group R. Clearly, the hypothetical transition state would be highly energetic in this case due to the 1,3-allylic strain (Cγ–R and Cα–H in the top pathway in Figure 3). The sluggish nature of reactions of enoate 1 are in agreement with this mechanistic interpretation.
4. Experimental section
4.1. General methods
Unless otherwise noted, all commercially obtained reagents were used without purification. THF was distilled from sodium benzophenone ketyl prior to use. Dichloromethane was distilled from calcium chloride. Reactions were carried out under a nitrogen atmosphere in oven-dried glassware using standard syringe, cannula and septa techniques. Reactions were monitored by TLC (Silica Gel 60 F254, 250 μm) and visualized with UV light and ceric ammonium molybdate solution. Aryl bromides f and g were prepared as previously described.16 Flash chromatography was performed on silica gel (32–63 μm, 60A° pore size). Optical rotations were measured with an Autopol III automatic polarimeter. 1H and 13C NMR spectra were recorded on JEOL 300 MHz spectrometer.
4.2. Preparation of enoate 1
4.2.1. 5-(tert-Butyldiphenylsilanyloxy)-4-dibenzylamino-pent-2-enoic acid methyl ester, 1
To oxalyl chloride (5.0 ml of 2M in CH2Cl2, 9.7 mmol) in dry CH2Cl2 (50 ml) at −78 °C was added DMSO (1.42 ml, 19.9 mmol) in CH2Cl2 (15 ml) over 10 min and the mixture was stirred for an additional 20 min. 3-(tert-Butyl-diphenyl-silanyloxy)-2-dibenzylamino-1-propanol12a,b (2.208 g, 4.3 mmol) in CH2Cl2 (15 ml) was added over 10 min and the mixture stirred for an additional 10 min. Triethylamine (3.37 ml, 23.9 mmol) in CH2Cl2 (15 ml) was added over 10 min and the white slurry was stirred for 20 min at −78 °C. To the cold reaction mixture was added methyl (triphenylphosphoranylidene) acetate (3.34 g, 9.7 mmol) in one portion and the resulting mixture was stirred for 16 h while it was allowed to warm up to rt. Water (150 ml) was added to reaction mixture, the two layers were separated and aqueous layer was extracted with CH2Cl2 (3 × 75 ml). The combined organic layers were dried over MgSO4 and solvent evaporated under reduced pressure. The residual oil was presorbed on silica gel and the product purified using chromatography with gradients 1% and 2% EtOAc/Hexane to afford enoate 1 (2.21 g, 90.6%) as a colorless oil. Rf 0.62 (10% EtOAc/Hexane); [α]D23 = −21.4 (c 0.14, CHCl3); 1H NMR (CDCl3) δ 7.63–7.19 (m, 20H), 7.03 (dd, 1H, J = 6.8, 15.6 Hz), 6.02 (dd, 1H, J = 1.3, 15.9 Hz), 4.02–3.8 (m, 3H), 3.78 (s, 1H), 3.77 (s, 3H), 3.62 (s, 1H), 3.58 (s, 1H), 3.51 (m, 1H), 1.03 (s, 9H); 13C NMR (CDCl3) δ 166.7, 146.1, 139.7, 135.6, 133.2, 129.8, 128.7, 128.5, 128.3, 128.2, 127.8, 127.0, 123, 63.9, 60.5, 54.5, 51.6, 26.8, 19.2; HRMS m/z (ESI) calcd for C36H41NO3Si (M+H)+ 564.2934, found 564.2921.
4.3. General procedure for the arylcuprate addition
Ca. 1 ml of a required aryl bromide (10.0 mmol) was added to crushed Mg turnings (10.0 mmol, 0.242 g) in THF (10 ml) under a nitrogen atmosphere. Once the reaction had started, the solution was warmed up and slightly darkened. The rest of the aryl bromide was added dropwise to allow a gentle reaction. The reaction mixture was allowed to cool to room temperature and was cannulated to a slurry of CuI (5 mmol, 0.952 g) in THF (10 ml) at − 78 °C. The mixture was stirred at − 78 °C for 40 min (in the synthesis of 3e and 4e the mixture was stirred at 0 °C for 2 h as no transmetalation occurred at − 78 °C). Me3SiCl (10.0 mmol, 1.08 g) and enoate 1 or 2 (1 mmol, in 10 ml of THF) was added sequentially at − 78 °C. The yellow brown solution was stirred overnight while slowly warming up to rt. The reaction mixture was quenched with a mixture of concd. NH4OH and satd. NH4Cl (1:9, 30 ml) and extracted with EtOAc (3 × 40 ml). The combined organic layers were washed with water and brine (2 × 5 ml) and dried over anhyd. MgSO4. The solution was concentrated under reduced pressure, the residue was absorbed on silica gel and purified by column chromatography (2 – 10% EtOAc/hexanes) to yield 3a-f (49–58%) and 4a-g (70–95%) as an oil.
4.3.1. 5-(tert-Butyldiphenylsilanyloxy)-4-dibenzylamino-3-phenylpentanoic acid methyl ester, 3a
58%; Rf 0.67 (10% EtOAc/Hexane); [α]D23 = −12.6 (c 0.15, CHCl3); 1H NMR (CDCl3) δ 7.9 – 6.91 (m, 25H), 4.18 (dd, 1H, J = 3.8, 11.2 Hz), 3.92 (dd, 1H, J = 3.8, 11.2 Hz), 3.8 (d, 2H, J = 13.7 Hz), 3.72 (m, 1H), 3.45 (s, 3H), 3.41 (br d, 2H), 2.99 (m, 1H), 2.7 (dd, 1H, J = 4.4, 15.1 Hz), 2.41 (dd, 1H, J = 10.2, 15.1 Hz), 1.22 (s, 9H); 13C NMR (CDCl3) δ 172.6, 142.1, 139.9, 136.0, 135.9, 133.4, 133.2, 131.0, 130.0, 129.1, 128.0, 126.7, 126.4, 61.3, 60.3, 54.7, 51.3, 41.8, 39.1, 27.1, 19.3; HRMS m/z (ESI) calcd for C42H47NO3Si (M+H)+ 642.3403, found 642.3383.
4.3.2. 5-(tert-Butyldiphenylsilanyloxy)-4-dibenzylamino-3-(4-methoxyphenyl)-pentanoic acid methyl ester, 3b
50%; Rf 0.57 (10% EtOAc/Hexane); [α]D24 = −10.7 (c 0.06, CHCl3); 1H NMR (CDCl3) δ 7.82 – 6.72 (m, 24H), 4.1 (dd, 1H, J = 3.8, 11.0 Hz), 3.87 (br dd, 1H), 3.83 (s, 3H), 3.79 – 3.72 (d, 2H, J = 13.7 Hz), 3.64 (m, 1H), 3.42 (s, 3H), 3.35 (d, 2H, J = 13.7 Hz), 2.91 (m, 1H), 2.62 (dd, 1H, J = 4.1, 15.1 Hz), 2.34 (dd, 1H, J = 10.4, 15.1 Hz), 1.17 (s, 9H); 13C NMR (CDCl3) δ 172.7, 158.1, 139.9, 135.9, 135.8, 134.2, 133.4, 133.1, 129.9, 129.0, 127.9, 127.8, 126.7, 113.3, 61.4, 60.4, 55.3, 54.7, 51.3, 41.2, 39.1, 27.1, 19.3; HRMS m/z (ESI) calcd for C43H49NO4Si (M+H)+ 672.3509, found 672.3484.
4.3.3. 5-(tert-Butyldiphenylsilanyloxy)-4-dibenzylamino-3-(4-fluorophenyl)-pentanoic acid methyl ester, 3c
55%; Rf 0.66 (10% EtOAc/Hexane); [α]D24 = −22.9 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 7.79 – 6.78 (m, 24H), 4.1 (dd, 1H, J = 3.6, 11.2 Hz), 3.83 (dd, 1H, J = 3.8, 11.3 Hz), 3.73 (d, 2H, J = 13.2 Hz), 3.62 (m, 1H), 3.40 (s, 3H), 3.33 (d, 2H, J = 13.2 Hz), 2.84 (m, 1H), 2.61 (dd, 1H, J = 4.1, 15.1 Hz), 2.26 (dd, 1H, J = 10.4, 14.8 Hz); 13C NMR (CDCl3) δ 172.1, 139.6, 137.9, 135.9, 135.8, 135.7, 133.3, 133.1, 130.4, 130.3, 130.1, 130.0, 128.9, 128.0, 127.9, 127.8, 126.8, 114.8, 114.5, 361.2, 60.0, 54.5, 51.3, 41.4, 41.1, 27.1, 19.2; HRMS m/z (ESI) calcd for C42H46FNO3Si (M + H)+ 660.3309, found 660.3304.
4.3.4. 5-(tert-Butyldiphenylsilanyloxy)-3-(4-chlorophenyl)-4-dibenzylamino-pentanoic acid methyl ester, 3d
58%; Rf 0.67 (10% EtOAc/Hexane); [α]D24 = −11.8 (c 0.1, CHCl3); 1H NMR (CDCl3) δ 7.82 – 6.85 (m, 24H), 4.2 (br dd, 1H), 3.86 (br dd, 1H), 3.77 (d, 2H, J = 13.5 Hz), 3.69 (m, 1H), 3.44 (s, 3H), 3.36 (d, 2H, J = 13.5 Hz), 2.90 (m, 1H), 2.65 (br dd, 1H, J = 3.8, 15.1 Hz), 2.31 (dd, 1H, J = 10.7, 15.1 Hz); 13C NMR (CDCl3) δ 172.3, 140.8, 139.6, 135.9, 135.8, 133.2, 133.0, 131.9, 130.4, 130.1, 130.0, 129.0, 128.0, 127.9, 127.9, 126.8, 61.2, 59.9, 54.6, 51.4, 41.5, 38.9, 27.1, 19.1; HRMS m/z (ESI) calcd for C42H46ClNO3Si (M+H)+ 676.3014, found 676.2995.
4.3.5. 5-(tert-Butyldiphenylsilanyloxy)-4-dibenzylamino-3-(3,4-dimethoxyphenyl)-pentanoic acid methyl ester, 3e
49%; Rf 0.52 (10% EtOAc/Hexane); [α]D24 = −13.6 (c 0.03, CHCl3); 1H NMR (CDCl3) δ 7.84 – 6.5 (m, 23H), 4.08 (br dd, 1H), 3.90 (m, 1H), 3.88 (br s, 3H), 3.74 (br d, 2H, J = 14.1 Hz), 3.60 (br s, 3H), 3.57 (br dd, 1H), 3.46 (br dd, 1H), 3.36 (br d, 2H, J = 14.1 Hz), 2.90 (br m, 1H), 2.64 (br dd, 1H, J = 4.7, 15.4 Hz), 2.42 (br dd, 1H, J = 10.2, 15.4 Hz), 1.14 (br s, 9H); 13C NMR (CDCl3) δ 172.7, 148.4, 147.5, 140.0, 135.9, 135.8, 134.7, 133.3, 133.1, 130.0, 129.9, 128.8, 128.0, 127.8, 126.7, 121.0, 111.7, 110.6, 61.7, 60.4, 55.9, 55.6, 54.9, 51.3, 41.6, 38.5, 27.2, 19.2; HRMS m/z (ESI) calcd for C44H51NO5Si (M+H)+ 702.3615, found 702.3609.
4.3.6. 3-Benzo[1,3]dioxol-5-yl-5-(tert-butyldiphenylsilanyloxy)-4-dibenzylamino-pentanoic acid methyl ester, 3f
52%; Rf 0.56 (10% EtOAc/Hexane); [α]D24 = −9.1 (c 0.01, CHCl3); 1H NMR (CDCl3) δ 7.82 – 6.21 (m, 23H), 5.90 (s, 2H), 4.09 (dd, 1H, J = 4.1, 11.2 Hz), 3.82 (dd, 1H, J = 3.8, 11.2 Hz), 3.73 (d, 2H, J = 13.7 Hz), 3.95 (m, 1H), 3.43 (s, 3H), 3.34 (d, 2H, J = 13.7 Hz), 2.84 (m, 1H), 2.57 (dd 1H, J = 4.1, 11.2), 2.27 (dd, 1H, J = 10.2, 15.4 Hz), 1.14 (s, 9H); 13C NMR (CDCl3) δ 172.5, 147.2, 145.9, 139.8, 135.9, 135.8, 135.7, 133.3, 133.1, 130.0, 129.0, 127.9, 127.7, 126.7, 122.3, 109.0, 107.6, 100.7, 61.4, 60.2, 54.6, 51.4, 41.8, 39.0, 29.7, 27.0, 19.3; HRMS m/z (ESI) calcd for C43H48NO5Si (M+H)+ 286.3302, found 286.3293.
4.3.7. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-phenylethyl)-2,2-dimethyloxazolidine-3-carboxylate, 4a
95%; Rf 0.47 (20% EtOAc/Hexane); [α]D21 = +35.7 (c 0.02, CHCl3); 1H NMR (CDCl3): δ 7.45-7.1 (m, 5H), 4.21 – 3.65 (br m, 3H), 3.6 – 3.46 (br m, 4H), 2.9 – 2.68 (br d, 2H),1.55 (br s, 6H), 1.45 (br s, 9H); 13C NMR (CDCl3) δ 173.0, 152.8, 140.4, 128.8, 128.5, 128.2, 127.7, 126.1, 94.5, 80.5, 63.8, 61.6, 51.7, 42.8, 32.1, 28.5, 26.5; HRMS m/z (ESI) calcd for C20H29NO5 (M+Na)+ 386.1943, found 386.1935.
4.3.8. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-(4-methoxyphenyl)ethyl)-2,2-dimethyloxazolidine-3-carboxylate, 4b
87%; Rf 0.43 (20% EtOAc/Hexane); [α]D21 = +40.0 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 7.2 (br d, 2H), 6.8 (br d, 2H), 4.14 – 3.65 (br m, 6H), 3.58 – 3.47 (br m, 4H), 2.86 – 2.68 (br d, 2H), 1.55 (br s, 6H), 1.48 (br s, 9H); 13C NMR (CDCl3) δ 165.0, 150.2, 144.4, 124.1, 120.9, 120.5, 106.4, 105.7, 86.5, 72.3, 55.7, 53.7, 47.1, 44.6, 33.9, 24.2, 20.4, 18.3; HRMS m/z (ESI) calcd for C21H31NO6 (M+Na)+ 416.2049, found 416.2053.
4.3.9. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-(4-fluorophenyl)ethyl)-2,2-dimethyloxazolidine-3-carboxylate, 4c
92%; Rf 0.5 (20% EtOAc/Hexane); [α]D21 = +33.6 (c 0.03, CHCl3); 1H NMR (CDCl3) δ 7.29 – 6.93 (br m, 5H), 4.14 – 3.63 (br m, 3H), 3.6 – 3.48 (br s, m, 4H), 2.9–2.68 (br d, 2H), 1.55 (br s, 6H), 1.46 (s, 9H); 13C NMR (CDCl3) δ 172.8, 163.4, 160.1, 135.9, 128.4, 115.5, 94.3, 80.4, 63.8, 61.6, 51.7, 42.4, 32.4, 28.4, 26.1; HRMS m/z (ESI) calcd for C20H28FNO5 (M+Na)+ 404.1849, found 404.1834.
4.3.10. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-(4-chlorophenyl)ethyl)-2,2-dimethyl-oxazolidine-3-carboxylate, 4d
83%; Rf 0.54 (20% EtOAc/Hexane); [α]D21 = +26.0 (c 0.03, CHCl3); 1H NMR (CDCl3) δ 7.33 – 7.12 (br dd, 4H), 4.14 – 3.68 (br m, 3H), 3.63 – 3.48 (br m, 4H), 2.9 – 2.7 (br d, 2H), 1.54 (br s, 6H), 1.46 (br s, 9H); 13C NMR (CDCl3) δ 172.8, 152.4, 138.8, 132.8, 129.7, 128.8, 94.7, 80.6, 63.9, 61.5, 51.8, 42.6, 33.0, 28.4, 26.6; HRMS m/z (ESI) calcd for C20H28ClNO5 (M+Na)+ 420.1554, found 420.1537.
4.3.11. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-(3,4-dimethoxyphenyl)ethyl)-2,2-dimethyl-oxazolidine-3-carboxylate, 4e
70%; Rf 0.33 (20% EtOAc/Hexane); [α]D21 = +38.2 (c 0.01, CHCl3); 1H NMR (CDCl3) δ 6.90 – 6.71 (br m, 3H), 4.14 – 3.66 (br m, 9H), 3.65 – 3.48 (br m, 4H), 2.9 – 2.71 (br s, 2H), 1.56 (br s, 6H), 1.48 (br s, 9H); 13C NMR (CDCl3) δ 173.2, 152.4, 149.0, 147.9, 132.8, 118.9, 111.3, 94.6, 80.5, 63.5, 61.8, 55.8, 51.7, 42.3, 32.8, 28.5, 26.6; HRMS m/z (ESI) calcd for C22H33NO7 (M+Na)+ 446.2155, found 446.2157.
4.3.12. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-(benzo[d][1,3]dioxol-6-yl)ethyl)-2,2-dimethyl-oxazolidine-3-carboxylate, 4f
92%; Rf 0.42 (20% EtOAc/Hexane); [α]D21 = +29.4 (c 0.02, CHCl3); 1H NMR (CDCl3) δ 6.84 – 6.6 (br m, 3H), 5.95 (br s, 2H), 4.14 – 3.66 (br m, 3H), 3.65 – 3.52 (br m, 4H), 2.74 (br d, 2H), 1.54 (br s, 6H), 1.48 (br s, 9H); 13C NMR (CDCl3) δ 173.0, 152.5, 147.8, 146.4, 134.2, 120.8, 108.4, 101.0, 92.6, 80.5, 63.5, 61.9, 51.7, 42.6, 32.5, 28.5, 26.4; HRMS m/z (ESI) calcd for C21H29NO7 (M+Na)+ 430.1842, found 430.1831.
4.3.13. (R)-tert-Butyl-4-((R)-2-(methoxycarbonyl)-1-(4-methoxybenzo[d][1,3]dioxol-6-yl)ethyl)-2,2-dimethyloxazolidine-3-carboxylate, 4g
86%; Rf 0.38 (20% EtOAc/Hexane); [α]D21 = +73.3 (c 0.01, CHCl3); 1H NMR (CDCl3) δ 6.41 (br d, 2H), 5.93 (br s, 3H), 4.14 – 3.67 (br m, 6H), 3.58 (br s, 3H), 3.54 (br m, 1H), 2.74 (br d, 2H), 1.50 (br s, 15H); 13C NMR (CDCl3) δ 173.3, 152.9, 152.0, 149.0, 143.7, 134.8, 128.9, 107.6, 101.4, 94.2, 91.3, 80.0, 63.8, 56.5, 42.8, 33.4, 29.9, 28.5, 26.7; HRMS m/z (ESI) calcd for C22H31NO8 (M+Na)+ 460.1947, found 460.2023.
4.4. General procedure for the removal of Boc and isopropylidene protection
A solution of HCl in MeOH was prepared by the careful addition of acetyl chloride 30 μl in 3 ml methanol at 0 °C. Compounds 4a-g (ca. 50 mg) were refluxed in this solution (3 ml) for 2 h at rt. The solvent was evaporated under reduced pressure and the residue was triturated with ethyl acetate and hexane. White to off white solids 5a-g were obtained in 85 – 90 % yield.
4.4.1. Methyl (3R,4R)-4-amino-5-hydroxy-3-phenylpentanoate, 5a
88%; 1H NMR (D2O) δ 7.49 – 7.31 (m, 5H), 4.0 (dd, 1H, J = 2.2, 9.9 Hz), 3.85 (dd, 1H, J = 5.5, 12.4 Hz), 3.67 (m, 1H), 3.58 – 3.4 (m, 4H), 3.01 (dd, 1H, J = 3.8, 15.1 Hz), 2.87 (dd, 1H, J = 10.7, 14.6 Hz); 13C NMR (D2O) δ 174.1, 137.4, 126.5 128.6, 128.3 59.2, 56.5, 52.3, 41.2, 37.0; HRMS m/z (ESI) calcd for C12H17NO3 (M+H)+ 224.1287, found 224.1242.
4.4.2. Methyl (3R,4R)-4-amino-5-hydroxy-3-(4-methoxyphenyl)pentanoate, 5b
90%; 1H NMR (D2O) δ 7.23 (d, 2 H, J = 7.2 Hz), 6.93 (d, 2H, J = 7.2 Hz), 3.89 (dd, 1H, J = 3.6, 9.4 Hz), 3.84 – 3.71 (m, 4H), 3.54 (m, 1H), 3.41 (s, 3H), 3.30 (m 1H), 2.88 (dd, 1H, J = 3.8, 14.1 Hz), 2.73 (br dd, 1H); 13C NMR (D2O) δ 174.2, 158.9, 129.7, 129.6, 114.9, 59.3, 56.8, 55.5, 52.4, 40.6, 37.3; HRMS m/z (ESI) calcd for C13H20NO4H (M+H)+ 254.1392, found 254.1339.
4.4.3. Methyl (3R,4R)-4-amino-3-(4-fluorophenyl)-5-hydroxypentanoate, 5c
88%; 1H NMR (D2O) δ 7.31 (m, 1H), 7.11 (m, 1H), 3.91 (dd, 1H, J = 3.0, 12.6 Hz), 3.78 (dd, 1H, J = 5.5, 12.6 Hz), 3.59 (m, 1H), 3.44 (s, 3H), 3.39 (m, 1H), 2.93 (dd, 1H, J = 4.4, 15.4 Hz), 2.76 (dd, 1H, J = 10.7, 15.4 Hz); 13C NMR (D2O) δ 174.4, 133.3, 130.3, 130.2, 116.4, 116.1, 59.2, 56.5, 52.4, 40.7, 37.2; HRMS m/z (ESI) calcd for C12H16FNO3 (M + H)+ 242.1192, found 242.1202.
4.4.4. Methyl (3R,4R)-4-amino-3-(4-chlorophenyl)-5-hydroxypentanoate, 5d
85%; 1H NMR (D2O): δ 7.36 (d, 2H, J = 8.5 Hz), 7.24 (d, 2H, J = 8.5 Hz), 3.89 (dd, 1H, J = 3.0, 12.6 Hz), 3.74 (dd, 1H, J = 5.5, 12.6 Hz), 3.72 (m, 1H), 3.41 (s, 3H), 3.34 (m, 1H), 2.89 (dd, 1H, J = 3.8, 15.4 Hz), 2.74 (br dd, 1H); 13C NMR (D2O) δ 174.0, 136.1, 133.8, 130.0, 129.5, 59.2, 56.4, 52.4, 40.9, 37.0; HRMS m/z (ESI) calcd for C12H17ClNO3 (M+H)+ 258.0897, found 258.0863.
4.4.5. Methyl (3R,4R)-4-amino-5-hydroxy-3-(3,4-dimethoxyphenyl)pentanoate, 5e
85%; 1H NMR (D2O): δ 6.92 (br d, 3H), 4.16 – 3.66 (br m, 8H), 3.63 – 3.25 (br m, 5H), 2.90 (br dd, 1H), 2.75 (br dd, 1H); 13C NMR (D2O) δ 174.2, 148.7, 148.1, 130.3, 121.4, 112.4, 111.6, 59.3, 55.8, 52.4, 48.9, 41.2, 37.2; HRMS m/z (ESI) calcd for C14H21NO5 (M+H)+ 284.1498, found 284.1506.
4.4.6. Methyl (3R,4R)-4-amino-3-(benzo[d][1,3]dioxol-6-yl)-5-hydroxypentanoate, 5f
90%; 1H NMR (D2O) δ 6.83 – 6.70 (m, 3H), 5.87 (s, 2H), 3.88 (dd, 1H, J = 2.3, 12.1 Hz), 3.71 (dd, 1H, J = 5.2, 12.6 Hz), 3.46 (s, 3H), 3.24 (m, 1H), 2.84 (br dd, 1H, J = 3.8, 14.8 Hz), 2.68 (br dd, 1H); 13C NMR (D2O) δ 174.0, 148.0, 147.2, 130.9, 122.0, 109.0, 108.1, 101.0, 59.1, 56.5, 52.3, 41.1, 37.2; HRMS m/z (ESI) calcd for C13H20NO4 (M+H)+ 268.1185, found 268.1105.
4.4.7. Methyl (3R,4R)-4-amino-5-hydroxy-3-(4-methoxybenzo[d][1,3]dioxol-6-yl)pentanoate, 5g
76%; 1H NMR (D2O): δ 6.57 (d, 2H, J = 8.8 Hz), 5.93 (br s, 2H), 3.95 – 3.73 (m, 5H), 2.89 (br dd, 1H), 2.76 (br dd, 1H); 13C NMR (D2O) δ 174.1, 149.2, 143.5, 134.9, 132.1, 108.2, 101.8, 59.2, 56.7, 56.6, 52.4, 41.5, 37.3; HRMS m/z (ESI) calcd for C14H19NO6 (M+H)+ 298.1291, found 298.1281.
4.5. General procedure for lactam formation
Compounds 5a-g (ca. 30 mg) were stirred in a freshly prepared solution of sodium methoxide in methanol (2 ml; pH ~ 9) for 1h at rt. The solvent was evaporated under reduced pressure. The residue was dissolved in CH2Cl2 (15 ml) and washed with water (3 × 3 ml), organic phase was dried with brine (3 ml) and over anhydrous MgSO4. After evaporation of CH2Cl2 under reduced pressure, white to pale yellow solids 6a-g were obtained in 70 – 85 % yield.
4.5.1. (4R,5R)-5-(Hydroxymethyl)-4-phenylpyrrolidin-2-one, 6a
80%; Rf 0.48 (5% MeOH/EtOAc); [α]D23 = −63.4 (c 0.01, CH3OH); 1H NMR (D2O) δ 7.31 (m, 5H), 4.04 (br m, 1H), 3.90 (dd, 1H, J = 9.4, 17.0 Hz), 3.27 (m, 1H), 3.16 (m, 1H), 2.82 (dd, 1H, J = 8.8, 17.0 Hz), 2.64 (dd, 1H, J = 8.9, 16.8 Hz); 13C NMR (D2O) δ 181.0, 138.1, 128.7, 128.0, 127.3, 61.7, 59.6, 41.5, 35.3; HRMS m/z (ESI) calcd for C11H13NO2 (M+H)+ 192.1025, found 192.1018.
4.5.2. (4R,5R)-5-(Hydroxymethyl)-4-(4-methoxyphenyl)pyrrolidin-2-one, 6b
85%; Rf 0.4 (10% MeOH/EtOAc); [α]D23 = −95.7 (c 0.02, CH3OH); 1H NMR (D2O) δ 7.15 (d, 2H, J = 8.5 Hz), 6.85 (d, 2H, J = 8.5 Hz), 3.90 (m, 1H), 3.77 – 3.65 (m, 4H), 3.16 (m, 1H), 3.06 (dd, 1H, J = 6.9, 10.4 Hz), 2.68 (dd, 1H, J = 9.3, 17.0 Hz), 2.50 (dd, 1H, J = 8.7, 16.8 Hz); 13C NMR (D2O) δ 180.9, 157.9, 130.5, 129.2, 114.0, 61.8, 59.6, 55.4, 41.0, 35.5. HRMS m/z (ESI) calcd for C12H15NO3 (M+H)+ 222.1130, found 222.1122.
4.5.3. (4R,5R)-4-(4-Fluorophenyl)-5-(hydroxymethyl)pyrrolidin-2-one, 6c
82%; Rf 0.49 (5% MeOH/EtOAc); [α]D23 = −105.7 (c 0.03, CH3OH); 1H NMR (D2O) δ 7.25 (br t, 2H), 7.05 (br t, 2H), 3.97 (m, 1H), 3.7 (br dd, 1H, J = 8.5, 17.3 Hz), 3.24 (br dd, 1H, J = 3.5, 13.6 Hz), 3.12 (br dd, 1H, J = 6.6, 11.5 Hz), 2.78 (br dd, 1H, J = 9.1, 16.7 Hz), 2.6 (br dd, 1H, J = 8.8, 16.8 Hz); 13C NMR (D2O) δ 180.9, 157.9, 129.2, 114.0, 61.8, 59.6, 55.4, 41.0, 35.5; HRMS m/z (ESI) calcd for C11H12FNO2 (M+H)+ 210.0930, found 210.0927.
4.5.4. (4R,5R)-4-(4-Chlorophenyl)-5-(hydroxymethyl)pyrrolidin-2-one, 6d
75%; Rf 0.44 (5% MeOH/EtOAc); [α]D23 = −113.7 (c 0.01, CH3OH); 1H NMR (D2O) δ 7.35 (br d, 2H, J = 7.9 Hz), 7.24 (br d, 2H, J = 8.2), 3.95 (m, 1H), 3.84 (dd, 1H, J = 7.98, 17.1 Hz), 3.2 (br dd, 1H, J = 3.0, 12.1 Hz), 3.08 (br dd, 1H, J = 6.0, 12.5 Hz), 2.74 (br dd, 1H, J = 8.8, 16.5 Hz), 2.56 (br dd, 1H, J = 8.8, 17.1 Hz); 13C NMR (D2O) δ 180.8, 138.8, 132.4, 129.6, 128.6, 61.5, 59.4, 41.2, 35.3; HRMS m/z (ESI) calcd for C11H12ClNO2 (M+H)+ 226.05, found 226.0643.
4.5.5. (4R,5R)-5-(Hydroxymethyl)-4-(3,4-dimethoxyphenyl)pyrrolidin-2-one, 6e
70%; Rf 0.47 (20% MeOH/EtOAc); [α]D21 = −110.0 (c 0.01, CH3OH); 1H NMR (D2O) δ 6.94 (br m, 3H), 3.89 (br s, 8H), 3.31 (br dd, 1H), 3.20 (br dd, 1H), 2.83 (br dd, 1H), 2.64 (br dd, 1H); 13C NMR (D2O) δ 181.0, 148.1, 147.3, 131.3, 120.5, 111.9, 61.8, 59.7, 55.9, 49.0, 41.5, 35.5; HRMS m/z (ESI) calcd for C13H17NO4 (M+H)+ 252.1236, found 252.1226.
4.5.6. (4R,5R)-4-(Benzo[d][1,3]dioxol-6-yl)-5-(hydroxymethyl)pyrrolidin-2-one, 6f
76%; Rf 0.35 (5% MeOH/EtOAc); [α]D23 = −104.2 (c 0.003, CH3OH); 1H NMR (D2O) δ 6.77 – 6.67 (m, 3H), 5.85 (s, 2H), 3.91 (m, 1H), 3.75 (dd, 1H, J = 8.5, 17.0 Hz), 3.19 (m, 2H), 2.70 (dd, 1H, J = 9.1, 16.7 Hz), 2.54 (dd, 1H, J = 8.8, 17.0 Hz); 13C NMR (D2O) δ 178.5, 148.0, 146.8, 131.7, 120.9, 108.3, 108.1, 101.2, 63.1, 59.7, 42.2, 36.2; HRMS m/z (ESI) calcd for C12H13NO4 (M+H)+ 236.0923, found 236.0921.
4.5.7. (4R,5R)-5-(Hydroxymethyl)-4-(4-methoxybenzo[d][1,3]dioxol-6-yl)pyrrolidin-2-one, 6g
70%; Rf 0.41(10% MeOH/EtOAc); [α]D24 = −123.4 (c 0.01, CH3OH); 1H NMR (D2O) δ 6.55 (d, 2H, J = 14.0 Hz), 5.93 (s, 2H), 3.96 (m, 1H), 3.85 (m, 4H), 3.30 (br m, 1H), 3.22 (br dd, 1H), 2.79 (dd, 1H, J = 9.1, 16.5 Hz), 2.63 (dd, 1H, J = 8.8, 1.8 Hz); 13C NMR (D2O) δ 180.8, 153.8, 146.1, 142.9, 107.7, 102.3, 101.7, 61.7, 59.6, 56.6, 41.9, 35.5; HRMS m/z (ESI) calcd for C13H15NO5 (M+H)+ 266.1028, found 266.1032.
4.6. Stereochemistry assignment by acetylation of 5b and 5f
Hydroxymethyl pyrrolidine-2-ones 5b and 5f (0.0185 g, 0.08 mmol) were dissolved in pyridine (1.5 ml) and stirred with Ac2O (0.224 ml, 2.37 mmol) in the presence of DMAP (0.5 mg) for 16 h at rt. The solution was concentrated under reduced pressure and the residual oil was presorbed on silica gel. The two acetylated products were purified by column chromatography (15% EtOAc/Hexane and 70 % EtOAc/Hexane).
4.6.1. (4R,5R)-1-Acetyl-4-(benzo[d][1,3]dioxol-6-yl)-5-(hydroxymethyl)pyrrolidin-2-one, 7f
45%; Rf 0.41 (50 % EtOAc/Hexane); 1H NMR δ 6.64 (m, 3H), 5.91 (s, 2H), 4.14 – 4.02 (m, 3H), 3.89 (dd, 1H, J = 3.3, 8.3 Hz), 3.79 (dd, 1H J = 8.3, 8.5 Hz), 3.54 (dd, 1H, J = 8.3, 11.3 Hz), 2.01 (s, 3H); 13C NMR (CDCl3) δ 176.9, 170.5, 148.2, 147.1, 131.0, 120.8, 108.5, 107.9, 101.3, 65.0, 56.4, 41.9, 35.6, 20.8; HRMS m/z (ESI) calcd for C14H15NO5 (M+H)+ 278.1028, found 278.1024.
4.6.2. ((2R,3R)-1-Acetyl-3-(4-methoxyphenyl)-5-oxopyrrolidin-2-yl)methyl acetate, 8b
55%; Rf 0.56, (60 % EtOAc/Hexane); 1H NMR δ 7.13 (d, 2H, J = 8.5 Hz), 6.88 (d, 2H, J = 8.5 Hz), 4.76 (m, 1H), 4.41 (dd, 1H, J = 2.8, 12.1 Hz), 3.8 (m, 4H), 3.62 (dd, 1H, J = 2.8, 12.1 Hz), 3.24 (dd, 1H, J = 15.2, 16.9 Hz), 2.72 (dd, 1H, J = 8.3, 16.9 Hz), 2.40 (s, 3H), 1.96 (s, 3H); 13C NMR (CDCl3) δ 174.5, 171.0, 170.0, 159.9, 128.5, 127.2, 114.4, 61.5, 58.7, 55.3, 39.5, 36.8, 25.4, 21.0; HRMS m/z (ESI) calcd for C16H19NO5 (M+Na)+ 328.1161, found 328.1149.
4.6.3. ((2R,3R)-1-Acetyl-3-(benzo[d][1,3]dioxol-6-yl)-5-oxopyrrolidin-2-yl)methyl acetate, 8f
25%; Rf 0.58, (50 % EtOAc/Hexane); 1H NMR δ 6.79 (d, 1H, J = 8.0 Hz), 6.69 – 6.66 (m, 2H), 5.97 (s, 2H), 4.73 (ddd, 1H, J = 2.8, 2.8, 8.2 Hz), 4.44 (dd, 1H, J = 2.8, 12.1 Hz), 3.75 (ddd, 1H, J = 8.2, 8.3, 15.2 Hz), 3.67 (dd, 1H, J = 2.8, 12.1 Hz), 3.20 (dd, 1H, J = 15.2, 16.9 Hz), 2.69 (dd, 1H, J = 8.3, 16.9 Hz); 13C NMR (CDCl3) δ 174.4, 169.9, 148.3, 147.2, 129.1,120.7, 108.6, 107.7, 101.3, 61.5, 58.7, 39.9, 36.9, 20.9; HRMS m/z (ESI) calcd for C21H29NO7 (M+Na)+ 342.0954, found 342.0946.
4.6.4. (4R,5R)-5-(Dibenzylamino)-tetrahydro-4-phenylpyran-2-one, 9a
Tetrabutylammonium fluoride (1M solution in THF, 0.498 ml, 0.5 mmol) was added dropwise to a solution of phenyl cuprate adduct 3a (62.5 mg, 0.1 mmol) in THF (3 ml). The reaction mixture was stirred for 16 h at rt. After completion of reaction (monitored by TLC) EtOAc (10 ml) was added and solution was washed with water (2 ml × 2) and with brine (2 ml × 2). This solution was dried over anhydrous MgSO4 and the solvent was concentrated under reduced pressure. The residue was presorbed on silica gel and purified by column chromatography (4% EtOAc//Hexane) to afford 9a (19.5 mg, 55% yield). Rf 0.38 (20% EtOAc/Hexane); 1H NMR (CDCl3) δ 7.44 - 6.99 (m, 15H), 4.47 (m, 2H), 3.75 (d, 2H, J = 13.8 Hz), 3.52 (d, 2H, J = 13.8 Hz), 3.35 (m, 2H), 2.82 (br dd, 1H), 2.60 (br dd, 1H); 13C NMR (CDCl3) δ 171.6, 142.1, 138.7, 128.8, 128.4, 128.3, 127.7, 127.3, 127.2; HRMS m/z (ESI) calcd for C25H25NO2 (M+H)+ 372.1964, found 372.1946.
4.6.5. (4R,5R)-5-Amino-tetrahydro-4-phenylpyran-2-one, 10a
To a solution of lactone 9a (15.2 mg, 0.041 mmol) in water/dioxane (2:1, 3 ml) were added 2 drops of AcOH and 10% Pd/C (10 mg). The solution was stirred under an H2 atmosphere (1 atm pressure) at rt for 24 h. The completion of the reaction was monitored by TLC. The reaction mixture was filtered through a celite pad and the filtrate was concentrated to give 4.7 mg of debenzylated lactone 10a (62% yield). 1H NMR (CDCl3) δ 7.41 (m, 5H), 3.96 (dd, 1H, J = 3.0, 12.7 Hz), 3.81 (dd, 1H, J = 5.8, 12.4 Hz), 3.64 (m, 1H), 3.37 (m, 1H), 2.94 (dd, 1H, J = 4.7, 15.4), 2.75 (dd, 1H, J = 10.5, 15.1); 13C NMR (CDCl3) δ 176.0, 137.8, 129.6, 128.6, 128.5, 59.4, 56.8, 41.5, 37.5; HRMS m/z (ESI) calcd for C11H13NO2·H2O (M+H)+ 210.1130, found 210.1133.
4.6.6. (4R,5R)-5-Hydroxymethyl-4-phenylpyrrolidin-2-one, 6a
Amino lactone 10a was stirred in a solution of freshly prepared NaOMe in MeOH (2 ml, pH ~ 9) at rt for 1 h. The reaction mixture was stirred with Amberlyst 15 (dry) ion-exchange resin (200 mg) at rt for 10 min. The solution was filtered and the filtrate was concentrated under reduced pressure. This afforded 3.2 mg of 6a (80 % yield), whose NMR spectra were identical to those recorded with the material obtained from 5a.
Acknowledgments
We thank Professor Patrick S. Mariano for his help with the preparation of the manuscript and the University of New Mexico Mass Spectrometry Facility for MS analyses. This work is supported by the US National Institutes of Health (RR-16480 and CA-99957) under the BRIN/INBRE and AREA programs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Martin SF. The amaryllidaceae alkaloids. In: Brossi AR, editor. The Alkaloids. Vol. 30. Academic Press; New York: 1987. pp. 251–376. [Google Scholar]
- 2.For recent reviews of synthetic work in this area, see: Polt R. Amaryllidaceae alkaloids with antitumor activity. In: Hudlicky T, editor. Organic Synthesis: Theory and Applications. Vol. 3. JAI Press; 1997. p. 109.Rinner U, Hudlicky T. Synlett. 2005:365–387.Chapleur Y, Chrétien F, Ibn Ahmed S, Khaldi M. Curr Org Syn. 2006;3:341–378.
- 3.Hoshino O. The amaryllidaceae alkaloids. In: Cordell GA, editor. The Alkaloids. Vol. 51. Academic Press; London: 1998. pp. 323–376. [Google Scholar]
- 4.(a) Pettit GR, Gaddamidi V, Herald DL, Singh SB, Cragg GM, Schmidt JM, Boettner FE, Williams M, Sagawa Y. J Nat Prod. 1986;49:995–1002. doi: 10.1021/np50048a005. [DOI] [PubMed] [Google Scholar]; (b) Pettit GR, Pettit GR, III, Backhaus RA, Boyd MR, Meerow AW. J Nat Prod. 1993;56:1682–1687. doi: 10.1021/np50100a004. [DOI] [PubMed] [Google Scholar]; (c) Liu J, Hu WX, He LF, Ye M, Li Y. FEBS Lett. 2004;578:245–250. doi: 10.1016/j.febslet.2004.10.095. [DOI] [PubMed] [Google Scholar]
- 5.Gabrielsen B, Monath TP, Huggins JW, Kefauver DF, Pettit GR, Groszek G, Hollingshead M, Kirsi JJ, Shannon WM, Shubert EM, Dare J, Ugarkar B, Ussery MA, Phelan MJ. J Nat Prod. 1992;55:1569–1581. doi: 10.1021/np50089a003. [DOI] [PubMed] [Google Scholar]
- 6.Quarzane-Amara M, Franetich J-F, Mazier D, Pettit GR, Meijer L, Doerig C, Desportes-Livage I. Antimicrob Agents Chemother. 2001;45:3409–3415. doi: 10.1128/AAC.45.12.3409-3415.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Yii S, Mikami S, Kitahara M, Yamazaki M. Immunopharmacology. 1998;40:151–162. doi: 10.1016/s0162-3109(98)00040-x. [DOI] [PubMed] [Google Scholar]; (b) Citoglu G, Tanker M, Gumusel B. Phytother Res. 1998;12:205–206. [Google Scholar]
- 8.(a) McLachlan A, Kekre N, McNulty J, Pandey S. Apoptosis. 2005;10:619–630. doi: 10.1007/s10495-005-1896-x. [DOI] [PubMed] [Google Scholar]; (b) Pandey S, Kekre N, Naderi J, McNulty J. Artificial Cells, Blood Substitutes, and Biotechnology. 2005;33:279–295. doi: 10.1081/bio-200066621. [DOI] [PubMed] [Google Scholar]; (c) Kekre N, Griffin C, McNulty J, Pandey S. Cancer Chemother Pharmacol. 2005;56:29–38. doi: 10.1007/s00280-004-0941-8. [DOI] [PubMed] [Google Scholar]
- 9.For illustration of various strategies to install the C10b stereocenter, see the published total syntheses of pancratistatin: Danishefsky S, Lee JY. J Am Chem Soc. 1989;111:4829–4837.Tian X, Königsberger K, Hudlicky T. J Am Chem Soc. 1995;117:3643–3644.Hudlicky T, Tian X, Königsberger K, Maurya R, Rouden J, Fan B. J Am Chem Soc. 1996;118:10752–10765.Trost BM, Pulley SR. J Am Chem Soc. 1995;117:10143–10144.Magnus P, Sebhat IK. J Am Chem Soc. 1998;120:5341–5342.Rigby JH, Maharoof USM, Mateo ME. J Am Chem Soc. 2000;122:6624–6628.Doyle TJ, Hendrix M, VanDerveer D, Javanmard S, Haseltine J. Tetrahedron. 1997;53:11153–11170.Pettit GR, Melody N, Herald DL. J Org Chem. 2001;66:2583–2587. doi: 10.1021/jo000710n.Kim S, Ko HJ, Kim E, Kim D. Org Lett. 2002;4:1343–1345. doi: 10.1021/ol0256419.Ko HJ, Kim E, Park JE, Kim D, Kim S. J Org Chem. 2004;69:112–121. doi: 10.1021/jo035371n.Li M, Wu A, Zhou P. Tetrahedron Lett. 2006;47:3707–3710.
- 10.(a) Manpadi M, Kornienko A. Tetrahedron Lett. 2005;46:4433–4437. [Google Scholar]; (b) Kireev AS, Manpadi M, Kornienko A. J Org Chem. 2006;71:2630–2640. doi: 10.1021/jo052383v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Reetz MT, Rohrig D. Angew Chem, Int Ed. 1989;28:1706–1709. [Google Scholar]; (b) Jako I, Uiber P, Mann A, Taddei M, Wermuth CG. Tetrahedron Lett. 1990;31:1011–1014. [Google Scholar]; (c) Hanessian S, Sumi K. Synthesis. 1991:1083–1089. [Google Scholar]; (d) Hanessian S, Wang W, Gai Y. Tetrahedron Lett. 1996;37:7477–7480. [Google Scholar]; (e) Hanessian S, Demont E, van Otterlo WAL. Tetrahedron Lett. 2000;41:4999–5003. [Google Scholar]; (f) Liang X, Andersch J, Bols M. J Chem Soc, Perkin Trans. 2001;1:2136–2157. [Google Scholar]; (g) Flamant-Robin C, Wang Q, Sasaki NA. Tetrahedron Lett. 2001;42:8483–8484. [Google Scholar]; (h) Flamant-Robin C, Wang Q, Chiaroni A, Sasaki NA. Tetrahedron. 2002;58:10475–10484. [Google Scholar]; (i) Kumar S, Flamant-Robin C, Wang Q, Chiaroni A, Sasaki A. J Org Chem. 2005;70:5946–5953. doi: 10.1021/jo050736k. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hulme AN, Montgomery CH, Henderson DK. J Chem Soc, Perkin Trans. 2000;1:1837–1841. [Google Scholar]; (b) Hulme AN, Curley KS. J Chem Soc, Perkin Trans. 2002;1:1083–1091. [Google Scholar]; (c) Williams L, Zhang Z, Shao F, Carroll PJ, Joullié MM. Tetrahedron. 1996;52:11673–11694. [Google Scholar]
- 13.For examples of such one-pot syntheses of γ-alkoxy-α,β-enoates, see: Ziegler FE, Wang Y. J Org Chem. 1998;63:426–427. doi: 10.1021/jo971998s.Ziegler FE, Wang Y. J Org Chem. 1998;63:7920–7930. doi: 10.1021/jo971998s.Nadein ON, Kornienko A. Org Lett. 2004;6:831–834. doi: 10.1021/ol049942p.Kireev AS, Nadein ON, Agustin VJ, Bush NE, Evidente A, Manpadi M, Ogasawara MA, Rastogi SK, Rogelj S, Shors ST, Kornienko A. J Org Chem. 2006;71:5694–5707. doi: 10.1021/jo0607562.
- 14.For reports of the synthesis and biological evaluation of aromatic analogues of pancratistatin, see: Rinner U, Hillebrenner HL, Adams DR, Hudlicky T, Pettit GR. Bioorg Med Chem Lett. 2004;14:2911–2915. doi: 10.1016/j.bmcl.2004.03.032.Rinner U, Hudlicky T, Gordon H, Pettit GR. Angew Chem Int Ed. 2004;43:5342–5346. doi: 10.1002/anie.200460218.Hudlicky T, Rinner U, Finn KJ, Ghiviriga I. J Org Chem. 2005;70:3490–3499. doi: 10.1021/jo040292c.Moser M, Sun X, Hudlicky T. Org Lett. 2005;7:5669–5672. doi: 10.1021/ol052372o.
- 15.(a) Roush WR, Lesur BM. Tetrahedron Lett. 1983;24:2231–2234. [Google Scholar]; (b) Yamamoto Y, Chounan Y, Nishii S, Ibuka T, Kitahara H. J Am Chem Soc. 1992;114:7652–7660. [Google Scholar]
- 16.(a) Gensler WJ, Stouffer JE. J Org Chem. 1958;23:908–910. [Google Scholar]; (b) Comber MF, Sargent MV. Aus J Chem. 1985;38:1481–1489. [Google Scholar]; (c) Iinima M, Tanaka T, Matsuura S. Yakugaku Zasshi. 1983;103:997–1000. doi: 10.1248/yakushi1947.103.9_994. [DOI] [PubMed] [Google Scholar]