

Abstract
Papillomavirus L2-based vaccines have generally induced low-level or undetectable neutralizing antibodies in standard in vitro assays yet typically protect well against in vivo experimental challenge in animal models. Herein we document that mice vaccinated with an L2 vaccine comprising a fusion protein of the L2 amino acids 11 to 88 of human papillomavirus type 16 (HPV16), HPV18, HPV1, HPV5, and HPV6 were uniformly protected from cervicovaginal challenge with HPV16 pseudovirus, but neutralizing antibodies against HPV16, -31, -33, -45, or -58 were rarely detected in their sera using a standard in vitro neutralization assay. To address this discrepancy, we developed a neutralization assay based on an in vitro infectivity mechanism that more closely mimics the in vivo infectious process, specifically by spaciotemporally separating primary and secondary receptor engagement and correspondingly by altering the timing of exposure of the dominant L2 cross-neutralizing epitopes to the antibodies. With the new assay, titers in the 100 to 10,000 range were measured for most sera, whereas undetectable neutralizing activities were observed with the standard assay. In vitro neutralizing titers measured in the serum of mice after passive transfer of rabbit L2 immune serum correlated with protection from cervicovaginal challenge of the mice. This “L2-based” in vitro neutralization assay should prove useful in critically evaluating the immunogenicity of L2 vaccine candidates in preclinical studies and future clinical trials.
INTRODUCTION
Clinical trials of human papillomavirus (HPV) L1 virus-like particle (VLP) prophylactic vaccines have demonstrated a high degree of safety, immunogenicity, and effectiveness at preventing infection and neoplastic disease caused by the vaccine-targeted types (reviewed in reference 24). Despite this success, vaccines based on the L2 minor capsid protein are attractive candidates for second-generation HPV prophylactic vaccines because, in contrast to L1 VLP vaccines, they induce broad cross-type protection as measured both with in vitro neutralization assays and with in vivo protection assays based on challenge with animal papillomavirus types or HPV pseudovirions (5, 14, 15, 16). For instance, immunization of rabbits with an HPV16-derived L2 peptide induced cross-protection against both cutaneous infection with cottontail rabbit papillomavirus (CRPV) and mucosal infection with rabbit oral papillomavirus (ROPV) (15). However, in vitro neutralization titers against homologous types induced by L2 immunogens have been much lower than the titers induced by L1 VLP-based vaccines, regardless of the adjuvant employed (34).
One possible explanation for the differences in neutralization titers is that an ordered multivalent display of epitopes on the VLP surface induces B cell activation and survival signals that cannot be matched by a monomeric antigen in conjunction with current adjuvants. However, virus-like display of L2 peptides has thus far not resulted in the induction of high titers of in vitro neutralizing antibodies (4, 35). A second possibility is that only a small proportion of the L2 antibodies generated by the vaccines can actually inhibit HPV infection. In this study, we have evaluated a third possibility, namely, that the current in vitro neutralization assays are insensitive measures of infection-inhibiting L2 antibodies and therefore underestimate the protective potential of L2 vaccines (29).
There are several reasons to believe that the latter explanation is at least partially responsible for this phenomenon. We have repeatedly observed that mice injected with L2-based polypeptide vaccines are fully protected from cervicovaginal challenge with HPV pseudovirions, although at the time of challenge, their sera do not contain in vitro neutralizing antibodies against the corresponding virus that are detectable in the standard neutralization assay. We believe that in vivo protection following L2 immunization is antibody mediated because passive transfer of L2 antibodies can fully protect against challenge (11). Additionally, L2 is not encoded by the pseudovirions used in the challenge studies (2). Finally, the main L2 cross-neutralization epitopes are poorly exposed on mature virus, and current neutralization assays, if they predominantly detect this subset of L2 epitopes, could substantially underestimate the potential neutralizing activity of a given L2 immune serum.
Our recent delineation of the in vivo infectious process, utilizing our murine cervicovaginal model, is relevant to the possible insensitivity of current in vitro neutralization assays for the measurement of L2 neutralizing antibodies (21, 32). In vivo, HPV pseudovirions bind initially to heparan sulfate proteoglycan (HSPG) moieties, located on the basement membrane (BM), that are exposed following disruption of epithelial integrity. This binding induces a conformational change in the capsid that causes exposure of a furin/proprotein convertase cleavage site located at the amino terminus of L2. Subsequent furin/PC cleavage leads to a second conformational change, which further exposes the L2 amino terminus, including the major cross-neutralization epitopes (21). According to our model, these events expose a previously occluded surface of L1 that can now engage an unidentified (or putative) cell surface receptor on the keratinocytes as they migrate over the exposed BM to repair the disrupted epithelium (10, 21). L2 neutralizing antibodies bind to capsids following furin cleavage, preventing infection because the bound antibodies prevent capsids from transferring to, or stably associating with, the keratinocyte cell surface receptor (11, 12). Importantly, the L2 cross-neutralizing epitopes are exposed for an exceptionally long period, several hours, prior to internalization by the cell.
In vitro infection depends on similar sequential changes but differs from the in vivo process in several key respects. Although cultured keratinocytes produce an extracellular matrix (ECM) with some similarities to the BM, the role of the ECM in HPV infection in vitro is not the same as that of the BM in vivo. Pseudovirions can bind to ECM in vitro, but this interaction is not critical for subsequent infection, since virus bound directly to cells in suspension can lead to infection (P. M. Day, unpublished observations). It is the HSPGs on the cell surface, rather than those on the ECM, that are critical for capsid binding and infection (13, 17). Furin cleavage and L2 epitope exposure also occur on the cell surface, not generally on the ECM (10). Therefore, all of the described conformational changes that occur prior to infectious entry take place on the cell surface in vitro, unlike the in vivo situation, where initial attachment and furin cleavage occur on the BM, independent of the cell surface. This topological difference may result in the in vivo situation offering a greater opportunity for antibodies directed against L2 to bind to exposed epitopes on the BM prior to any interaction of the capsids with cell surfaces.
Given this new understanding of the differences between the mechanisms of in vivo and in vitro HPV infection, we sought to devise a more sensitive in vitro assay for measuring neutralizing L2 antibodies. This improved assay is based on the construction of an in vitro infection process that closely mimics our mechanistic model for in vivo infection. In the current study, we present the developmental steps in the formulation of this neutralization assay and demonstrate its sensitivity, relative to a traditional pseudovirion-based neutralization assay, in the detection of potentially protective L2 antibodies.
MATERIALS AND METHODS
Pseudovirions.
All pseudovirions were produced as previously described (3), utilizing the “ripcord” variation for in vitro assays and the “standard” variation for in vivo assays as detailed on the laboratory website (http://home.ccr.cancer.gov/lco/default.asp). The capsid-encoding plasmids p16Llw, p31Llw, p45Llw, p52Llw, p18Llw, p58Llw, and p11Lw and p11lw were utilized. For in vivo imaging, the plasmid pCLucf (a firefly luciferase expression plasmid) was encapsidated. For neutralization assays, either pYSeAP (a secreted alkaline phosphatase [SeAP] expression plasmid) or pfwB (a green fluorescent protein [GFP] expression plasmid) were encapsidated. All plasmids are described on the laboratory website.
Cell lines.
The HeLa cell line was obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin (P/S). 293TT cells, which were previously described, were cultured in the same medium (1). 804G cells were a kind gift of Jonathan Jones (31) and were also cultured in DMEM-FBS and P/S. The pgsa-745 cell line was also obtained from ATCC and cultured in DMEM-FBS, P/S, and 10 mM proline. The same media were utilized for propagation of FD11 cells and CHOΔfurin (Δfurin) cells obtained from Steven Leppla (NIAID, NIH) and David Fitzgerald (NCI, NIH), respectively. MCF10A cells, obtained from ATCC were cultured in DMEM/F12 medium supplemented with 5% horse serum, P/S, 500 μg/ml hydrocortisone (Sigma H-0888), 10 μg/ml insulin (Sigma I-1882), and 20 μg/ml epidermal growth factor (100-15; Peprotech).
Animal studies.
Six- to eight-week-old female BALB/cAnNCr mice were obtained from the National Cancer Institute and housed and handled in accordance with the NCI-approved guidelines, and all protocols were approved by the NCI's Animal Care and Use Committee. Animals were immunized subcutaneously with 5 μg of HPV16 VLPs or 25 μg of His6-tagged L2 11-88×5 peptide (19) precipitated onto Imject Alum (77161; Thermo) in a final volume of 100 μl. Animals were immunized three times, with each injection spaced 2 weeks apart, and sera were collected 2 weeks after the final immunization in order to create the serum pools. Polyclonal rabbit serum was generated against the L2 11-88×5 polypeptide by Lampire Biological Laboratories. Three immunizations with 300 μg peptide at 3-week intervals were performed. The first immunization was in complete Freund's adjuvant. The following two were in incomplete Freund's adjuvant. The exsanguination bleed was 2 weeks following the final immunization.
In vivo pseudovirus delivery and infection.
The mice received 3 mg of Depo-Provera (NDC 0009-0626-01; Pfizer) 5 days prior to infection. Animals were pretreated with 4% nonoxynol-9 approximately 5 h prior to administration of pseudovirus, as previously described (8, 20, 32). A pseudovirus inoculum of 10 μg, based on L1 content, was used regardless of HPV type, since this would equalize the potential number of target epitopes for the neutralizing antibodies. For those mice receiving the passively transferred rabbit sera, the indicated volume of sera was diluted in 1× phosphate-buffered saline (PBS) to a final volume of 100 μl and was administered intraperitoneally 24 h prior to infection. Infection was measured 48 h after pseudovirus delivery by intravaginally instilling 20 μl of d-luciferin–K+ salt (0.3 mg, 122796; Caliper Life Sciences), followed by imaging with an Ivis 100 system (Caliper Life Sciences) at 4°C, as previously described (8, 20). Raw data were computed using the Living Image software program (Caliper Life Sciences). An identical region of interest (ROI) was drawn around the area from which the luciferase signal was emitted for each mouse, and the average radiance within the ROI was determined. Data represent the mean luminescence for 5 mice per group.
L1-based neutralization assay.
The L1-based neutralization assay was performed as previously described (28). Briefly, pYSeAP-containing pseudovirions were incubated with immune serum on ice for 60 min prior to addition to previously plated 293TT cells. SeAP production was measured using the Great EscAPe kit (631737; Clontech) 72 h following pseudovirus addition.
L2-based neutralization assay.
MCF10A cells were plated at a concentration of 2 × 104/well in a 96-well assay plate in a volume of 100 μl of growth medium and propagated for 24 h. Following this incubation, which allows for deposition of the ECM, the cells were washed twice with PBS and lysed by incubation for 5 min at 37°C with 50 μl of prewarmed lysis buffer (PBS containing 0.5% [vol/vol] Triton X-100–20 mM NH4OH). Following lysis, 100 μl of PBS was added to each well, and a 100-μl volume was removed. This gentle washing was repeated two more times. Then the entire remaining volume was gently removed. The ECM-containing wells were then washed twice further with 100 μl of PBS. A pseudovirion (with the pfwb plasmid packaged) solution, prepared in conditioned medium from CHOΔfurin cells in a total volume of 120 μl/well, was added to the prepared ECM-containing wells following removal of the final PBS wash. This pseudovirus solution also contained 5 μg/ml of heparin (Sigma H-4784). Addition of this small amount of heparin results in an improved consistency of antibody titers (data not shown). This virus-furin-heparin mixture was incubated overnight at 37°C. The next day, the medium was removed and the wells were washed twice with PBS. The final wash was replaced with an antibody dilution series made in pgsa-745 growth medium at a volume of 100 μl/well. The plate was then incubated at 37°C for 6 h to allow efficient antibody binding to target epitopes. This incubation can be shortened if desired. No change in antibody titers was observed with incubation times of 1 h, 2 h, 4 h, or 6 h (data not shown). Following this incubation, pgsa-745 cells were added directly to the antibody-containing wells at a concentration of 8 × 103/well in a volume of 50 μl. Infection was allowed to continue for 2 days at 37°C, and individual cells were scored by flow cytometric analysis for GFP expression as a measure of infection. Although pseudovirus with a packaged GFP expression plasmid was used for the development of the assay to facilitate enumeration of individual infectious events, the method should be readily transferable to other described pseudovirion-based expression assay systems (e.g., secreted alkaline phosphatase and Gaussia luciferase).
All pseudovirions were titrated in the assay to determine the amount of virus needed to yield 20 to 30% GFP-positive cells. This amount was used for the neutralization assays. Conditioned medium from CHOΔfurin cells was prepared by plating 1 × 106 cells in 17 ml of growth medium 4 days prior to harvesting. For all experiments, 3-fold dilution series of antisera were made starting with an initial dilution of 1/50. Data were analyzed, and 50% inhibitory concentrations (IC50s) were determined using the GraphPad Prism software program. Where indicated, furin inhibitor (calbiochem, 344930) was added at a final concentration of 5 μM.
Western blot.
Fifty nanograms of capsids were added to ECM extracted from cultured cells as described above or 1 μg of Cultrex EHS-derived ECM (Trevigen 3432-005-01) and allowed to attach at 37°C overnight. Following this incubation, wells were washed three times with PBS. Bound capsids were liberated directly into Laemmli SDS-PAGE dye, run on a 4 to 12% Tris-glycine gel, and transferred to a polyvinylidene difluoride (PVDF) membrane for Western analysis. HPV16 and HPV18 were detected with Camvir-1 antibody (Abcam ab69). All other HPV types were detected with rabbit polyclonal sera generated against the respective virus-like particles (sera were generated as previously described) (33).
RESULTS
Examination of binding to various ECMs.
The first step in the development of an in vitro infection assay that more closely mimicked in vivo events was to identify an ECM source that could support strong capsid association for multiple HPV types. We examined the binding of HPV16, HPV11, HPV18, HPV31, HPV45, HPV52, and HPV58 capsids to ECM derived from HaCaT cells, MDF10A cells, 804G cells, and Cultrex, a commercial ECM preparation derived from Engelbreth-Holm-Swarm murine sarcoma cells. These cell lines were examined because MCF10A and 804G were reported to secrete an ECM that is rich in laminin 332 (22, 26), a proposed ECM receptor for HPV11 (9), and we have previously observed a strong association of HPV16 capsids with HaCaT-derived ECM (10). All HPV types bound well to the ECM produced by HaCaT and MCF10A cells. The binding to 804G-derived ECM was weaker for all HPV types examined, and binding to the Cultrex ECM preparation was weak for all types except HPV16 (Fig. 1). The factors responsible for these observed differences are unclear and are under investigation.
Fig 1.
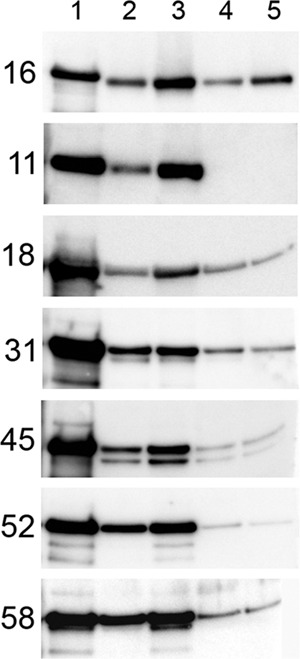
Pseudovirion binding to extracellular matrices. Pseudovirions were bound overnight to the indicated matrices at 37°C. Unbound capsids were removed by washing. Bound capsids were solubilized in SDS-PAGE loading dye, separated on a 4 to 12% acrylamide gel, and processed for Western analysis. The HPV type is indicated on the left of each row. Input virus was loaded in lane 1. Virus bound to ECM preparations is shown as follows: lane 2, HaCaT; lane 3, MCF10A; lane 4, 804G; lane 5, Cultrex, a commercial ECM preparation derived from EHS cells.
Examination of ECM-based infection.
As noted earlier, furin cleavage is required for papillomavirus infection in vivo and in cultured cells, although the location of the furin cleavage occurs on the basement membrane in vivo and on the cell surface in standard cell culture-based infectivity assays (11, 21, 30). To confirm that infection resulting from virus adhered to ECM prior to addition of cells was also furin dependent, we bound GFP-expressing HPV16 pseudovirus to ECM produced by MCF10A cells, one of the two ECM substrates that strongly bound the various HPV types, in the presence of conditioned medium from a furin-deficient CHO cell line, FD11, or a furin-secreting cell line, CHOΔfurin, which is also derived from CHO (6, 18). Therefore, these cell lines presumably differ only in furin production. Following overnight virus adsorption, media and unbound virus were removed, and wells were washed thoroughly. Susceptibility to infection was evaluated for three different target cells, HeLa, FD11, and the HSPG-deficient cell line pgsa-745, which differ with respect to their expression of furin and of cell surface HSPG. The cells were added to the ECM-virus-containing wells, and infection was allowed to proceed for 48 h. Additionally, for one experimental set, the saturation of capsid binding to the ECM substrate was estimated by placing the unbound virus and media from the wells into a new plate, to which HeLa cells were added.
As shown in Fig. 2A, the addition of Δfurin supernatant greatly increased infection in all cases. Furthermore, it was essential for infection of the FD11 cells, as expected, since these cells are deficient for endogenous furin production, although they express HSPGs. The results confirm the requirement for furin for HPV infection. The pgsa-745 cell line expresses furin but lacks HSPG, due to a deficiency of xylosyltransferase (25). Therefore, infection of this line is also highly dependent on the Δfurin supernatant, since it is robustly infected only after the requirement for HSPG has been bypassed by furin precleavage of the capsids (12). Since HeLa cells express both HSPG and furin, they can be infected without addition of exogenous furin. However, the finding that exogenous furin increases infection of HeLa cells indicates that furin is a limiting factor for in vitro infection of this commonly employed cell line. The examination of virus transferred to new wells and tested with HeLa cells indicates that infection by the transferred virus was consistently about one-half that of the direct HeLa infection, regardless of the amount of input virus. Therefore, the ECM binding was not saturable under the conditions employed, although the lower titer of the transferred virus may imply a relatively unstable interaction with the ECM.
Fig 2.
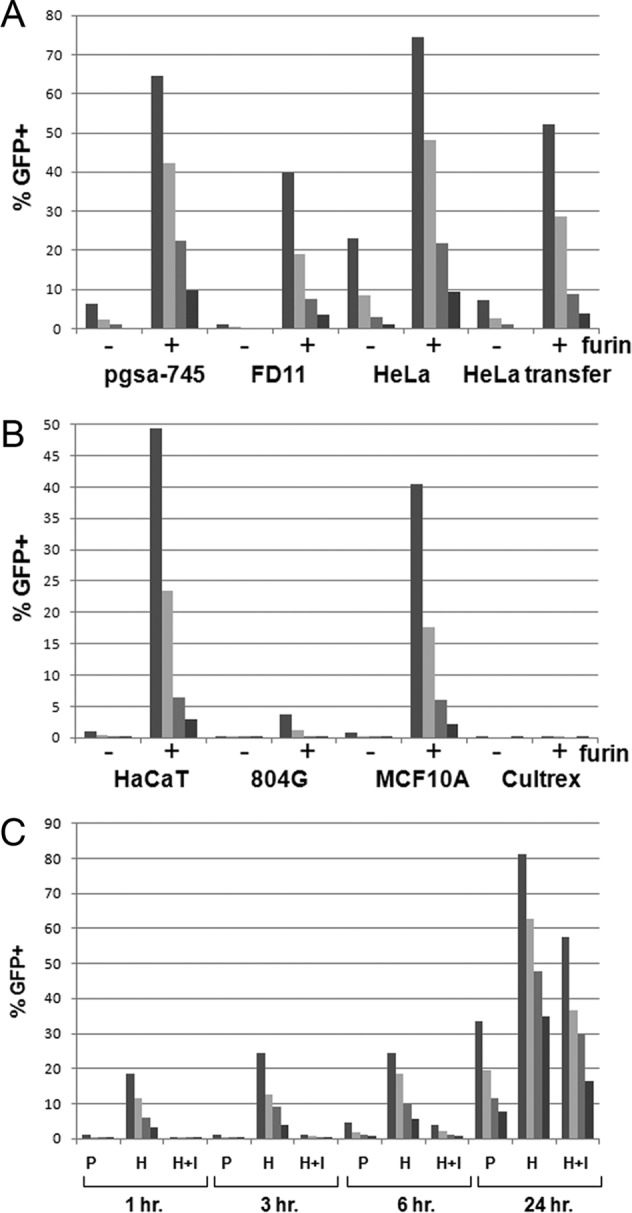
Furin-dependent infection following ECM attachment. (A) A titration series of HPV16 pseudovirus containing a packaged GFP expression plasmid was applied to MCF10A-derived ECM in conditioned medium from FD11 cells or CHOΔfurin cells (indicated as −/+ furin, respectively). Unbound virus and medium were removed by washing, the indicated target cells were added, and infection was allowed to proceed for 2 days. Pseudovirus infection was determined by flow cytometric analysis of GFP+ cells. The HeLa transfer condition shows the infection derived from unbound pseudovirus that was transferred to empty wells. HeLa cells were then added, and infection was allowed to proceed as described above. (B) A titration series of HPV16 pseudovirus containing a packaged GFP expression plasmid was applied to the matrices indicated in conditioned medium from either FD11 cells or CHOΔfurin cells (indicated as −/+ furin). Unbound virus and medium were removed by washing, pgsa-745 target cells were added, and infection was allowed to proceed for 2 days. Pseudovirus infection was determined by flow cytometric analysis of GFP+ cells. (C) A titration series of HPV16 pseudovirus containing a packaged GFP expression plasmid was applied to MCF10A-derived ECM in conditioned medium from CHOΔfurin cells for the indicated time. Unbound virus and medium were removed by washing, the target cells were added, and infection was allowed to proceed for 2 to 3 days, depending upon the time of virus removal. The target cells were either pgsa-745 (P), HeLa (H), or HeLa and 5 μM furin inhibitor (H+I). Pseudovirus infection was determined by flow cytometric analysis of GFP+ cells.
We next determined how the panel of ECM preparations utilized initially (Fig. 1) would perform in this infectivity assay, using pgsa-745 cells as the target cell line and HPV16 as a model pseudovirus. As shown in Fig. 2B, we found that high infectivity was obtained after binding HPV16 capsids to the ECM produced by HaCaT and MCF10A. In all cases, infectivity was greatly increased in the presence of Δfurin supernatant compared to results with FD11 supernatant. Surprisingly, neither the 804G-derived ECM nor the Cultrex ECM preparation supported subsequent infection, although, as shown in Fig. 1, HPV16 capsids bound reasonably well to these matrices. We speculate that binding to these matrices does not induce, or inhibits, the conformational changes required for furin cleavage and/or the exposure of the secondary receptor binding site.
For subsequent experiments, we chose to use ECM produced by MCF10A cells as the binding substrate, since all the HPV types examined bound best to this ECM and HPV16 binding resulted in high infectivity of target cells. To maximize secondary receptor-dependent infection, the assay was performed in the presence of Δfurin supernatant and utilized pgsa-745 as the target cell (12). Because our in vivo studies have demonstrated that the furin-dependent exposure of the major L2 neutralization epitopes is a slow process (21), we decided to examine this parameter within the constraints of this in vitro assay. To address this, we adsorbed HPV16 pseudovirus to MCF10A-derived ECM in the presence of Δfurin supernatant for 1 h, 3 h, 6 h, or 24 h. Following the incubation, unbound pseudovirus and furin were removed, and infection of pgsa-745 cells, HeLa cells, or HeLa cells in the presence of furin inhibitor was assessed. As seen in Fig. 2C, the infection of both pgsa-745 and HeLa cells with furin inhibitor was poor until the 24-h time point. This indicates that, similar to the situation in vivo, in vitro exposure of the L2 furin cleavage site is a slow process. We also observe an increase in untreated HeLa cells at 24 h, following furin cleavage, similar to that shown in Fig. 2A. Therefore, the long initial incubation of pseudovirus and ECM in the presence of furin is essential for subsequent infection of HSPG- or furin-deficient cell lines.
A simplified schematic of the finalized assay is shown in Fig. 3. We refer to this newly developed assay as the “L2-based neutralization assay,” as opposed to the original “L1-based neutralization assay.”
Fig 3.
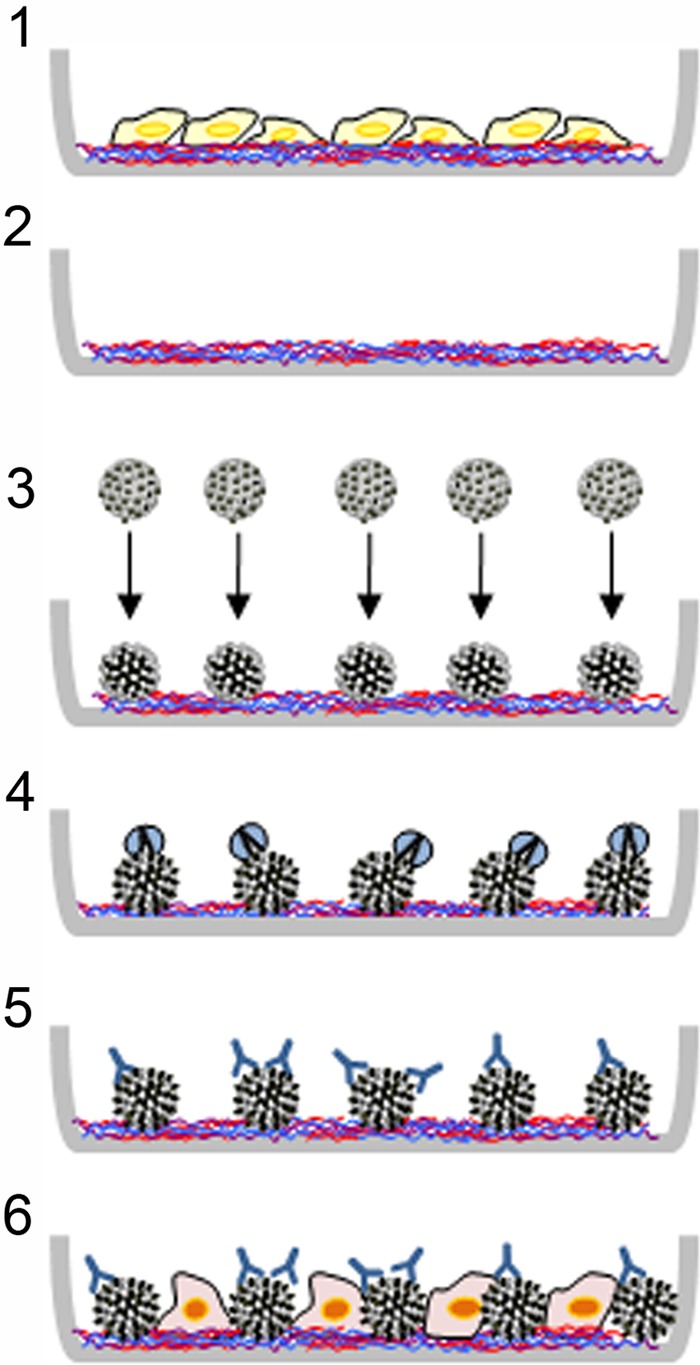
Schematic of L2-based neutralization assay. The simplified diagram illustrates the main steps in this assay. 1, MCF10A cells are plated in a 96-well plate and grown for 24 h to allow for ECM deposition; 2, the ECM remains on the plastic following lysis of the MCF10A cells; 3, a pseudovirus preparation is plated onto the ECM; this interaction results in a conformational change in the capsid that exposes the N terminus of L2; 4, exogenous furin is applied with the pseudovirions to allow cleavage of the conserved N-terminal furin cleavage site in L2; this results in exposure of the major cross neutralization epitopes in L2; 5, following removal of unbound pseudovirus, an antibody titration series is applied to the plate and incubated at 37°C for 6 h; 6, HSPG-deficient pgsa-745 cells are plated into the virus/antibody-containing wells. Infection is assessed following a 48-h incubation.
Examination of neutralization titers.
To initiate a comparison of the performance of the L2-based and L1-based neutralization assays, we analyzed sera from animals immunized with an L2 immunogen comprising a fusion polypeptide composed of amino acids 11 to 88 from HPV16, HPV18, HPV1, HPV5, and HPV6, designated L2 (11-88×5) (19). This immunogen is a candidate for clinical development because it induces relatively high titers of cross-neutralizing antibodies in the L1-based neutralization assay and protects against challenge by homologous and heterologous HPV types in a mouse model (11). We determined neutralizing activity against HPV types 16, 31, 33, 45, and 58. These types were selected for the following reasons: HPV16 is a component of the L2 (11-88×5) candidate vaccine, as well as the commercial HPV VLP vaccines; HPV31, -33, and -58 are phylogenetically related to HPV–16 but are progressively more divergent from it and are not included in either the L2- or VLP-based vaccines; the L2 (11-88×5) vaccine protects against HPV58, in contrast to the VLP vaccine (27); and HPV45 is closely related to HPV18.
A polyclonal rabbit serum was tested first, because rabbits generally generate higher titers of in vitro neutralizing antibodies against L2 immunogens than mice. As shown in Table 1, the mean titer against HPV16 was 120-fold higher in the L2-based assay, and similar increases in titers were observed against the other tested types, ranging from 66- to 114-fold. These results encouraged us to examine the sera from L2-immunized mice.
Table 1.
Comparison of neutralization titers obtained with the L2-based assay versus those obtained with the L1-based assaya
| Parameter | Value for virus |
||||
|---|---|---|---|---|---|
| HPV16 | HPV31 | HPV33 | HPV45 | HPV58 | |
| Neutralization titer | |||||
| Nonimmune | <50 | <50 | <50 | <50 | <50 |
| Rabbit anti-L2 L1 assay | 12,150 | 1,350 | 4,050 | 12,150 | 4,050 |
| Rabbit anti-L2 L2 assay | 1.5 × 106 | 1.2 × 105 | 4.6 × 105 | 9.0 × 105 | 2.7 × 105 |
| Fold increase | 120 | 85 | 114 | 74 | 66 |
The L1-based neutralization assay was compared to the new L2-based neutralization assay using a rabbit polyclonal serum raised against L2 (11-88×5) as a sample immune serum. Serum from a nonimmune rabbit was used as a control. The fold increase in sensitivity between the two assays is indicated.
Given the higher titers of the L2 (11-88×5)-immune rabbit serum in the L2-based neutralization assay, we examined sera from mice that had been immunized with the L2 (11-88×5) antigen and shown to be protected against experimental cervicovaginal challenge with HPV16, -31, -45, or -58 pseudovirus (11). Using pools of sera obtained from 7 groups of animals (5 mice/group) immunized with L2 (11-88×5), in vitro neutralizing titers were measured by the L2-based assay. This assay gave positive results with HPV16, -45, and -58 for all 7 pools and positive results with HPV31 and -33 for 6 of the 7 pools (Table 2, L2-based assay). The titers for the heterologous HPV types ranged from 2 × 102 to 2.7 × 104, with titers of >103 for 19 of the 26 positives.
Table 2.
In vitro neutralization titers for sera from immune animalsa
| Assay and serum pool | Neutralization titer for virus |
||||
|---|---|---|---|---|---|
| HPV16 | HPV31 | HPV33 | HPV45 | HPV58 | |
| L2-based assay | |||||
| PBS, pool 1 | <50 | <50 | <50 | <50 | <50 |
| PBS, pool 2 | <50 | <50 | <50 | <50 | <50 |
| HPV16 VLP, pool 1 | 172,080 | <50 | <50 | <50 | <50 |
| HPV16 VLP, pool 2 | 306,550 | <50 | <50 | <50 | <50 |
| L2 11-88×5, pool 1 | 7,500 | 50 | 4,315 | 2,780 | 5,370 |
| L2 11-88×5, pool 2 | 13,760 | 765 | 4,130 | 2,640 | 4,025 |
| L2 11-88×5, pool 3 | 4,095 | <50 | <50 | 365 | 700 |
| L2 11-88×5, pool 4 | 10,730 | 7,865 | 27,145 | 7,805 | 13,820 |
| L2 11-88×5, pool 5 | 20,900 | 2,600 | 1,605 | 5,890 | 3,395 |
| L2 11-88×5, pool 6 | 27,630 | 480 | 210 | 5,005 | 10,940 |
| L2 11-88×5, pool 7 | 3,420 | 190 | 22,385 | 1,375 | 1,140 |
| L1-based assay | |||||
| PBS, pool 1 | <50 | <50 | <50 | <50 | <50 |
| PBS, pool 2 | <50 | <50 | <50 | <50 | <50 |
| HPV16 VLP, pool 1 | 7,600 | <50 | <50 | <50 | <50 |
| HPV16 VLP, pool 2 | 60,605 | <50 | <50 | <50 | <50 |
| L2 11-88×5, pool 1 | <50 | <50 | <50 | <50 | <50 |
| L2 11-88×5, pool 2 | <50 | <50 | <50 | <50 | <50 |
| L2 11-88×5, pool 3 | <50 | <50 | <50 | <50 | <50 |
| L2 11-88×5, pool 4 | <50 | <50 | 50 | 50 | <50 |
| L2 11-88×5, pool 5 | <50 | <50 | <50 | 50 | <50 |
| L2 11-88×5, pool 6 | <50 | <50 | 50 | <50 | <50 |
| L2 11-88×5, pool 7 | <50 | <50 | 150 | 50 | <50 |
| P value, 1-7 (L2-based vs L1-based assay) | 0.001 | 0.011 | 0.012 | 0.002 | 0.001 |
Cross-neutralization titers obtained with “L1-based” and “L2-based” assays. Sera were obtained from mice that had been immunized with L2 (11-88×5) and shown to be immune to in vivo challenge with HPV16, -31, -45, or -58. Sera from 5 animals that underwent identical treatment were pooled and assessed for in vitro neutralization with the newly described L2-based neutralization assay or the “L1-based” assay. As specificity controls, pooled sera from HPV16 VLP-immunized animals were also assayed. The assay was performed in duplicate, and the average titer is shown. Triplicate experiments were not possible due to limited sample volume. The bottom row indicates the P value of the difference between the two groups of 7 pools for each virus type. Titers that were found to be <50 were assigned a value of 25 for statistical analysis. The P value is based on the Mann-Whitney test with Gaussian approximation.
In contrast, the L2-based assay gave negative cross-neutralization results with pooled sera of mice immunized with HPV16 VLPs, confirming the specificity of the cross-neutralization observed with the anti-L2 sera in the L2-based assay. Furthermore, the L2-based assay was much more sensitive than the L1-based assay for detecting the L2 cross-neutralizing antibodies, since most of the pooled sera from the L2 (11-88×5)-immunized mice were negative for the heterologous HPV types in the L1-based assay, and the titers of those that were positive never exceeded 150 (Table 2, L1-based assay). The L2-based assay produced higher titers for sera from animals immunized with HPV16 VLPs, but the sensitivity was increased by 1 order of magnitude, compared with an increase of at least 2 orders of magnitude for antibodies to L2.
It is not surprising that L1 neutralizing antibodies are also efficiently detected in the new assay. Although previous in vitro studies have shown that high levels of neutralizing anti-L1 sera prevent L2 epitope exposure (10), these sera are also able to completely neutralize furin-precleaved pseudovirus (unpublished data). This additional mechanism of neutralization is consistent with the well-described ability of anti-L1 antibodies to prevent in vitro infection after cell surface binding (7). Additionally, in vivo analysis revealed that neutralization at lower anti-L1 antibody concentrations occurred by a mechanism that permitted BM binding and L2 epitope exposure but prevented stable association with the keratinocytes (11).
Passive transfer of L2 (11-88×5) antisera: in vivo and in vitro correlation.
To further validate the L2-based neutralization assay, we assessed the correlation between the in vitro neutralizing titers of passively transferred L2 antibodies in the circulation of mice at the time of cervicovaginal challenge and the protection of the mice from cervicovaginal infection. The mice were given an intraperitoneal injection of a dilution series of the rabbit polyclonal serum raised against L2 (11-88×5) and were challenged 24 h later with HPV16 pseudovirions.
In vivo protection from infection was observed at the two lowest dilutions of immune rabbit serum (100 μl and 20 μl) but lost at the two highest dilutions (2 μl and 0.2 μl) (Fig. 4A). When in vitro neutralization titers were determined for the individual mice with sera taken at the time of challenge, all animals that had been protected from in vivo challenge possessed detectable neutralization titers in the L2-based assay (Table 3). The average in vitro neutralization titers for these animals is shown in Table 3. One striking example of the generally good correlation between the two assays is that the high outlier seen in the in vivo experiment at the 20-μl dose had an exceptionally low neutralization titer measured in vitro (Table 3, mouse 4). The correlation between the in vitro titers and the in vivo inhibition, which is shown in Fig. 4B, had a Spearman coefficient correlation of 0.8376 with a two-tailed P value of <0.0001.
Fig 4.
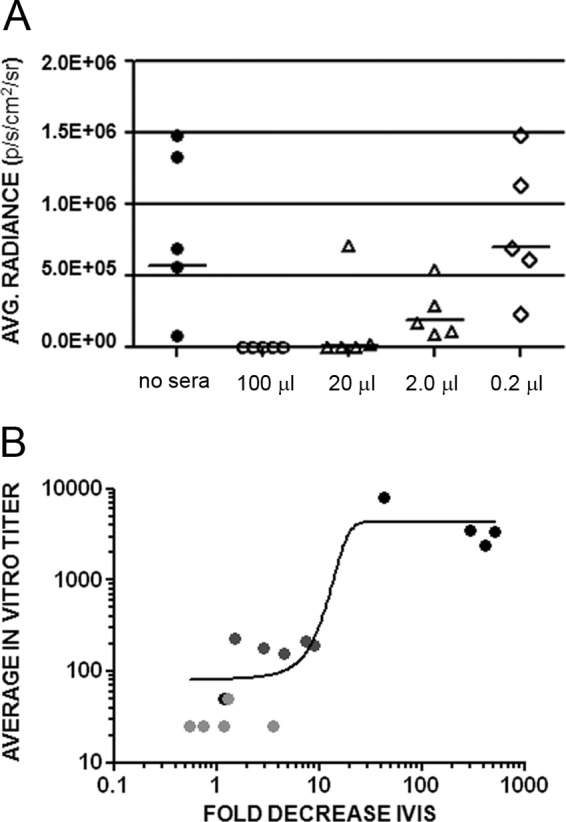
In vivo protection and correlation with in vitro neutralization titers. (A) In vivo infection of animals that were passively transferred with the indicated volume of rabbit anti-L2 (11–88×5) serum was compared to control HPV16 infection (no sera). The volume of serum transferred per animal is indicated. All sera were diluted to a total volume of 100 μl/animal. Each symbol represents an individual animal. (B) The correlation of these values with the in vitro neutralization titers (Table 3) obtained for the circulating antibodies from these animals obtained immediately prior to challenge. The fold decrease in the in vivo infection is indicated on the horizontal axis. The average in vitro titer is indicated on the vertical axis. Each circle represents an individual animal. The black circles represent the 20-μl dose, dark gray represents the 2-μl dose, and light gray represent the 0.2-μl dose.
Table 3.
In vitro neutralization titers of sera obtained from animals that had passive transfer of immune seruma
| Serum (immunization dose [μl]) | In vitroneutralization titer | Ivis value | % inh. |
|---|---|---|---|
| Preimmune 1 | <50 | 1.3 × 106 | |
| Preimmune 2 | <50 | 1.5 × 106 | |
| Mouse 1 (20) | 2,419 | 2.0 × 103 | 100 |
| Mouse 2 (20) | 3,497 | 2.8 × 103 | 100 |
| Mouse 3 (20) | 3,350 | 1.6 × 103 | 100 |
| Mouse 4 (20) | 50 | 7.1 × 105 | 14 |
| Mouse 5 (20) | 8,100 | 1.9 × 104 | 98 |
| Mouse 6 (2.0) | 211 | 1.1 × 105 | 87 |
| Mouse 7 (2.0) | 154 | 1.8 × 105 | 82 |
| Mouse 8 (2.0) | 190 | 9.3 × 104 | 89 |
| Mouse 9 (2.0) | 183 | 2.9 × 105 | 65 |
| Mouse 10 (2.0) | 228 | 5.4 × 105 | 35 |
| Mouse 11 (0.2) | <50 | 2.3 × 105 | 70 |
| Mouse 12 (0.2) | 50 | 6.4 × 105 | 23 |
| Mouse 13 (0.2) | <50 | 1.1 × 106 | −33 |
| Mouse 14 (0.2) | <50 | 1.5 × 106 | −80 |
| Mouse 15 (0.2) | <50 | 7.0 × 105 | 16 |
Comparison of in vitro neutralization titers and in vivo protection. The sera from the experiment shown in Fig. 4A (passive transfer of rabbit immune serum) were used to obtain in vitro neutralization titers with the L2-based assay. The assay was performed in duplicate, and the average titer is shown. The IVIS value indicates the average radiance value indicating in vivo infection from Fig. 4A. Percent inhibition (% inh.) is determined based on the average in vivo infection for the five animals shown in Fig. 4A (8.3 × 105, average radiance). A negative value for percent inhibition indicates a value over the average radiance of the control conditions.
DISCUSSION
In this study, we have developed an in vitro neutralization assay that appears to be at least 100-fold more sensitive for the detection of infection-inhibiting L2 antibodies than the standard neutralization assay, which was developed to measure neutralizing antibodies elicited by L1 VLP vaccines. It is likely that this new assay will be critical for immunogenicity analyses of the anticipated early-phase clinical trials of L2-based vaccines. Since antibody responses to subunit vaccines, e.g., L1 VLPs, are often quantitatively similar in mice and humans, it would be difficult to move forward with clinical testing of an L2 vaccine that generated low or undetectable in vitro neutralizing activity in preclinical studies, as has generally been found using the in vitro L1-based assay. It should also be useful in preclinical studies to reevaluate the potential of some of the many published and unpublished L2 candidates that might have been abandoned because they generated low or no neutralizing antibodies, as measured in the standard L1-based assay.
The rationale for development of the L2-based assay was based on the recognition that passive transfer of sera from animals vaccinated with L2-based immunogens conferred protection against genital HPV challenge, although the sera lacked detectable neutralizing activity in the standard L1-based in vitro neutralization assay. The passive transfer results are encouraging for L2 immunogens because they indicate that protective antibodies are induced. However, such in vivo assays are too cumbersome for routine use in the immunogenicity analyses of a clinical trial.
Development of the L2-based assay was enabled by the new understanding of the early in vivo steps involved in HPV infection, especially those that occur on the basement membrane and lead to exposure of the L2 cross-neutralization epitopes. The assay was based on the hypothesis that generating a process of in vitro infection that more closely mimics the model of in vivo infection would increase the sensitivity with which L2 neutralizing antibodies can be detected. The fact that this hypothesis was correct provides strong support for the broad outlines of the model, particularly as it relates to the exposure of the L2 cross-neutralization epitopes (21).
The passive transfer experiments indicate a correlation for the rabbit serum between in vivo protection and the in vitro titer obtained with the L2-based assay. Complete in vivo protection (98% to 100%, with the exception of one outlier) was observed following transfer of 20 μl of serum. In vitro titers in the range of 2,419 to 8,100 were obtained for the protected animals. Transfer of 2 μl of serum resulted in intermediate in vivo protection (35% to 89%) and in vitro titers from 154 to 228. In vitro titers were below the limit of detection for 4/5 animals at the 0.2-μl dose, whereas 3 animals were partially protected in vivo (16, 23, and 70% protection). Therefore, the in vivo assay may still be slightly more sensitive when considering serum concentrations that offer a lower level of protection. However, the in vitro assay is not subject to the intrinsic variability associated with animal experimentation.
However, it should also be noted that the pools of L2 (11-88×5) immune sera demonstrated rather large variations in titers against specific HPV types, and the titers measured for a specific pool against one type did not predict the titers measured against other types (Table 2, L2-based assay). For instance, pool 7 had the lowest titer against the homologous type HPV16 (3,420) but had an exceptionally high titer against heterologous type HPV33 (22,385). Pool 3 was exceptional in that it had no measurable titer against HPV31 and -33 in the new assay, despite a titer of over 4,000 against HPV16. In contrast, pool 4 had rather consistent titers against all types tested, in the 8,000 to 27,000 range. In future studies, it will be important to compare L2 candidate vaccines for consistency in the induction of neutralizing antibodies among individuals and across types.
A recent mouse study identified substantial differences in the relative sensitivities of the in vivo genital HPV challenge assay and the standard L1-based in vitro assay as measures of protective L1 antibodies (23). These authors demonstrated that the in vivo detection of protection was slightly more sensitive (30-fold) than the in vitro assay for a murine L1-specific monoclonal antibody. However, examination of a polyclonal murine anti-Gardasil serum indicated that the in vivo assay was 500-fold more sensitive. It is possible that multiple mechanisms of neutralization are at play in the polyclonal response in vivo and that these are not equally measurable in the in vitro L1 neutralization assay. In support of this conjecture, we have previously shown that passively transferred anti-L1 serum could neutralize by different mechanisms when present at low concentrations versus high concentrations (11). In contrast, passively transferred anti-L2 serum neutralized by the same mechanism at both high and low concentrations, which might provide an explanation for our results showing a closer alignment of in vivo and in vitro assays.
In future work, it will also be important to assess the degree to which the species source of the serum may affect the in vivo response and its correspondence to in vitro titers. In the current study, an immune rabbit serum was used for the passive transfer experiments. There may exist some species-specific differences in transudation of transferred serum or Fc receptor-mediated mechanisms of neutralization.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
We gratefully acknowledge the gifts of the 804G cell line from Jonathan Jones, the FD11 cell line from Steven Leppla, and the Δfurin cell line from David Fitzgerald.
D. R. Lowy and J. T. Schiller are inventors for intellectual property owned by the U.S. government for the L2 vaccine.
Footnotes
Published ahead of print 16 May 2012
REFERENCES
- 1. Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78: 751– 757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 119: 445– 462 [DOI] [PubMed] [Google Scholar]
- 3. Buck CB, Thompson CD. 2007. Production of papillomavirus-based gene transfer vectors. Curr. Protoc. Cell Biol. Chapter 26: Unit 26.1 [DOI] [PubMed] [Google Scholar]
- 4. Caldeira Jdo C, et al. 2010. Immunogenic display of diverse peptides, including a broadly cross-type neutralizing human papillomavirus L2 epitope, on virus-like particles of the RNA bacteriophage PP7. Vaccine 28: 4384– 4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chandrachud LM, et al. 1995. Vaccination of cattle with the N-terminus of L2 is necessary and sufficient for preventing infection by bovine papillomavirus-4. Virology 211: 204– 208 [DOI] [PubMed] [Google Scholar]
- 6. Chiron MF, Fryling CM, FitzGerald D. 1997. Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J. Biol. Chem. 272: 31707– 31711 [DOI] [PubMed] [Google Scholar]
- 7. Christensen ND, Cladel NM, Reed CA. 1995. Postattachment neutralization of papillomaviruses by monoclonal and polyclonal antibodies. Virology 207: 136– 142 [DOI] [PubMed] [Google Scholar]
- 8. Cuburu N, et al. 2009. Sublingual immunization with nonreplicating antigens induces antibody-forming cells and cytotoxic T cells in The female genital tract mucosa and protects against genital papillomavirus infection. J. Immunol. 183: 7851– 7859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Culp TD, Budgeon LR, Marinkovich MP, Meneguzzi G, Christensen ND. 2006. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 80: 8940– 8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Day PM, Gambhira R, Roden RB, Lowy DR, Schiller JT. 2008. Mechanisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J. Virol. 82: 4638– 4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Day PM, et al. 2010. In vivo mechanisms of vaccine-induced protection against HPV infection. Cell Host Microbe 8: 260– 270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 82: 12565– 12568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Day PM, et al. 2007. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J. Virol. 81: 8784– 8792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Embers ME, Budgeon LR, Pickel M, Christensen ND. 2002. Protective immunity to rabbit oral and cutaneous papillomaviruses by immunization with short peptides of L2, the minor capsid protein. J. Virol. 76: 9798– 9805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gambhira R, et al. 2007. Protection of rabbits against challenge with rabbit papillomaviruses by immunization with the N terminus of human papillomavirus type 16 minor capsid antigen L2. J. Virol. 81: 11585– 11592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gambhira R, et al. 2007. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J. Virol. 81: 13927– 13931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 75: 1565– 1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. 1995. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 63: 82– 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jagu S, et al. 2009. Concatenated multitype L2 fusion proteins as candidate prophylactic pan-human papillomavirus vaccines. J. Natl. Cancer Inst. 101: 782– 792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson KM, et al. 2009. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 83: 2067– 2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kines RC, Lowy TCDR, Schiller JT, Day PM. 2009. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. U. S. A. 106: 20458– 20463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langhofer M, Hopkinson SB, Jones JC. 1993. The matrix secreted by 804G cells contains laminin-related components that participate in hemidesmosome assembly in vitro. J. Cell Sci. 105 (Pt 3): 753– 764 [DOI] [PubMed] [Google Scholar]
- 23. Longet S, Schiller JT, Bobst M, Jichlinski P, Nardelli-Haefliger D. 2011. A murine genital-challenge model is a sensitive measure of protective antibodies against human papillomavirus infection. J. Virol. 85: 13253– 13259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lowy DR, Schiller JT. 2006. Prophylactic human papillomavirus vaccines. J. Clin. Invest. 116: 1167– 1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lugemwa FN, Esko JD. 1991. Estradiol beta-D-xyloside, an efficient primer for heparan sulfate biosynthesis. J. Biol. Chem. 266: 6674– 6677 [PubMed] [Google Scholar]
- 26. Martin KJ, et al. 1998. Down-regulation of laminin-5 in breast carcinoma cells. Mol. Med. 4: 602– 613 [PMC free article] [PubMed] [Google Scholar]
- 27. Paavonen J, et al. 2009. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 374: 301– 314 [DOI] [PubMed] [Google Scholar]
- 28. Pastrana DV, et al. 2004. Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology 321: 205– 216 [DOI] [PubMed] [Google Scholar]
- 29. Pastrana DV, et al. 2005. Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2. Virology 337: 365– 372 [DOI] [PubMed] [Google Scholar]
- 30. Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. U. S. A. 103: 1522– 1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riddelle KS, Green KJ, Jones JC. 1991. Formation of hemidesmosomes in vitro by a transformed rat bladder cell line. J. Cell Biol. 112: 159– 168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roberts JN, et al. 2007. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat. Med. 13: 857– 861 [DOI] [PubMed] [Google Scholar]
- 33. Roden RB, et al. 1996. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 70: 5875– 5883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roden RB, et al. 2000. Minor capsid protein of human genital papillomaviruses contains subdominant, cross-neutralizing epitopes. Virology 270: 254– 257 [DOI] [PubMed] [Google Scholar]
- 35. Schellenbacher C, Roden R, Kirnbauer R. 2009. Chimeric L1-L2 virus-like particles as potential broad-spectrum human papillomavirus vaccines. J. Virol. 83: 10085– 10095 [DOI] [PMC free article] [PubMed] [Google Scholar]