

Abstract
Mallory-Denk bodies (MDBs) are hepatocyte cytoplasmic inclusions found in several liver diseases and consist primarily of the cytoskeletal proteins, keratins 8 and 18 (K8/K18). Recent evidence indicates that the extent of stress-induced protein misfolding, a K8>K18 overexpression state, and transglutaminase-2 activation promote MDB formation. In addition, the genetic background and gender play an important role in mouse MDB formation, but the effect of aging on this process is unknown. Given that oxidative stress increases with aging, the authors hypothesized that aging predisposes to MDB formation. They used an established mouse MDB model—namely, feeding non-transgenic male FVB/N mice (1, 3, and 8 months old) with 3,5 diethoxycarbonyl-1,4-dihydrocollidine for 2 months. MDB formation was assessed using immunofluorescence staining and biochemically by demonstrating keratin and ubiquitin-containing crosslinks generated by transglutaminase-2. Immunofluorescence staining showed that old mice had a significant increase in MDB formation compared with young mice. MDB formation paralleled the generation of high molecular weight ubiquitinated keratin-containing complexes and induction of p62. Old mouse livers had increased oxidative stress. In addition, 20S proteasome activity and autophagy were decreased, and endoplasmic reticulum stress was increased in older livers. Therefore, aging predisposes to experimental MDB formation, possibly by decreased activity of protein degradation machinery.
Keywords: Mallory-Denk body, aging, oxidative stress, proteasome activity, autophagy, endoplasmic reticulum stress
Mallory-Denk bodies (MDBs) are characteristic hepatocellular inclusions observed in multiple liver diseases, including alcoholic and non-alcoholic steatohepatitis (ASH and NASH, respectively) (Zatloukal et al. 2007). MDBs are defined by their morphological appearance and molecular composition and consist primarily of the intermediate filament (IF) proteins, keratins 8 and 18 (K8/K18), together with ubiquitin (Ub), p62, and heat shock proteins (Hsps). Recent studies demonstrated that MDB formation requires a K8>K18 overexpression state and transamidation via transglutaminase-2 (TG2), resulting in generation of keratin crosslinks in response to liver injury (Omary et al. 2009). In addition, genetic background and gender have been shown to play an important role in MDB formation (Hanada et al. 2008, 2010). For example, male mice are significantly more susceptible to MDB formation as compared with female mice (Hanada et al. 2010), and mouse strains show a wide range of MDB formation (e.g., C3H have low propensity, C57BL have high propensity, and FVB are intermediate) (Hanada et al. 2008).
Aging is characterized by a progressive and irreversible decline of various physiological functions of an organism, resulting in a decreased resistance to multiple forms of stress, as well as an increased susceptibility to numerous diseases. Although the mechanisms of aging have been poorly understood, oxidative stress is now considered to play a role in this process (Golden et al. 2002). Recent evidence indicates that oxidative stress also relates to the pathogenesis of many liver diseases, including ASH and NASH (Tanikawa and Torimura 2006). Therefore, we hypothesized that aging may influence MDB formation in hepatocytes.
MDBs can be experimentally induced in livers of mice chronically fed griseofulvin or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC), and these established models have been instrumental in elucidating essential aspects relating to the pathogenesis of MDBs (Zatloukal et al. 2007). In this study, we investigated the relationship between aging and MDB formation in response to DDC as determined by immunofluorescence staining and detection of keratin and Ub-crosslinks using different age groups of mice (1, 3, and 8 months old). We also investigated factors that might contribute to MDB formation upon aging by detection of oxidative stress-related protein adducts and measurement of 20S proteasome activity and autophagy in these mouse livers.
Materials and Methods
Antibodies
The following antibodies (Abs) were used: rat anti-K8 monoclonal Ab (Troma І; Developmental Studies Hybridoma Bank, Iowa City, IA); rabbit anti-mouse/human K8 and K18 Ab-8592, rabbit anti-mouse/human K18 Ab-4668, mouse anti-Ub Ab, rabbit anti-XBP-1 Ab, and rabbit anti-GRP78 Ab (Santa Cruz Biotechnology; Santa Cruz, CA); rabbit anti-TG2 Ab, mouse anti-Hsp60 Ab, and mouse anti-human K8 Ab (TS1) (Labvision; Fremont, CA); rabbit anti-p62 Ab (MBL; Nagoya, Japan); rabbit anti-LC3 Ab (Novus Biologicals; Littleton, CO); and mouse anti-4- hydroxy-2-nonenal (HNE)–modified protein Ab and mouse anti-malondialdehyde (MDA) Ab (JaICA, Shizuoka, Japan).
Animal experiments
Non-transgenic FVB/N male mice (1, 3, and 8 months old) were used (CLEA Japan; Tokyo, Japan). To induce MDBs, eight mice/age group were fed a powdered chow (MF Certified Diet; Oriental Yeast, Tokyo, Japan) containing 0.1% DDC (Sigma-Aldrich; St. Louis, MO) for 2 months. Age-matched males (n=12, 4 mice/age group) were kept on a same chow without DDC and used as controls. Mice were euthanized by CO2 inhalation, and blood was collected by intracardiac puncture for subsequent measurement of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total bilirubin (TB). Livers were removed, weighed, and divided into 1- to 2-mm slices and then apportioned for fixation in 10% formaldehyde and then hematoxylin and eosin (HE) staining and histological analysis, snap-freezing in liquid nitrogen for biochemical analysis, or embedding in optimal-cutting temperature compound and then frozen for subsequent immunofluorescence staining. In the experiments testing the effect of DDC on keratin crosslinking, human K8-overexpressing transgenic mice (Ku and Omary 2006) were fed with a DDC-containing diet. All mice received humane care, and their use was approved by the Kurume University Institutional Animal Care and Use Committee.
Histological analysis
HE-stained liver sections were assessed by an experienced clinical hepatopathologist (J.A.) for the parameters shown in Table 1. Lesions of interest were semi-quantitatively scored as follows (Hanada et al. 2008): ductular reaction (0–4: 0, none; 1, rare; 2, moderate; 3, frequent; and 4, abundant) and hepatocyte ballooning (0–3: 0, none; 1, rare; 2, frequent; 3, abundant). Periductal fibrosis was scored as absent (0) or present (1), and the number of acidophil bodies per 10 power fields (20×) was counted.
Table 1.
Serology of Different Age Groups of Mice before DDC Treatment
| Age | |||
|---|---|---|---|
| 1 Month | 3 Months | 8 Months | |
| Liver/body weight ratio, % | 6 ± 0.3 | 5.3 ± 0.3 | 5.5 ± 0.9 |
| AST, IU/L | 195 ± 113 | 150 ± 50 | 194 ± 109 |
| ALT, IU/L | 91 ± 89 | 49 ± 7 | 51 ± 18 |
| TB, mg/dl | 0.03 ± 0.1 | 0.08 ± 0.1 | 0.07 ± 0.1 |
Values presented as mean ± SD. ALT, alanine aminotransferase; AST, aspartate aminotransferase; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; TB, total bilirubin.
Immunofluorescence staining
Liver tissues were sectioned (6 µm) and fixed with acetone for 10 min (–20C). Nonspecific binding was blocked with a buffer containing 5% bovine serum albumin, followed by incubation with the primary and secondary Abs as described previously (Ku et al. 2004). Stained sections were viewed using confocal microscopy (Fluoview FV 300; Olympus, Tokyo, Japan). To quantify the extent of MDB formation, a 20× lens was used and the number of cells with K8/K18 and Ub-positive aggregates was counted (10 fields were analyzed per liver specimen).
Immunoperoxidase staining for MDA
After preparation of tissue sections with xylenes followed by hydration with graded alcohols, sections were washed with phosphate-buffered saline (PBS). Endogenous peroxidase activity was quenched by application of 0.5% hydrogen peroxide, and the slides were washed in PBS again. Blocking serum was applied to the slides, and then they were incubated for 60 min with primary Ab followed by incubation with VECTASTAIN ABC Kit (Vector Laboratories; Burlingame, CA). Diaminobenzidine (DAB) was used to detect MDA-positive hepatocytes, and slides were counterstained with hematoxylin.
Protein isolation and analysis
Total liver lysates were prepared using a homogenization buffer (0.187 M Tris-HCl [pH 6.8], 3% SDS, and 5 mM EDTA), and samples were subsequently diluted to a desired protein concentration with 4× reducing Laemmli sample buffer. Equal amounts of proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. After blotting, antigen-Ab complexes were detected by enhanced chemiluminescence (ECL Advance Western Blotting Detection Kit; GE Healthcare UK, Little Chalfont, UK). In the experiments of K8, Ub, and LC3 blotting, the relative intensities of immunoreactive bands were quantified by densitometry (Multi Gauge; Fujifilm, Tokyo, Japan) and the LC3-II/I ratio was calculated.
20S proteasome activity assay
Frozen livers were homogenized with a buffer containing 50 mM HEPES (pH 7.5), 5 mM EDTA, 150 mM NaCl, and 1% Triton X-100. After centrifugation, samples were prepared by adding 2 mM adenosine triphosphate (ATP) into the supernatant. Proteasomal activity was determined using a kit (Chemicon; Temecula, CA). The assay is based on detection of the fluorophore 7-amino-4-methylcoumarin (AMC) after cleavage from the peptide-labeled substrate LLVY-AMC by the proteasome. The free AMC fluorescence was quantified using a fluorometer.
Statistical analysis
Values are expressed as the mean ± SD. Statistical analyses in Table 2 were done to compare the values of each parameter between the groups. Data were analyzed using the Kruskal-Wallis H-test. The post hoc test was performed by the Mann-Whitney U-test with Bonferroni correction. A p-value <0.05 was considered statistically significant.
Table 2.
Serology and Liver Histology of Different Age Groups of Mice after DDC Treatment
| Age | |||
|---|---|---|---|
| 1 Month | 3 Months | 8 Months | |
| Liver/body weight ratio, % | 12.1 ± 1.3*,** | 13.8 ± 0.9* | 13.4 ± 0.7** |
| AST, IU/L | 1761 ± 338 | 1267 ± 612 | 1624 ± 493 |
| ALT, IU/L | 2206 ± 428 | 1601 ± 618 | 1896 ± 548 |
| TB, mg/dL | 1.2 ± 1.2** | 0.3 ± 0.4** | 0.6 ± 0.4 |
| MDBs | 11.6 ± 12.3** | 19.2 ± 16.2 | 35.6 ± 19.8** |
| Ductular reaction | 2.5 ± 0.8 | 3 ± 0 | 2.9 ± 0.4 |
| Ballooning | 1.3 ± 0.5 | 1 ± 0 | 1.3 ± 0.5 |
| Acidophil bodies | 1 ± 0.8 | 0.6 ± 0.8 | 1 ± 0.8 |
Values presented as mean ± SD. ALT, alanine aminotransferase; AST, aspartate aminotransferase; DDC, 3,5-diethoxycarbonyl-1,4-dihydrocollidine; MDB, Mallory-Denk body; TB, total bilirubin.
0.01. **p<0.05.
Results
DDC-induced liver hypertrophy and hepatic injury
To address the potential effect of aging on predisposition to MDB formation, three mouse groups of different ages (1, 3, and 8 months old; eight mice/age group) were fed a DDC-containing diet for 2 months to induce MDBs. The non-DDC-fed animals of all ages had livers of similar size (Table 1), and no obvious abnormal liver histology was detected (not shown). Average levels of ALT and TB were also similar under basal conditions between the groups (Table 1).
As described previously (Zatloukal et al. 2007), DDC induced hepatomegaly and pronounced liver injury (Table 1). The extent of hepatomegaly, which is represented as liver/body weight ratio, significantly increased in old mice (8 months old) compared with young mice (1 month old) (13.4 ± 0.7 vs 12.1 ± 1.3; p<0.05). Serological analysis showed that DDC-fed young mice had increased AST and ALT levels compared with 3- or 8-month-old mice, but there was no statistical significance between them (p=0.06 in ALT levels between 1 and 3 months old). TB level was significantly increased in 1-month-old mice as compared with 3-month-old mice (1.2 ± 1.2 vs 0.3 ± 0.4; p<0.05).
Analysis of MDB formation and histological evaluation
To analyze MDB formation, immunofluorescence staining for K8/K18 and Ub (the major MDB constituents) was performed (Fig. 1). Total MDB counts were estimated by counting cells with all dots/aggregates that double-stained with Ub and K8/K18. Using this method of MDB quantification, 8-month-old mice had significantly increased MDB formation compared with 1-month-old mice (35.6 ± 19.8 vs 11.6 ± 12.3; p<0.05) (Table 2). There was no statistical significance in the number of cells with MDBs between 1- and 3-month-old mice or 3- and 8-month-old mice.
Figure 1.

Assessment for Mallory-Denk body (MDB) formation using immunofluorescence staining for K8/K18 and Ub in livers of 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)–fed mice. Livers were double-labeled with Abs to K8/K18 (red) and Ub (green). MDBs are seen as yellow clumps due to colocalization of keratins and Ub (arrows). Eight-month-old mouse livers had the highest number of MDBs as compared with livers of younger mice (A–D) and had no MDBs under basal condition (E, F). Scale bars: (A–C, E) 200 µm; (D) 50 µm; (F) 20 µm.
We also performed histological analysis using HE-stained livers (Table 2). Although ductular reaction was frequently seen in DDC-fed mouse livers, there was no significant difference upon aging. Previously, we reported that MDB formation correlates with hepatocyte ballooning (Hanada et al. 2008). However, ballooned hepatocytes did not correlate with the extent of MDB formation. Acidophil body formation also showed no difference between the groups, and periductal fibrosis was not seen in all livers.
Biochemically observed K8-containing high molecular weight complexes correlate with the extent of MDB formation
Transglutaminases catalyze protein crosslinking by forming a covalent linkage between the ϵ-amino group of lysine and γ-amino group of glutamine (Lismaa et al. 2009). The excess K8 provides an excellent TG2 substrate, and keratin crosslinking is required for MDB formation and can be assessed biochemically (Strnad et al. 2007). An example of the crosslinked K8-containing species after DDC feeding is shown in Figure 2A. According to the data obtained by densitometry, although there was no statistical significance, there was a trend that old mouse livers had an increased level of K8-positive high molecular weight complexes (arrow pointing to band at the top of the stacking gel) compared with that from young mouse livers (1.2 ± 0.3 in 1-month-old mice vs 1.8 ± 0.9 in 8-month-old mice; Fig. 2B), and this observation is in line with the results of immunofluorescence staining. The expression of p62 protein, which is a Ub-binding protein known to promote MDB formation (Stumptner et al. 2002, 2007), was also increased in DDC-treated old mouse livers. Protein expression of K18 and TG2 was almost similar in these livers.
Figure 2.

Biochemical evidence for age-related formation of Mallory-Denk bodies (MDBs) in the livers of 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)–fed mouse livers. (A) Human K8 transgenic mice were fed a DDC-containing diet or a regular diet, and liver homogenates were isolated and blotted with an antibody to human K8. Crosslinked human K8-containing complexes are highlighted by the arrows. (B) Liver homogenates were isolated from 1-, 3-, and 8-month-old mice fed a DDC-containing diet. Four independent livers were analyzed from each age group. The homogenates were blotted with Abs to the indicated antigens. The immunoblots show the presence of high molecular weight K8- and Ub-containing complexes (arrows) at the top of the stacking gel (S). Relative intensity of these bands was quantified by densitometry. (C) Duplicate samples to those used in panel B were analyzed for the expression of K18, p62, TG2, HNE, and Hsp60 (used as a loading control). Arrowheads highlight HNE-modified proteins that manifest increased intensity in livers of the older mice.
Oxidative stress and protein degradation machinery–related alterations during MDB formation
Several lines of evidence demonstrate that oxidative damage accumulates with age, and the rate of accumulation of oxidative damage scales with life span (Beckman and Ames 1998). In addition, oxidative stress is now considered to play a key role in the pathogenesis of many liver diseases (Arteel 2003; Tanikawa and Torimura 2006; Cohen et al. 2011). Therefore, the extent of oxidative stress was examined by testing for the presence of modified proteins by HNE, the major membrane lipid peroxidation product (Forman et al. 2008). DDC treatment induced production of HNE-modified proteins, and notably, these proteins were observed preferentially in the 8-month-old mice (arrowheads, Fig. 2C). Immunohistochemistry for MDA-modified proteins was also increased in old mouse livers (Fig. 3). Next we measured the liver 20S proteasome activity and observed that this activity, as measured by detection of fluorophore cleavage by the proteasome, was similar between different age groups under basal conditions (Fig. 4A). In contrast, DDC administration induced a significant decrease in proteasome activity in all livers (from ~20,000 to 12,000 relative units; compare the y-axis in Fig. 4A with Fig. 4B) but with a more prominent effect in the livers of 8-month-old DDC-fed mice (11,982 ± 202 in 1-month-old vs 8415 ± 366 in 8-month-old mice; p=0.006). This observation led us to examine the activity of another protein degradation system, autophagy, because prior reports demonstrated that loss of autophagy induced inclusion formation in hepatocytes and neurons (Hara et al. 2006; Komatsu et al. 2005, 2006). The extent of autophagy activation was examined by immunoblotting for LC3, a homologue of yeast Atg8 that localizes to autophagosomal membranes upon conversion from the LC3-I to the LC3-II forms (Mizushima and Yoshimori 2007). Densitometric analysis showed that in 8-month-old mouse livers, the LC3-II/I ratio was significantly decreased compared with those from 3-month-old mice (0.35 ± 0.1 vs 0.57 ± 0.06; p<0.05) (Fig. 4C, D), and a similar trend was observed between the livers from 8-month-old and 1-month-old mice (0.35 ± 0.1 vs 0.54 ± 0.16; p=0.06). The endoplasmic reticulum (ER) has a crucial quality-control mechanism for protein folding, and ER stress relates to MDB-like inclusion formation in hepatocytes (Hanada et al. 2007). Therefore, we analyzed the expression of ER stress markers, GRP78 and XBP-1, and observed that these proteins were increased in DDC-treated old mouse livers (Fig. 5).
Figure 3.

Immunohistochemistry for malondialdehyde (MDA)–modified proteins in the liver. Liver sections from the control and 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)–treated mice were stained with an Ab to MDA-modified proteins and visualized by diaminobenzidine (DAB) staining. MDA-positive hepatocytes were increased in old mouse livers (C). Scale bars: (A–C) 100 µm.
Figure 4.
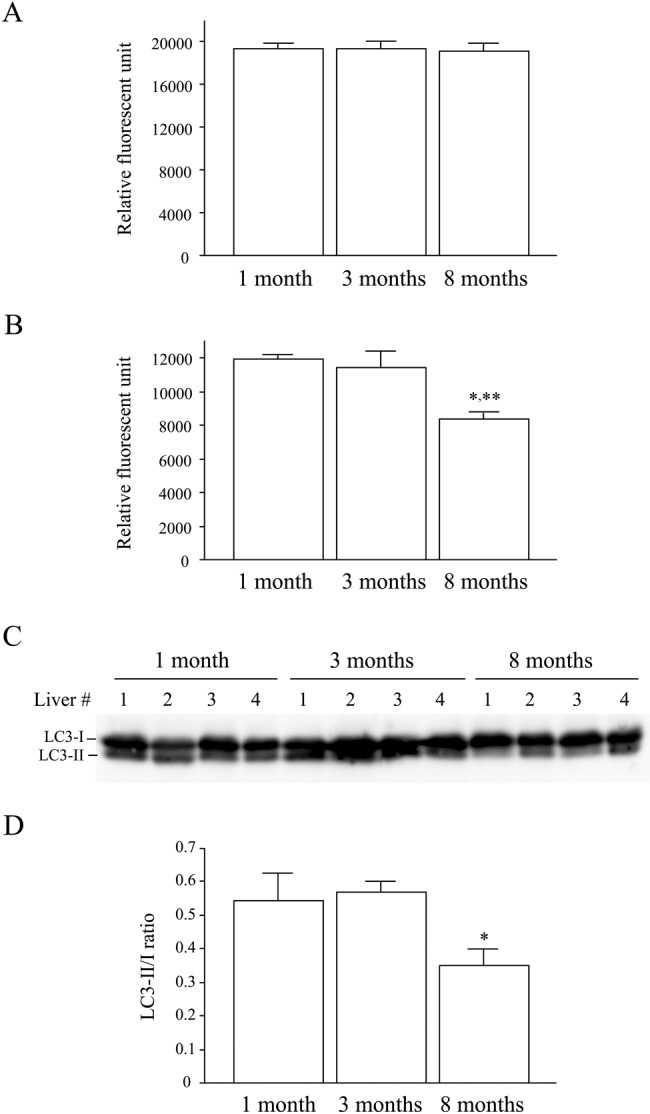
Decreased proteasome and autophagy activity upon aging in response to 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) administration. (A, B) Proteasome activity in liver homogenates was measured by detection of the fluorophore after cleavage of the substrate as described in Materials and Methods. The activity was measured in the livers of 1-, 3-, and 8-month-old mice under basal conditions (A) and after DDC feeding (B). (C, D) Autophagy activity was determined using the LC3-II/I ratio as a surrogate marker. For this, liver homogenates from DDC-fed mice with feeding starting at the indicated ages were immunoblotted with antibodies to LC3-I/II. Note that the LC3-II/I ratio showed a significant decrease in 8-month-old mouse livers. Values in panels A, B, and D represent the mean ± SD (n=4). *p<0.05 compared with 3-month-old group (B, D). **p<0.01 compared with 1-month-old group.
Figure 5.
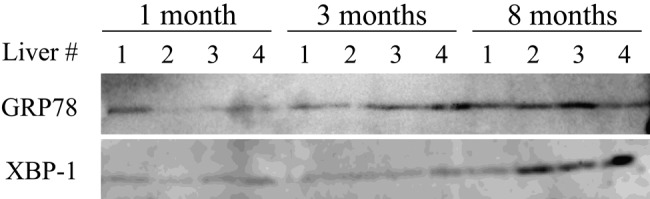
Increased endoplasmic reticulum (ER) stress upon aging in response to 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) administration. Liver homogenates were isolated from 1-, 3-, and 8-month-old mice fed a DDC-containing diet. Four independent livers were analyzed from each age group. The homogenates were blotted with Abs to ER stress markers, GRP78 and XBP-1. Note that the expression of these proteins was increased in 8-month-old mouse livers.
Discussion
We previously reported that both genetic background and gender influence MDB formation, likely via gene modifiers related to oxidative stress or drug metabolism of the compound that induces MDB formation (Hanada et al. 2008, 2010). The present study shows that aging also affects MDB formation in DDC-treated mouse livers. Proteasome activity was significantly decreased in old mouse livers, which suggests that increased hepatocyte production of misfolded proteins promotes MDB formation. In support of this, the inability to degrade cellular proteins by the proteasome is considered responsible, at least in part, for MDB formation because inhibition of this enzyme induces formation of aggregates in vitro (Harada et al. 2003; Bardag-Gorce et al. 2004). Moreover, MDB-like inclusions are observed after administration of proteasome inhibitors in vivo (French et al. 2001; Harada et al. 2008). In addition, intragastric infusion of ethanol to rats, as a model for alcoholic liver injury in humans, decreased proteasome activity possibly via ethanol-induced oxidative stress (Donohue et al. 2004). These results may explain why MDBs are commonly observed in human alcoholic liver injury. Previously we reported that MDB formation correlates with hepatocyte ballooning (Hanada et al. 2008). However, ballooned hepatocytes did not correlate with the extent of MDB formation in this study. One possible reason for this observation is that the extent of these morphological changes was relatively small compared with that shown previously, possibly because of a short duration of DDC intoxication (2 months vs 3 months). Generally, detection of ballooning is more difficult than MDB detection, and therefore, the difference in sensitivity for detecting a small amount of them might influence the results. Moreover, generally immunofluorescence staining is a more sensitive method than HE staining to detect morphological characteristics, which might also affect the results.
Recent evidence showed that autophagy helps protect from MDB formation and that its activation even promotes resolution of preformed MDBs (Harada et al. 2008). Therefore, we assessed autophagy activity and observed that LC3-II induction was decreased in old mouse livers. Moreover, ER stress, which affects correct folding of misfolded proteins, was increased in these livers. Together with the results of proteasome activity, our observations suggest that impairment of the protein degradation machinery by aging relates to MDB formation. In this regard, chaperones, which have a crucial role for protein refolding, are likely to be involved because induction of these proteins is impaired by DDC, ethanol, and aging (Soti and Csermely 2002; Carbone et al. 2005; Strnad et al. 2008).
Oxidative stress is increased not only by DDC administration but also by aging (Beckman and Ames 1998; Hanada et al. 2010; Hoare et al. 2010). Indeed, HNE- and MDA-modified proteins were increased in old mouse livers compared with those of young mice. In addition, oxidative stress results in decreased proteolysis by the proteasome (Grune et al. 1995). Therefore, it is possible that the decreased proteasome activity observed in old mouse livers was affected by increased oxidative stress. This stress also promotes p62 protein induction, which suggests that oxidative stress also influences MDB formation via p62 induction given that p62 is able to form aggregates even in the absence of K8/18 and may accelerate MDB formation (Geetha and Wooten 2002; Komatsu et al. 2007; Stumptner et al. 2007). Alternatively, increased oxidative stress may promote MDB formation by induction of ER stress (Hanada et al. 2007).
K8-overexpressed mice or K18 knockout mice are known to develop MDB formation spontaneously upon aging (Magin et al. 1998; Nakamichi et al. 2005). In humans, it is notable that the histological features of pediatric patients with NASH do not include the formation of MDBs, likely because MDBs are rarely seen in this population, in contrast to the situation in adult NASH (Barshop et al. 2008; Brunt 2010). The findings in experimental MDB formation provide support to the human observations.
In conclusion, we investigated the relationship between MDB formation and aging and showed that old mice are more susceptible to MDB formation. MDB formation in older mice was accompanied by increased oxidative stress. In addition, decreased proteasome and autophagy activity and increased ER stress were observed in these livers. Our findings add aging to the known predisposition factors that promote MDB formation.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
References
- Arteel GE. 2003. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 124:778–790 [DOI] [PubMed] [Google Scholar]
- Bardag-Gorce F, Riley NE, Nan L, Montgomery RO, Li J, French BA, Lue YH, French SW. 2004. The proteasome inhibitor, PS-341, causes cytokeratin aggresome formation. Exp Mol Pathol. 76:9–16 [DOI] [PubMed] [Google Scholar]
- Barshop NJ, Sirlin CB, Schwimmer JB, Lavine JE. 2008. Review article: epidemiology, pathogenesis and potential treatments of paediatric non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 28:13–24 [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN. 1998. The free radical theory of aging matures. Physiol Rev. 78:547–581 [DOI] [PubMed] [Google Scholar]
- Brunt EM. 2010. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 7:195–203 [DOI] [PubMed] [Google Scholar]
- Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. 2005. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 315:8–15 [DOI] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH. 2011. Human fatty liver disease: old questions and new insights. Science. 332:1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue TM, Jr, Kharbanda KK, Casey CA, Nanji AA. 2004. Decreased proteasome activity is associated with increased severity of liver pathology and oxidative stress in experimental alcoholic liver disease. Alcohol Clin Exp Res. 28:1257–1263 [DOI] [PubMed] [Google Scholar]
- Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna Levy S. 2008. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys. 477:183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French BA, van Leeuwen F, Riley NE, Yuan QX, Bardag-Gorce F, Gaal K, Lue YH, Marceau N, French SW. 2001. Aggresome formation in liver cells in response to different toxic mechanisms: role of the ubiquitin-proteasome pathway and the frameshift mutant of ubiquitin. Exp Mol Pathol. 71:241–246 [DOI] [PubMed] [Google Scholar]
- Geetha T, Wooten MW. 2002. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett. 512:19–24 [DOI] [PubMed] [Google Scholar]
- Golden TR, Hinerfeld DA, Melov S. 2002. Oxidative stress and aging: beyond correlation. Aging Cell. 1:117–123 [DOI] [PubMed] [Google Scholar]
- Grune T, Reinheckel T, Joshi M, Davies KJ. 1995. Proteolysis in cultured liver epithelial cells during oxidative stress: role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 270:2344–2351 [DOI] [PubMed] [Google Scholar]
- Hanada S, Harada M, Kumemura H, Omary MB, Koga H, Kawaguchi T, Taniguchi E, Yoshida T, Hisamoto T, Yanagimoto C, et al. 2007. Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol. 47:93–102 [DOI] [PubMed] [Google Scholar]
- Hanada S, Snider NT, Brunt EM, Hollenberg PF, Omary MB. 2010. Gender dimorphic formation of mouse Mallory-Denk bodies and the role of xenobiotic metabolism and oxidative stress. Gastroenterology. 138:1607–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada S, Strnad P, Brunt EM, Omary MB. 2008. The genetic background modulates susceptibility to mouse liver Mallory-Denk body formation and liver injury. Hepatology. 48:943–952 [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 441:885–889 [DOI] [PubMed] [Google Scholar]
- Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. 2008. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 47:2026–2035 [DOI] [PubMed] [Google Scholar]
- Harada M, Kumemura H, Omary MB, Kawaguchi T, Maeyama N, Hanada S, Taniguchi E, Koga H, Suganuma T, Ueno T, et al. 2003. Proteasome inhibition induces inclusion bodies associated with intermediate filaments and fragmentation of the Golgi apparatus. Exp Cell Res. 288:60–69 [DOI] [PubMed] [Google Scholar]
- Hoare M, Das T, Alexander G. 2010. Aging, telomeres, senescence, and liver injury. J Hepatol. 53:950–561 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 441:880–884 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 131:1149–1163 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 169:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku NO, Omary MB. 2006. A disease- and phosphorylation-related nonmechanical function for keratin 8. J Cell Biol. 174:115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku NO, Toivola DM, Zhou Q, Tao GZ, Zhong B, Omary MB. 2004. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 78:489–517 [DOI] [PubMed] [Google Scholar]
- Lismaa SE, Mearns BM, Lorand L, Graham RM. 2009. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev. 89:991–1023 [DOI] [PubMed] [Google Scholar]
- Magin TM, Schröder R, Leitgeb S, Wanninger F, Zatloukal K, Grund C, Melton DW. 1998. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8-positive aggregates. J Cell Biol. 140:1441–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy. 3:542–545 [DOI] [PubMed] [Google Scholar]
- Nakamichi I, Toivola DM, Strnad P, Michie SA, Oshima RG, Baribault H, Omary MB. 2005. Keratin 8 overexpression promotes mouse Mallory body formation. J Cell Biol. 171:931–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Strnad P, Hanada S. 2009. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest. 119:1794–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soti C, Csermely P. 2002. Chaperones come of age. Cell Stress Chaperones. 7:186–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P, Harada M, Siegel M, Terkeltaub RA, Graham RM, Khosla C, Omary MB. 2007. Transglutaminase 2 regulates Mallory body inclusion formation and injury-associated liver enlargement. Gastroenterology. 132:1515–1526 [DOI] [PubMed] [Google Scholar]
- Strnad P, Tao GZ, So P, Lau K, Schilling J, Wei Y, Liao J, Omary MB. 2008. “Toxic memory” via chaperone modification is a potential mechanism for rapid Mallory-Denk body reinduction. Hepatology. 48:931–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumptner C, Fuchsbichler A, Heid H, Zatloukal K, Denk H. 2002. Mallory body—a disease-associated type of sequestosome. Hepatology. 35:1053–1062 [DOI] [PubMed] [Google Scholar]
- Stumptner C, Fuchsbichler A, Zatloukal K, Denk H. 2007. In vitro production of Mallory bodies and intracellular hyaline bodies: the central role of sequestosome 1/p62. Hepatology. 46:851–860 [DOI] [PubMed] [Google Scholar]
- Tanikawa K, Torimura T. 2006. Studies on oxidative stress in liver diseases: important trends in liver research. Med Mol Morphol. 39:22–27 [DOI] [PubMed] [Google Scholar]
- Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M, Omary MB. 2007. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 313:2033–2049 [DOI] [PubMed] [Google Scholar]