

Abstract
In the title molecule, C11H6F2N2O2, the benzene and pyridine rings form a dihedral angle of 32.57 (6)°. The nitro group is tilted with respect to the pyridine ring by 12.26 (9)°. An intramolecular C—H⋯F hydrogen bond is present. In the crystal, molecules interact through π–π stacking interactions [centroid–centroid distances = 3.7457 (14) Å], forming columnar arrangements along the b axis. The crystal packing is further enforced by intermolecular C—H⋯O and C—H⋯N hydrogen bonds.
Related literature
For general background to organic light-emitting diodes (OLEDs), see: Baldo et al. (2000 ▶); Flamigni et al. (2007 ▶); Yang et al. (2007 ▶); Yersin (2008 ▶). For luminescent IrIII complexes containing 2-phenylpyridine or its derivatives, see: Nazeeruddin et al. (2003 ▶); Dedeian et al. (2007 ▶); Chin et al. (2007 ▶); Shen et al. (2011 ▶).
Experimental
Crystal data
C11H6F2N2O2
M r = 236.18
Orthorhombic,
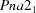
a = 22.185 (4) Å
b = 3.7457 (6) Å
c = 11.894 (2) Å
V = 988.4 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.14 mm−1
T = 296 K
0.14 × 0.12 × 0.08 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.981, T max = 0.989
6331 measured reflections
1750 independent reflections
1450 reflections with I > 2σ(I)
R int = 0.032
Refinement
R[F 2 > 2σ(F 2)] = 0.034
wR(F 2) = 0.081
S = 1.06
1750 reflections
155 parameters
1 restraint
H-atom parameters constrained
Δρmax = 0.14 e Å−3
Δρmin = −0.12 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT (Bruker, 2005 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812024713/rz2765sup1.cif
Supplementary material file. DOI: 10.1107/S1600536812024713/rz2765Isup2.mol
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812024713/rz2765Isup3.hkl
Supplementary material file. DOI: 10.1107/S1600536812024713/rz2765Isup4.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C10—H10A⋯O1i | 0.93 | 2.56 | 3.306 (3) | 138 |
| C8—H8A⋯N1ii | 0.93 | 2.58 | 3.448 (3) | 156 |
| C4—H4A⋯F1 | 0.93 | 2.40 | 2.893 (3) | 113 |
Symmetry codes: (i) 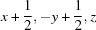 ; (ii)
; (ii) 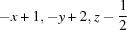 .
.
Acknowledgments
We are grateful to the Open Project Program of the State Key Laboratory of Materials-Oriented Chemical Engineering, China (grant No. KL10–14) and the National Natural Science Foundation of China (grant No. 21171093) for financial support.
supplementary crystallographic information
Comment
In recent years, IrIII cyclometalated complexes have received considerable attention because of their outstanding photochemical and photophysical properties, which make this class of complexes widely suitable to a variety of photonic applications and promising emissive materials in organic light-emitting diodes (OLEDs) (Baldo et al., 2000; Flamigni et al., 2007; Yang et al., 2007; Yersin, 2008). IrIII complexes containing 2-phenylpyridine (ppy) and its derivatives are known to exhibit high triplet quantum yields due to mixing the singlet and the triplet excited states via spin-orbit coupling, leading to high phosphorescence efficiencies (Nazeeruddin et al., 2003; Dedeian et al., 2007; Chin et al., 2007). It has been concluded that ppy-containing IrIII complexes can emit lights covering a full range of visible colors by introducing electron-donating or -withdrawing groups to the pyridyl or phenyl rings, which can adjust the HOMO-LUMO energy gaps of the complexes (Shen et al., 2011). As a contribution to this research field, we report herein the synthesis and crystal structure of the title compound. The electron-withdrawing fluoro and nitro groups have been introduced on the phenyl and pyridine rings, respectively, of the title compound, and investigations on IrIII complexes containing the title compound will be carried out soon.
The X-ray analysis of the title compound (Fig. 1) shows that the molecule is non-planar, the phenyl and pyridine rings forming a dihedral angle of 32.57 (6)°. The nitro group is slightly skewed with respect to the pyridine ring with a dihedral angle of 12.26 (9)%. An intramolecular C—H···F hydrogen bond (Table 1) stabilizes the molecular conformation. In the crystal structure (Fig. 2), π–π stacking interactions involving overlapping benzene and pyridine rings with centroid-to-centroid distances of 3.7457 (14) Å pack the molecules in columnar arrays running parallel the b axis. Furthermore, the columns interact via intermolecular C—H···O and C—H···N hydrogen bonds (Table 1).
Experimental
2-Chloro-5-nitropyridine (3.18 g, 20.0 mmol), 2,4-difluorophenylboric acid (4.00 g, 25.0 mmol) and triphenylphosphine (0.524 g, 2.0 mmol) were dissolved in THF (50 ml). After an aqueous solution of sodium carbonate (2 M, 30 ml) and palladium diacetate (0.122 g, 0.5 mmol) were added in, the mixture was refluxed under argon atmosphere for 24 h. After being cooled to room temperature, the reacted mixture was poured into water (50 ml) and was further extracted with dichloromethane (50 ml × 3). The combined extract was washed with saturated brine, dried over magnesium sulfate, and then evaporated to dryness. The crude product was purified by silica gel column chromatography (eluant: petroleum ether/ethyl acetate, 6:1 v/v), and colourless crystals of the title compound were at last obtained by recrystallization from ethanol in a yield of 70.5% (3.32 g).
Refinement
All H atoms were positioned geometrically and refined using a riding model with C—H = 0.93 Å for phenyl and pyridyl H–atoms. The Uiso(H) were allowed at 1.2Ueq(C).
Figures
Fig. 1.

The molecular structure of the title compound, showing 50% probability displacement ellipsoids.
Fig. 2.

Partial packing diagram of the title compound showing the hydrogen bonding network and π···π interactions as red dashed lines.
Crystal data
| C11H6F2N2O2 | F(000) = 480 |
| Mr = 236.18 | Dx = 1.587 Mg m−3 |
| Orthorhombic, Pna21 | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: P 2c -2n | Cell parameters from 24 reflections |
| a = 22.185 (4) Å | θ = 1.9–26.7° |
| b = 3.7457 (6) Å | µ = 0.14 mm−1 |
| c = 11.894 (2) Å | T = 296 K |
| V = 988.4 (3) Å3 | Block, colourless |
| Z = 4 | 0.14 × 0.12 × 0.08 mm |
Data collection
| Bruker APEXII CCD diffractometer | 1750 independent reflections |
| Radiation source: fine-focus sealed tube | 1450 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.032 |
| ω scans | θmax = 25.0°, θmin = 1.8° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −24→26 |
| Tmin = 0.981, Tmax = 0.989 | k = −4→4 |
| 6331 measured reflections | l = −14→14 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.034 | w = 1/[σ2(Fo2) + (0.0417P)2] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.081 | (Δ/σ)max < 0.001 |
| S = 1.06 | Δρmax = 0.14 e Å−3 |
| 1750 reflections | Δρmin = −0.12 e Å−3 |
| 155 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 1 restraint | Extinction coefficient: 0.020 (2) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), 823 Friedel pairs |
| Secondary atom site location: difference Fourier map | Flack parameter: 1.3 (9) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | 0.43717 (7) | 0.8814 (4) | 0.19399 (11) | 0.0706 (5) | |
| F2 | 0.64096 (6) | 0.8761 (5) | 0.29027 (14) | 0.0783 (5) | |
| N1 | 0.39001 (8) | 0.6832 (5) | 0.52314 (14) | 0.0468 (5) | |
| N2 | 0.23174 (9) | 0.3925 (7) | 0.5480 (2) | 0.0604 (6) | |
| C1 | 0.33559 (10) | 0.6124 (6) | 0.56308 (18) | 0.0487 (6) | |
| H1A | 0.3278 | 0.6546 | 0.6388 | 0.058* | |
| C2 | 0.29008 (9) | 0.4788 (6) | 0.4966 (2) | 0.0461 (5) | |
| C3 | 0.29993 (10) | 0.4202 (6) | 0.38367 (19) | 0.0499 (6) | |
| H3A | 0.2696 | 0.3316 | 0.3376 | 0.060* | |
| C4 | 0.35623 (9) | 0.4973 (6) | 0.34139 (19) | 0.0480 (6) | |
| H4A | 0.3644 | 0.4647 | 0.2654 | 0.058* | |
| C5 | 0.40050 (9) | 0.6236 (5) | 0.41293 (17) | 0.0392 (5) | |
| C6 | 0.46353 (9) | 0.6929 (5) | 0.37757 (17) | 0.0406 (5) | |
| C7 | 0.48022 (10) | 0.8139 (6) | 0.27144 (19) | 0.0457 (6) | |
| C8 | 0.53882 (12) | 0.8781 (6) | 0.2407 (2) | 0.0529 (6) | |
| H8A | 0.5485 | 0.9618 | 0.1693 | 0.064* | |
| C9 | 0.58240 (10) | 0.8137 (6) | 0.3194 (2) | 0.0524 (6) | |
| C10 | 0.57000 (11) | 0.6967 (7) | 0.4255 (2) | 0.0552 (7) | |
| H10A | 0.6007 | 0.6581 | 0.4773 | 0.066* | |
| C11 | 0.51028 (9) | 0.6372 (6) | 0.45341 (19) | 0.0468 (6) | |
| H11A | 0.5011 | 0.5571 | 0.5254 | 0.056* | |
| O1 | 0.19644 (9) | 0.2185 (6) | 0.49287 (19) | 0.0910 (7) | |
| O2 | 0.22205 (9) | 0.4973 (7) | 0.6427 (2) | 0.1016 (8) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0672 (10) | 0.0979 (13) | 0.0468 (8) | 0.0059 (9) | −0.0017 (7) | 0.0135 (8) |
| F2 | 0.0529 (8) | 0.1004 (11) | 0.0818 (11) | −0.0123 (8) | 0.0206 (8) | −0.0020 (9) |
| N1 | 0.0453 (11) | 0.0560 (12) | 0.0390 (11) | −0.0035 (9) | 0.0008 (8) | −0.0029 (9) |
| N2 | 0.0466 (13) | 0.0676 (13) | 0.0671 (16) | −0.0070 (11) | 0.0044 (12) | 0.0016 (11) |
| C1 | 0.0491 (13) | 0.0584 (14) | 0.0387 (13) | −0.0033 (12) | 0.0016 (10) | −0.0016 (10) |
| C2 | 0.0423 (12) | 0.0445 (12) | 0.0514 (15) | 0.0006 (10) | 0.0018 (11) | 0.0024 (11) |
| C3 | 0.0468 (13) | 0.0535 (13) | 0.0494 (14) | −0.0034 (11) | −0.0103 (11) | −0.0086 (12) |
| C4 | 0.0531 (14) | 0.0548 (14) | 0.0360 (12) | 0.0004 (11) | −0.0040 (11) | −0.0040 (11) |
| C5 | 0.0446 (12) | 0.0343 (11) | 0.0386 (11) | 0.0014 (10) | −0.0010 (9) | 0.0006 (9) |
| C6 | 0.0492 (14) | 0.0339 (11) | 0.0387 (12) | 0.0036 (9) | 0.0022 (11) | −0.0021 (10) |
| C7 | 0.0543 (15) | 0.0437 (13) | 0.0392 (12) | 0.0042 (11) | 0.0012 (11) | 0.0027 (11) |
| C8 | 0.0614 (16) | 0.0494 (16) | 0.0480 (13) | −0.0010 (12) | 0.0133 (12) | 0.0017 (11) |
| C9 | 0.0449 (14) | 0.0506 (14) | 0.0617 (16) | −0.0035 (11) | 0.0146 (13) | −0.0053 (12) |
| C10 | 0.0486 (15) | 0.0605 (16) | 0.0564 (16) | 0.0036 (11) | 0.0012 (12) | 0.0028 (13) |
| C11 | 0.0443 (13) | 0.0487 (14) | 0.0474 (13) | 0.0022 (10) | 0.0016 (11) | 0.0038 (11) |
| O1 | 0.0561 (11) | 0.1191 (18) | 0.0978 (18) | −0.0343 (12) | −0.0064 (12) | −0.0072 (13) |
| O2 | 0.0777 (15) | 0.151 (2) | 0.0764 (15) | −0.0335 (14) | 0.0293 (12) | −0.0233 (16) |
Geometric parameters (Å, º)
| F1—C7 | 1.351 (3) | C4—C5 | 1.383 (3) |
| F2—C9 | 1.365 (2) | C4—H4A | 0.9300 |
| N1—C1 | 1.324 (3) | C5—C6 | 1.483 (3) |
| N1—C5 | 1.350 (3) | C6—C11 | 1.390 (3) |
| N2—O1 | 1.212 (3) | C6—C7 | 1.391 (3) |
| N2—O2 | 1.213 (3) | C7—C8 | 1.372 (3) |
| N2—C2 | 1.467 (3) | C8—C9 | 1.367 (4) |
| C1—C2 | 1.377 (3) | C8—H8A | 0.9300 |
| C1—H1A | 0.9300 | C9—C10 | 1.364 (4) |
| C2—C3 | 1.378 (3) | C10—C11 | 1.384 (3) |
| C3—C4 | 1.377 (3) | C10—H10A | 0.9300 |
| C3—H3A | 0.9300 | C11—H11A | 0.9300 |
| C1—N1—C5 | 118.19 (19) | C11—C6—C7 | 116.04 (19) |
| O1—N2—O2 | 124.2 (2) | C11—C6—C5 | 119.54 (19) |
| O1—N2—C2 | 117.6 (2) | C7—C6—C5 | 124.4 (2) |
| O2—N2—C2 | 118.2 (2) | F1—C7—C8 | 117.1 (2) |
| N1—C1—C2 | 122.4 (2) | F1—C7—C6 | 119.44 (19) |
| N1—C1—H1A | 118.8 | C8—C7—C6 | 123.5 (2) |
| C2—C1—H1A | 118.8 | C9—C8—C7 | 117.1 (2) |
| C1—C2—C3 | 120.1 (2) | C9—C8—H8A | 121.4 |
| C1—C2—N2 | 119.2 (2) | C7—C8—H8A | 121.4 |
| C3—C2—N2 | 120.7 (2) | C10—C9—F2 | 118.8 (2) |
| C4—C3—C2 | 117.8 (2) | C10—C9—C8 | 123.2 (2) |
| C4—C3—H3A | 121.1 | F2—C9—C8 | 118.0 (2) |
| C2—C3—H3A | 121.1 | C9—C10—C11 | 117.8 (2) |
| C3—C4—C5 | 119.4 (2) | C9—C10—H10A | 121.1 |
| C3—C4—H4A | 120.3 | C11—C10—H10A | 121.1 |
| C5—C4—H4A | 120.3 | C10—C11—C6 | 122.3 (2) |
| N1—C5—C4 | 122.1 (2) | C10—C11—H11A | 118.9 |
| N1—C5—C6 | 114.14 (19) | C6—C11—H11A | 118.9 |
| C4—C5—C6 | 123.72 (19) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C10—H10A···O1i | 0.93 | 2.56 | 3.306 (3) | 138 |
| C8—H8A···N1ii | 0.93 | 2.58 | 3.448 (3) | 156 |
| C4—H4A···F1 | 0.93 | 2.40 | 2.893 (3) | 113 |
Symmetry codes: (i) x+1/2, −y+1/2, z; (ii) −x+1, −y+2, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: RZ2765).
References
- Baldo, M. A., Thompson, M. E. & Forrest, S. R. (2000). Nature (London), 403, 750–753. [DOI] [PubMed]
- Bruker (2005). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Chin, C. S., Eum, M.-S., Kim, S. Y., Kim, C. & Kang, S. K. (2007). Eur. J. Inorg. Chem. pp. 372–375.
- Dedeian, K., Shi, J., Forsythe, E. & Morton, D. C. (2007). Inorg. Chem. 46, 1603–1611. [DOI] [PubMed]
- Flamigni, L., Barbieri, A., Sabatini, C., Ventura, B. & Barigelletti, F. (2007). Top. Curr. Chem. 281, 143–203.
- Nazeeruddin, Md. K., Humphry-Baker, R., Berner, D., Rivier, S., Zuppiroli, L. & Graetzel, M. (2003). J. Am. Chem. Soc. 125, 8790–8797. [DOI] [PubMed]
- Sheldrick, G. M. (1996). SADABS. University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shen, X., Wang, F.-L., Sun, F., Zhao, R., Wang, X., Jing, S., Xu, Y. & Zhu, D.-R. (2011). Inorg. Chem. Commun. 14, 1511–1515.
- Yang, C.-H., Cheng, Y.-M., Chi, Y., Hsu, C.-J., Fang, F.-C., Wong, K.-T., Chou, P.-T., Chang, C.-H., Tsai, M.-H. & Wu, C.-C. (2007). Angew. Chem. Int. Ed. 46, 2418–2421. [DOI] [PubMed]
- Yersin, H. (2008). In Highly Efficient OLEDs with Phosphorescent Materials Weinheim: Wiley-VCH.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812024713/rz2765sup1.cif
Supplementary material file. DOI: 10.1107/S1600536812024713/rz2765Isup2.mol
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812024713/rz2765Isup3.hkl
Supplementary material file. DOI: 10.1107/S1600536812024713/rz2765Isup4.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report