

Abstract
Recent findings in molecular biology implicate the involvement of proprotein convertase subtilisin/kexin type 9 (PCSK9) in low-density lipoprotein receptor (LDLR) protein regulation. The cholesterol-lowering potential of anti-PCSK9 antisense oligonucleotides (AONs) modified with bridged nucleic acids (BNA-AONs) including 2′,4′-BNA (also called as locked nucleic acid (LNA)) and 2′,4′-BNANC chemistries were demonstrated both in vitro and in vivo. An in vitro transfection study revealed that all of the BNA-AONs induce dose-dependent reductions in PCSK9 messenger RNA (mRNA) levels concomitantly with increases in LDLR protein levels. BNA-AONs were administered to atherogenic diet-fed C57BL/6J mice twice weekly for 6 weeks; 2′,4′-BNA-AON that targeted murine PCSK9 induced a dose-dependent reduction in hepatic PCSK9 mRNA and LDL cholesterol (LDL-C); the 43% reduction of serum LDL-C was achieved at a dose of 20 mg/kg/injection with only moderate increases in toxicological indicators. In addition, the serum high-density lipoprotein cholesterol (HDL-C) levels increased. These results support antisense inhibition of PCSK9 as a potential therapeutic approach. When compared with 2′,4′-BNA-AON, 2′,4′-BNANC-AON showed an earlier LDL-C–lowering effect and was more tolerable in mice. Our results validate the optimization of 2′,4′-BNANC-based anti-PCSK9 antisense molecules to produce a promising therapeutic agent for the treatment of hypercholesterolemia.
Keywords: antisense, BNA, hypercholesterolemia, LNA, PCSK9
Introduction
Statins are lipid-lowering drugs that achieve a strong reduction of serum low-density lipoprotein cholesterol (LDL-C) mainly via the indirect activation of LDL receptor (LDLR)-mediated hepatic uptake of LDL from the blood.1,2 The development of drugs that directly regulate the expression of hepatic LDLR would thus be a compelling strategy to obtain the efficacy of statin-induced LDL-C reduction while compensating for potential weaknesses of statin therapy, such as severe adverse effects (e.g., myopathy). The molecular basis of LDLR regulation as well as cholesterol maintenance has been enthusiastically elucidated,2,3,4,5,6,7 and several causative molecules of hypercholesterolemia relevant to the direct regulation of LDLR function have recently been identified.8,9,10,11 Proprotein convertase subtilisin/kexin type 9 (PCSK9), which was recently identified as the third gene relevant to autosomal dominant hypercholesterolemia,10 is involved in the maintenance of cholesterol balance. A number of human mutations in PCSK9 have been reported. Gain-of-function mutations are associated with autosomal dominant hypercholesterolemia, whereas loss-of-function mutations are relevant to low blood levels of LDL-C.12 Recent findings have suggested the involvement of PCSK9 in LDLR regulation. PCSK9 is synthesized mainly in the liver, small intestine, and kidney as a 72-kDa soluble zymogen that subsequently undergoes autocatalytic cleavage into an active form. The active 63-kDa enzyme in complex with the cleaved prodomain is secreted into the bloodstream. Secreted PCSK9 directly binds to an extracellular part of the LDLR. The LDLR–PCSK9 complex is transported from the cell surface to the endosomal system for digestion. PCSK9 forms a stable complex with LDLR in lysosomes, which disturbs the recycling of LDLR to reduce LDL uptake.4,13,14 PCSK9 would be a pivotal regulator of LDLR and an attractive target for lipid-lowering therapy, although some molecular functions of PCSK9 remain unknown.
To achieve PCSK9 inhibition, several “molecular-targeted” approaches have been attempted. To our knowledge, berberine, an isoquinoline plant alkaloid, is the sole small molecule that achieves suppression of PCSK9 expression at the transcriptional level.15,16,17 An antibody against secreted PCSK9 efficiently reduced the serum LDL-C levels of mice and monkeys.18 Small interfering RNA formulated in a lipidoid nanoparticle can induce liver-specific reduction of PCSK9 messenger RNA (mRNA) and serum total cholesterol levels in wild-type mice.19 These proof-of-concept studies demonstrate the therapeutic promise of PCSK9-targeted therapies. Antisense inhibition of PCSK9 is superior to the aforementioned strategies because antisense oligonucleotide (AON) molecules can deactivate intrahepatic mRNA as well as proteins in the blood; in addition, they target the liver via a simple delivery methodology. Graham et al. showed that 2′-O-methoxyethyl-modified phosphorothioate oligonucleotide (MOE) (100 mg/kg/week) administered for a 6-week period to mice fed a high-fat diet reduced hepatic PCSK9 mRNA and serum LDL-C. However, a large dose of MOE is necessary to obtain sufficient efficacy. More recently, Gupta et al. demonstrated that a reduced amount of 2′,4′-bridged nucleic acid (BNA) (also called as locked nucleic acid (LNA))-modified gapmer efficiently suppresses PCSK9 mRNA and induces an increase in LDLR protein levels both in vitro and in vivo.20 Due to the high-affinity binding of 2′,4′-BNA–modified AON molecules, in many cases, 2′,4′-BNA–modified gapmer shows improved antisense potency in vivo as compared to MOE-based gapmer. However, in some cases, the repeated administration of 2′,4′-BNA–modified gapmer causes hepatotoxicity.21 The development of more potent and less toxic antisense molecules is necessary for clinical usage.22 We have developed a series of 2′,4′-BNAs such as 2′,4′-BNA and 2′,4′-BNANC, which have chemical bridges between the 2′ and 4′ positions of the ribose rings; 2′,4′-BNANC–modified oligonucleotides retain high-affinity binding to RNA and higher nuclease stability than 2′,4′-BNA–modified oligonucleotides.23,24,25 Therefore, 2′,4′-BNANC–modified anti-PCSK9 AONs would be expected to possess distinct cholesterol-lowering potency and toxicological risks in vivo. Actually, 2′,4′-BNANC–modified phosphatase and tensin homolog deleted from chromosome ten (PTEN) inhibitor showed high potency without the onset of hepatotoxicity.26 In this study, we present the effective gene silencing and cholesterol-lowering effects of both 2′,4′-BNA– and 2′,4′-BNANC–based anti-PCSK9 AON. In addition, we showed the toxicological characteristics of 2′,4′-BNA– and 2′,4′-BNANC–based AONs.
Results
Physicochemical properties of a 2′,4′-BNA–modified anti-PCSK9 AON in vitro
The 2′,4′-BNA–modified phosphorothioate oligonucleotides (P900SL) and a conventional phosphorothioate AON (P900S) were designed based on the previously identified potential sequence.27 The control sequence was CR01S, which has the same conventional phosphorothioate backbone as P900S, but does not target any specific genes in mice (Table 1). Upper and lower case letters in the sequences represent 2′,4′-BNA and DNA, respectively. We additionally selected another sequence, a consensus sequence between the mouse and human sequences to dispense with the sequence translation to adapt to human clinical trials. P901SL and P901SNC both possess five gaps, and nine of 20 nucleotides are substituted by 2′,4′-BNA and 2′,4′-BNANC (Table 1, Figure 1). Note that the sequence, length, and composition of AONs have not been fully optimized yet here. The melting temperature (Tm) values of P900S, P900SL, P901S, P901SL, and P901SNC with their target RNA were determined by UV-melting experiments. AONs modified with 2′,4′-BNA and 2′,4′-BNANC showed an excellent target affinity compared to conventional phosphorothioate AONs (Table 1).
Table 1. Properties of modified oligonucleotides used in this study.
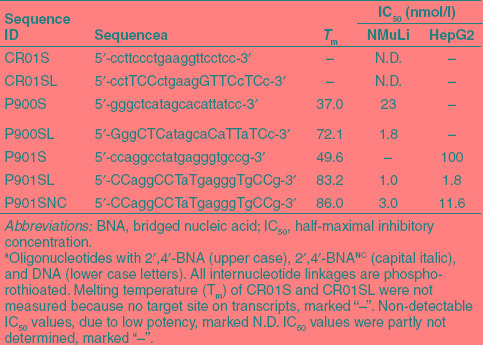
Figure 1.

Structure of bridged nucleic acids (BNA) used in this study. LNA, locked nucleic acid.
In vitro gene silencing properties
We next evaluated in vitro gene silencing properties of AONs by using mice hepatic NMuLi cells (Figure 2a–c) treated with 1–50 nmol/l of P900SL by means of lipofection. Real-time reverse transcription-PCR revealed a dose-dependent reduction of the PCSK9 mRNA level as compared to the GAPDH mRNA level (half-maximal inhibitory concentration (IC50) = 3.0 nmol/l) (Figure 2a). By contrast, no such effective reduction of the mRNA level occurred in P900S-treated cells (IC50 = 41.2 nmol/l) (Figure 2a), and treatment with CR01S and CR01SL did not yield any silencing (Figure 2b). To investigate whether suppression of PCSK9 mRNA by P900SL affects the PCSK9 protein level, we performed western blotting experiments. A dose-dependent reduction of the PCSK9 protein level occurred in P900SL-treated NMuLi cells (Figure 2a). On the other hand, the protein levels of LDLR in P900SL-treated cells were increased in a dose-dependent manner, showing the inverse relationship between PCSK9 and LDLR protein levels. PCSK9 protein was thoroughly inhibited by P900SL at a concentration of ~10 nmol/l. The increase in LDLR protein simultaneously reached a plateau at this concentration. Gene silencing properties of P901S, P901SL, and P901SNC were demonstrated both in murine and human hepatic cell lines. P901SL and P901SNC showed a similar silencing efficacy as P900SL in NMuLi cells (Figure 2a). Both P901SL and P901SNC showed far more efficient mRNA inhibitory activity than P901S in HepG2 cells (Figure 2d). The reduction rate of PCSK9 protein levels also supported this trend (Figure 2e).
Figure 2.

In vitro silencing properties of AONs (CR01S, CR01SL, P900S, P900SL, P901SL, and P901SNC). AONs were transfected into NMuLi cells. (a,b) After a 24-hour incubation, cells were collected and the expression levels of PCSK9 mRNA were determined. Data represent means ± SD. (c) PCSK9 and LDLR proteins were also detected by western blotting. AONs (P901S, P901SL, and P901SNC) were transfected into HepG2 cells. (d) After a 24-hour incubation, HepG2 cells were collected and the expression levels of PCSK9 mRNA were determined. Data represent mean values ± SD. (e) PCSK9 and β-actin proteins were detected by western blotting. AON, antisense oligonucleotide; LDLR, low-density lipoprotein receptor; mRNA, messenger RNA.
The level of intrahepatic AONs in normal chow-fed mice after a single treatment with BNA-modified AONs
Next, we examined whether BNA-modified AONs can also be good inhibitors of PCSK9 in mice. A naked AON (P900SL) was intravenously (i.v.), subcutaneously (s.c.), and intraperitoneally (i.p.) injected into normal chow-fed C57BL/6J mice (Figure 3). After 72 hours, the hepatic PCSK9 mRNA levels were measured, and the half-maximal effective dose (ED50) values were determined. Intravenous administration showed the most efficient hepatic reduction of PCSK9 mRNA (ED50 = 7.5 mg/kg) (Figure 3a). Subcutaneous and intraperitoneal injections resulted in ED50 values of 8.8 and 12.1 mg/kg, respectively (Figure 3c,d). Next, we measured the amount of intact P900SL that accumulated in the liver after i.v. administration by using a previously described ELISA method.28 The intrahepatic content of P900SL was directly proportional to the applied doses, and the saturation of accumulation was not observed even at the highest dose of 70 mg/kg, which is consistent with a previous report29 (Figure 3b).
Figure 3.

Single administration of P900SL to normal-chow fed C57BL/6J mice. (a,b) Hepatic PCSK9 messenger RNA (mRNA) level and P900SL content 72 hours after a single intravenous (i.v.) administration were expressed as a function of dose level. (c,d) Liver PCSK9 mRNA expression level was measured and presented for each dose 72 hours after subcutaneous (s.c.) and intraperitoneal (i.p.) injection, respectively. Data represent mean values ± SD. n = 4–7.
Repeated administration of BNA-modified AONs to atherogenic diet-fed mice
To determine the pharmacological effects of BNA-modified AONs, we monitored the serum cholesterol change upon repeated administration of AONs. Naked AONs (P900S and P900SL) were i.p. injected into atherogenic diet (cholesterol content, 1.25%)-fed C57BL/6J mice twice during 5 days at a dosage of 10 mg/kg per administration. Injections were performed on days 1 and 4. On day 5, livers were harvested to measure gene expression, and blood was collected for the lipid component analysis and toxicity evaluation. As shown in Figure 4a, a significant silencing effect of the hepatic PCSK9 mRNA level was only observed in the P900SL-treated arm. The increase in hepatic LDLR protein was shown by western blotting analysis (Figure 4c). The average serum LDL-C levels were concomitantly reduced by ~30%, although no statistically significant differences were observed (Figure 4b). Collectively, P900SL showed a mild cholesterol-lowering effect in this short-term experiment with the lack of serum elevation of liver transaminases, aspartate aminotransferase and alanine aminotransferase (ALT) (Figure 4d).
Figure 4.

Short-term effects of P900S and P900SL. Atherogenic diet-fed mice received intraperitoneal administration of P900S or P900SL at a dose of 10 mg/kg twice during 4 days. (a) Liver PCSK9 mRNA levels 4 days after the first injection. (b) Relative serum LDL-C levels 4 days after the first injection. (c) Hepatic LDLR protein expression levels were estimated by western blotting. (d) Serum liver transaminases (AST, ALT) were analyzed. Data represent mean values (± SD). *P < 0.05. ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; mRNA, messenger RNA.
To obtain cholesterol-lowering efficacy profiles and toxicological information, we next performed longer-term and multiple-dose experiments in mice. P900SL was i.p. injected twice weekly at a dosage of 2–40 mg/kg/week for 6 weeks into high-cholesterol–loaded mice. After 4 weeks of treatment, blood samples were collected from tail veins, and the precise cholesterol profiles in serum lipoproteins were analyzed by the high-performance liquid chromatography method. Supplementary Table S1 shows the raw serum total cholesterol levels and the cholesterol levels of fractionated lipoprotein components. Figure 5a,b show raw data plots of the high-performance liquid chromatography analysis of serum and the serum LDL-C ratio. Total cholesterol levels did not show a cholesterol-lowering effect or any other specific trends with an increase of doses. However, a precise lipoprotein component analysis revealed that P900SL had induced a dose-dependent LDL-C reduction, and the serum LDL-C reduction of 43% was achieved at a dose of 40 mg/kg/week, whereas serum HDL-C levels increased inversely with LDL-C levels. After a 6-week dosing regimen, livers and blood were harvested to analyze gene and protein expression and toxicities. A dose-dependent decrease of LDL-C, as seen at week 4, was continuously observed. Intrahepatic cholesterol content showed no significant difference between arms (Supplementary Figure S1 and Supplementary Materials and Methods). Hepatic gene expression levels were analyzed by the real-time reverse transcription-PCR method. Hepatic PCSK9 mRNA was efficiently reduced in a dose-dependent manner (Figure 5c). And, the reduction of PCSK9 mRNA did not affect LDLR mRNA expression levels (Figure 5e). However, LDLR protein levels increased concomitantly with decreased LDL-C levels (Figure 5d). Hepatic LDLR protein levels were significantly increased by ~1.5-fold in a dose-dependent manner. Note that we here collected sera and analyzed lipid profiles 2 weeks ahead of sacrifice in order to distribute the stressors associated with treatments, which strongly disturbs serum cholesterol levels.
Figure 5.

Dose-dependent responses of physiological parameters to P900SL dosing. (a) Changes in cholesterol fractions upon administration of P900SL analyzed by HPLC. (b) Ratio of serum LDL-C levels after 4 weeks of treatment to before treatment were arranged in order of doses. (c) Liver PCSK9 mRNA levels were measured at all dose levels after 6 weeks of treatment. (d) Hepatic LDLR protein levels were determined 6 weeks after treatment started. (e) The expression levels of genes regulating lipid homeostasis in liver were analyzed. Data represent mean values ± SD. *P < 0.05. n = 5. HDL, high-density lipoprotein; HPLC, high performance liquid chromatography; LDL, low-density lipoprotein; LDL-C, LDL cholesterol; LDLR, LDL receptor; mRNA, messenger RNA; VLDL, very LDL.
Related gene expression associated with repeated dosing
We further investigated the effect of BNA-modified AONs on molecules related to the cholesterol metabolism (Figure 5e). Knockdown of PCSK9 mRNA induced a reduction in the gene expression of sterol regulatory element-binding protein 2 (SREBP2), a transcriptional factor controlling sterol synthesis; hydroxymethylglutaryl-CoA (HMG-CoA) synthase, an enzyme in the cholesterol synthesis pathway; hepatic triglyceride lipase (LIPC); and ATP-binding cassette transporter (ABCA1), a key enzyme for HDL production. On the other hand, scavenger receptor class B type 1 (SR-B1), a receptor for HDL particles, and cholesterol acyltransferase (ACAT1) showed no significant change, and the expression level of cholesterol 7α-hydroxylase (CYP7A1), the first and rate-limiting enzyme in bile acid synthesis, dose-dependently increased.
Toxicological characteristics of BNA-AONs associated with repeated dosing
Toxicological characteristics of P900SL upon subchronic dosing were estimated by an analysis of blood biochemistry and a histopathological analysis (Figure 6). Because phosphorothioate AONs accumulate mainly in the kidney and liver,30,31,32 hepatotoxicity and renal toxicity are the most likely types of toxicity; 2′,4′-BNA–based AONs have the potential to induce hepatotoxicity.21 However, our experiments showed only moderate increases in liver transaminases (even at higher doses of P900SL) and a dose-dependent mild increase in serum blood urea nitrogen levels (Figure 6a). Histopathologically, no treatment-related changes were seen in the liver or kidneys. As shown in Figure 6b, although the majority of mice treated with saline or therapeutic regimes showed cloudy swelling of hepatocytes, no cellular damage was found in the centrilobular and perilobular hepatocytes, which frequently experience toxicological insults. In addition, granulomas or inflammation were sporadically observed in the saline group and treated groups (Supplementary Table S2).
Figure 6.

Changes in toxicological parameters upon P900SL dosing. (a) Serum liver transaminases (AST and ALT) and BUN levels were measured. Data represent mean values ± SD. *P < 0.05. (b) Representative H&E stain images of liver of saline- and P900SL-treated mice. Bar indicates 100 µm. AST, aspartate aminotransferase; ALT, alanine aminotransferase; BUN, blood urea nitrogen; CV, central vein; H&E, hematoxylin and eosin; PV, portal vein.
Repeated administration of 2′,4′-BNANC–modified AONs to atherogenic diet-fed mice with the least toxicity
To examine the effect of 2′,4′-BNANC–modified AONs, P901SNC as well as P901SL were applied to atherogenic diet-fed mice to investigate their potencies and toxicity profiles. P901SL and P901SNC were i.p. administered twice weekly for 6 weeks at a dose of 1–20 and 1–10 mg/kg/injection, respectively. On week 4, peripheral blood was collected from tail veins, and the cholesterol content of each lipoprotein fraction was measured. P901SL showed no significant reduction of LDL-C levels, whereas P901SNC reduced serum LDL-C levels by ~30% at the highest dose (10 mg/kg/injection), and the reduction occurred in a dose-dependent manner (Figure 7a). We observed a slower onset of the reduction of LDL-C of P901SL 6 weeks after the first treatment (Supplementary Figure S2). Meanwhile, hepatic PCSK9 mRNA expression was suppressed and the hepatic LDLR protein was increased accompanied by AON treatment with relatively large deviations (Figure 7b,c). PCSK9 mRNA was inhibited by P901SL and P901SNC as efficiently as P900SL. LDLR protein responded maximally to 20 mg/kg/injection of P901SL and 10 mg/kg/injection of P901SNC. The increasing rates of these two arms were nearly the same (~1.5-fold increase). The lack of severe hepatotoxicities or kidney toxicities was confirmed by serum chemistry in both the P901SL- and P901SNC-treated arms (Supplementary Table S3). Collectively, 2′,4′-BNANC–based AON, P901SNC, was as safe as the 2′,4′-BNA–based counterpart, P901SL.
Figure 7.

Dose-dependent and chemistry-dependent differences in serum LDL-C levels, hepatic LDLR protein, and PCSK9 mRNA levels after treatment of P901SL and P901SL. (a) Raw values of serum LDL-C were obtained at 4th week of schedule. (b) Hepatic LDLR protein expression levels were measured after the end of the schedule. (c) Liver PCSK9 mRNA levels were measured at all dose levels after 6 weeks of treatment. Data represent mean values ± SD. *P < 0.05, **P < 0.001 (versus a saline-treated control arm). n = 5. LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; mRNA, messenger RNA.
Discussion
We demonstrated in this article that BNA-based antisense therapeutics successfully inhibited hepatic PCSK9 expression, resulting in a strong reduction of the serum LDL-C levels of mice. Our findings support the hypothesis that PCSK9 is a potential therapeutic target for hypercholesterolemia. To the best of our knowledge, this is the first time that we were able to show that BNA-based AONs induced cholesterol-lowering action in hypercholesterolemic mice. P900SL yielded high inhibitory activity in vivo as well as in vitro, and i.v. administration gave the highest peak inhibitory activity of the three representative routes. Nevertheless, i.p. and s.c. injections are alternative routes to i.v. injection, because they yielded adequate dose-responsive suppression of PCSK9 mRNA. In fact, Mipomersen for the treatment of hypercholesterolemia is given s.c. rather than i.p. due to practicality.33,34 The rationality of s.c. injection is also supported by the pharmacokinetic nature and disposition properties of 2′-O-methyl phosphorothioate AON.35 The peak tissue levels of 2'-O-methyl phosphorothioate AON after i.v. injection were higher than after s.c. and i.p. administration, and i.v. injection gives the peak of exon skipping efficiency. However, the bioavailability of 2′-O-methyl phosphorothioate AON was similar among all routes, and durable effects were observed after s.c. administration; therefore, the investigators selected s.c. administration for a preclinical study in mice. On the other hand, the hepatic accumulation of P900SL associated with a single i.v. injection increased directly with dose. This result was totally consistent with a 2′,4′-BNA gapmer targeting apolipoprotein B.29 Further research to elucidate the pharmacokinetics of BNA-AONs is now underway.
The in vivo silencing properties of 2′,4′-BNA–based AON that targets PCSK9 mRNA were previously reported by Gupta et al.20 They achieved the silencing of PCSK9 concomitantly with the upregulation of hepatic LDLR in female mice. However, they did not mention the cholesterol-lowering effect of the 2′,4′-BNA–based AON. We observed a dose-dependent decrease of LDL-C levels and a dose-dependent increase of HDL-C levels (Figure 5). There seems to be no trend in the net changes of serum total cholesterol levels (Supplementary Table S1). The increase in LDLR protein levels reported by Gupta et al.20 were a little higher than those in Figure 5d. The likely explanation is the difference of experimental conditions in which they have used female NMRI mice with standard maintenance diet, whereas we used male C57BL/6J mice with atherogenic diet which contains 1.25% of cholesterol, 0.5% of cholic acid, and so on. Hepatic influx of these ingredients in the diet are known to strongly induce the downregulation of LDLR and cholesterol biosynthetic enzymes such as HMG-CoA reductase by downregulating SREBPs, important transcriptional factors, which results in LDLR reduction.13,36,37 We also observed dose-dependent moderate increases of aspartate aminotransferase, ALT, and blood urea nitrogen levels, whereas histopathological analysis revealed no severe hepatic toxicities (Figure 6, Supplementary Table S2). Collectively, a continuous dosing of P900SL showed potent LDL-C reduction in mice without severe side effects, and this report provides the experimental proof of the cholesterol-lowering effects of BNA-based AONs.
We also achieved a dose-dependent decrease in serum LDL-C levels by using a 2′,4′-BNANC–based AON (P901SNC) (Figure 7a). In this case, serum HDL-C levels and the levels of liver and kidney toxicity indicators were not elevated (Supplementary Table S3). When compared with P901SNC, a delayed decrease of LDL-C levels was observed in the P901SL-treated arm (Supplementary Figure S2). We also showed here that a 2′,4′-BNANC–based AON (P901SNC) has greater potential to inhibit PCSK9 and to reduce serum cholesterol levels with no toxicity than a conventional 2′,4′-BNA–based AON. The high-potency and low-toxicity characteristics of a 2′,4′-BNANC–based AON were previously reported to effectively inhibit PTEN mRNA without elevation of the serum ALT level, whereas elevated serum ALT was observed in the 2′,4′-BNA counterpart-treated arm.26 Thus, we conclude that 2′,4′-BNANC–based AONs can be a promising therapeutic agent for antisense therapy. However, the structure-activity relationship of BNA-based AONs still remains to be elucidated.
As previously reported, LDLR mRNA expression was independent of PCSK9 inhibition.20,27 The coordinate repression of SREBP2, HMG-CoA synthase, and ABCA1 is in agreement with a response to a transient massive influx of cholesterol in the liver (Figure 5e). HMG-CoA synthase and ABCA1 are both known to be regulated by SREBP2, whereas SR-B1 and ACAT1 are atypical factors regulated by SREBP2.3,4 The mRNA expression levels of the latter two factors were unchanged by P900SL, while an increased expression level of CYP7A1 was induced by P900SL. CYP7A1 is the first and rate-limiting enzyme in bile acid synthesis; bile acids are versatile signaling molecules that play critical roles in maintaining the homeostasis of cholesterol, lipid, glucose, and energy.38,39 CYP7A1 deficiency in humans is associated with dyslipidemia, the formation of gallstones, and atherosclerosis.40 A previous report showed that C57BL/6J mice overexpressing CYP7A1 are less susceptible to atherogenic diet-induced elevations of very low-density lipoprotein, intermediate-density lipoprotein, and LDL-C and to reductions of HDL-C and triglyceride and less susceptible to the formation of gallstones and atherosclerosis than their nontransgenic littermates.41,42 Similarly, under atherogenic conditions, we observed a reduction in the serum LDL-C level and induction of HDL-C in P900SL-treated mice; these effects may be partly due to the induction of CYP7A1 to maintain intrahepatic cholesterol homeostasis. Collectively, we observed the strong induction of CYP7A1 mRNA associated with the strong suppression of PCSK9. Further investigation of underlying mechanisms of PCSK9 inhibition in atherogenic diet-fed mice are necessary to extrapolate the cholesterol-lowering efficacy of PCSK9 inhibitors in humans.
In conclusion, we found that the strong inhibition of PCSK9 by BNA-based AONs can greatly reduce serum LDL-C levels in atherogenic diet-fed mice via a novel mechanism of CYP7A1 upregulation. These results indicate that PCSK9 is an excellent drug target for the treatment of hypercholesterolemia. Although a structure of AON has not been fully optimized yet here, the 2′,4′-BNANC–based AON shows higher potency than its 2′,4′-BNA counterpart and like 2′,4′-BNA shows no toxicity under the conditions tested. Meanwhile, a recent report from Gupta et al. demonstrated the superiority of a shorter AON with a smaller number of 2′,4′-BNA modifications over a conventional 20-mer 2′,4′-BNA-gapmer in an antisense potency. They have used a 13-mer AON with five 2′,4′-BNAs and eight gaps that efficiently inhibit the expression of PCSK9 mRNA and protein. We suppose that 2′,4′-BNANC–based AON also has its own unique optimal structure and a further optimization of sequence, length, and composition of 2′,4′-BNANC–based AONs would provide a more reliable 2′,4′-BNANC–based PCSK9 inhibitor.
Materials and Methods
AONs. Two types of modified nucleic acids, 2′,4′-BNA and 2′,4′-BNANC, were partially incorporated into 20-mer phosphorothioated oligodeoxyribonucleotides with various sequences. CR01S and CR01SL were 20-mer phosphorothioate DNA and 2′,4′-BNA–modified AON, respectively, designed not to target any gene in mice. P900S and P900SL were designed to target murine PCSK9 mRNA, and their sequences have been reported previously. P901S, P901SL, and P901SNC target an identical consensus sequence on mice and human PCSK9 mRNA but have different chemistries. The exact sequences of molecules used in this study are presented in Table 1. A large-scale synthesis of 2′,4′-BNANC units was conducted by BNA (Osaka, Japan). All 2′,4′-BNANC monomer units were obtained from BNA. All modified oligonucleotides were provided by Gene Design (http://www.genedesign.co.jp/). The syntheses were conducted by standard phosphoramidite procedures, and products were carefully processed under aseptic conditions and purified. All products were endotoxin-free and contained low levels of residual salts for in vivo usage.
In vitro transfection procedures. For AON transfection experiments, NMuLi cells or HepG2 cells were seeded at 5.0 × 105 cells per well in 6-well plates. AONs were transfected by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's procedures. After a 4-hour transfection, cells were continuously incubated for an additional 20 hours at 37 °C. After incubation, cells were collected and subjected to subsequent analyses.
In vivo pharmacological experiments. All animal procedures were performed in accordance with the guidelines of the Animal Care Ethics Committee of the National Cerebral and Cardiovascular Center Research Institute (Osaka, Japan). All animal studies were approved by an institutional review board. C57BL/6J mice were obtained from CLEA Japan (Tokyo, Japan). All mice were male, and studies were initiated when animals were 6–8 weeks of age. Mice were maintained on a 12-hour light/dark cycle and fed ad libitum. Mice were fed a normal chow (CE-2; CLEA Japan) or an artery-hardening food, F2HFD1 (Oriental Yeast, Tokyo, Japan) for 2 weeks before the first treatment and during treatment. Mice received single or multiple treatments of AONs administered i.v, i.p. or s.c. in the dose range of 1–70 mg/kg/injection. Peripheral blood was collected from a tail vein in BD Microtainers (BD, Franklin Lakes, NJ) for separation of serum. Lipid component analysis of serum was performed by Skylight Biotech (http://www.skylight-biotech.com/). At the time of sacrifice, mice were anesthetized with diethyl ether (Wako, Osaka, Japan). Livers were harvested and snap-frozen until subsequent analysis. Whole blood was collected and subjected to serum separation for subsequent analysis.
mRNA quantification. Total RNA was isolated from cultured cells or mouse liver tissues by using TRIzol Reagent (Invitrogen) according to the manufacturer's procedure. Gene expression was evaluated by a two-step quantitative reverse transcription-PCR method. Reverse-transcription of RNA samples was performed by using a High Capacity cDNA Reverse-Transcription Kit (Applied Biosystems, Foster City, CA), and quantitative PCR was performed by a Fast SYBR Green System or TaqMan Gene Expression Assays (Applied Biosystems). The mRNA levels of target genes were normalized to the GAPDH mRNA level. The following primer sets were used for quantitative PCRs. Ror murine PCSK9; forward: 5′-TCAGTTCTGCACACCTCCAG-3′, reverse: 5′-GGG TAAGGTGCGGTAAGTCC-3′ and forward: 5′-GCTCAACTG TCAAGGGAAGG-3′, reverse: 5′-CGTTGAGGATGCGGCTA TAC-3′. For human PCSK9; forward: 5′-AAGGGAAGGG CACGGTTAG-3′, reverse: 5′-GAGTAGAGGCAGGCATCG TC-3′. For murine GAPDH; forward: 5′-GTGTGAACGGATTT GGCCGT-3′, reverse: 5′-GACAAGCTTCCCATTCTCGG-3′ and for human GAPDH; forward: 5′-GAGTCAACGG ATTTGGTCGT-3′, reverse: 5′-GACAAGCTTCCCGTTCTC AG-3′. For murine LDLR, SREBP2, SR-BI, LIPC, HMGCS2, CYP7A1, ABCA1, and ACAT1, TaqMan Gene Expression Assays were used; assay IDs: Mm00440169_m1, Mm01306297_g1, Mm00450236_m1, Mm01147313_m1, Mm00550050_m1, Mm00484152_m1, Mm01350760_m1, Mm00507463_m1, respectively.
Western blotting analysis. Cultured cells and frozen liver tissues were suspended in lysis buffer (150 mmol/l NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mmol/l Tris, pH 8.0, 20 × Complete Mini protease inhibitor cocktail 1:20 (Roche, Indianapolis, IN)) and homogenized with TissueLyser II (Qiagen, Valencia, CA). Total protein concentrations were measured with a detergent compatible assay kit (Bio-Rad, Hercules, CA). Solutions were subjected to electrophoresis on 16 or 6% Tris-glycine gels (Invitrogen) and transferred to a polyvinylidene difluoride membrane (Bio-Rad). PCSK9 western blotting was performed at room temperature for 1 hour with a primary anti-rabbit PCSK9 antibody (1:200; Abcam, Cambridge, UK). Additional analyses were performed by using anti-LDLR antibody (R&D Systems, Minneapolis, MN) and anti-β actin antibody (Cell Signaling Technology, Danvers, MA). Membranes were washed three times with phosphate-buffered saline containing 0.3% Tween 20. Blots were labeled by using horseradish peroxidase-conjugated secondary antibodies, either goat anti-rabbit or donkey anti-goat antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Chemiluminescent detection was performed by using an ECL plus Western blot detection kit (Amersham Biosciences, Buckinghamshire, UK), and bands were visualized by using an LAS-4000mini image analyzer (Fuji Film, Tokyo, Japan). β-Actin expression levels were used as an internal standard.
The determination of P900SL content in liver
Materials and reagents. The template DNA was a 29-mer DNA (5′-gaatagcgaggataatgtgctatgagccc-3′), which is complementary to P900SL, with biotin at the 3′-end. The ligation probe DNA was a 9-mer DNA (5′-tcgctattc-3′) with phosphate at the 5′-end and digoxigenin at the 3′-end. The template DNA and the ligation probe DNA were purchased from Japan Bio Services (Saitama, Japan). Reacti-Bind NeutrAvidin-coated polystyrene strip plates were purchased from Thermo Fisher Scientific (Waltham, MA) (nunc immobilizer streptavdin F96 white, 436015). The template DNA solution (100 nmol/l) was prepared in hybridization buffer containing 60 mmol/l Na2HPO4 (pH 7.4), 0.9 mol/l NaCl, and 0.24% Tween 20. The ligation probe DNA solution (200 nmol/l) was prepared in 1.5 units/well of T4 DNA ligase (TaKaRa, Shiga, Japan) with 66 mmol/l Tris-HCl (pH 7.6), 6.6 mmol/l MgCl2, 10 mmol/l DTT, and 0.1 mmol/l ATP.
The washing buffer used throughout the assay contained 25 mmol/l Tris-HCl (pH 7.2), 0.15 mol/l NaCl, and 0.1% Tween 20. Anti-digoxigenin-AP antibody (Fab fragments conjugated with alkaline phosphatase) was obtained from Roche Diagnostics. A 1:2,000 dilution of the antibody with 1:10 super block buffer in TBS (Pierce, Rockford, IL) was used in the assay. The alkaline phosphatase luminous substrate was prepared in 250 µmol/l CDP-Star (Roche) with 100 mmol/l Tris-HCl (pH 7.6) and 100 mmol/l NaCl.
Sample preparation. Frozen liver tissue was collected in a 2-ml tube with 1 ml of phosphate-buffered saline and a zirconia ball (ø 5 mm; Irie, Tokyo, Japan) and mechanically homogenized for 2 minutes at 30 oscillations per second by a TissueLyser II apparatus (Qiagen). Total protein concentrations were measured with a detergent compatible assay kit (Bio-Rad) and adjusted to 8 mg/l with phosphate-buffered saline. The assay was performed at the concentration range of 128 pmol/l–400 nmol/l in duplicate. For the standard curve, 10 standard solutions were prepared. To AON-untreated mice, liver homogenates were added to P900SL solutions to prepare 10 standard samples at a range of 128 pmol/l–400 nmol/l.
Assay procedures. The template DNA solution (100 µl) and standard solution (10 µl) or liver homogenates (10 µl) containing the P900SL were added to Reacti-Bind Neutr Avidin-coated polystyrene strip 96-well plates and incubated at 37 °C for 1 hour to allow the binding of biotin to streptavidin-coated wells and hybridization. After hybridization, the plate was washed three times with 200 µl of washing buffer. Then, ligation probe DNA solution (100 µl) was added, and the plate was incubated at room temperature (15 °C) for 3 hours. The plate was then washed three times with the washing buffer. Subsequently, 200 µl of a 1:2,000 dilution of anti-digoxigenin-AP was added, and the plate was incubated at 37 °C for 1 hour. After washing three times with the washing buffer, CDP-Star solution was added to the plate, and finally the luminescence intensity was determined by using a Centro XS3 luminometer (Berthold Technologies, Bad Wildbad, Germany) one second after the addition of CDP-Star. The linear range of 128 pmol/l–400 nmol/l in this ELISA system was determined as r > 0.99.
Serum chemistry and hematoxylin and eosin staining. Serum from blood collected from the inferior vena cava upon sacrifice was subjected to serum chemistry. Assay kits (WAKO) were used to measure serum levels of aspartate aminotransferase, ALT, blood urea nitrogen, and creatinine, which are biomarkers for hepatic and kidney toxicities. Formalin-fixed liver samples (20% formalin; WAKO) were sliced by microtome (Leica Microsystems, Wetzlar, Germany), embedded in Histsec (Merck, Darmstadt, Germany) and stained with Carrazzi's hematoxylin and Tissue-Tek eosin solutions.
Statistical analysis. Pharmacological studies were performed with 5–7 mice per treatment group. A Student's t-test was performed for comparison of two arms. P < 0.05 or P < 0.01 was considered to be of statistical significance.
Acknowledgments
We thank Eiko Shibata, Mai Inoue, Megumu Morimoto, and Manami Sone for their technical support, who are affiliated with National Cerebral and Cardiovascular Center Research Institute. A part of this work was supported by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO) and a research grant from the Ministry of Health, Labor, and Welfare (H23-seisakutansaku-ippan-004). T.Y. thanks the Research Fellowship from the Japan Society for the Promotion of Science (JSPS) for Young Scientists. T.I. is a CEO of BNA Inc., the company that produces 2′,4′-BNANC monomers. The other authors declared no conflict of interest.
Supplementary Material
Comparison of intrahepatic cholesterol levels between control and 20 mg/kg/injection of P900SL-treated arms.
Relation between given dose and serum LDL-C levels of P901SL 6 weeks after treatment started.
Serum raw cholesterol levels in atherogenic diet-fed mice after 4 weeks of P900SL treatment.
Summary of histopathological findings.
Toxicological parameters.
References
- Jones P, Kafonek S, Laurora I., and, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study) Am J Cardiol. 1998;81:582–587. doi: 10.1016/s0002-9149(97)00965-x. [DOI] [PubMed] [Google Scholar]
- Goldstein JL., and, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL., and, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS.et al. (2003Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes Proc Natl Acad Sci USA 10012027–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong WJ, Liu J., and, Jiang JD. Human low-density lipoprotein receptor gene and its regulation. J Mol Med. 2006;84:29–36. doi: 10.1007/s00109-005-0717-6. [DOI] [PubMed] [Google Scholar]
- Issandou M. Pharmacological regulation of low density lipoprotein receptor expression: current status and future developments. Pharmacol Ther. 2006;111:424–433. doi: 10.1016/j.pharmthera.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Zelcer N., and, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M.et al. (2001Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein Science 2921394–1398. [DOI] [PubMed] [Google Scholar]
- Harada-Shiba M, Takagi A, Miyamoto Y, Tsushima M, Ikeda Y, Yokoyama S.et al. (2003Clinical features and genetic analysis of autosomal recessive hypercholesterolemia J Clin Endocrinol Metab 882541–2547. [DOI] [PubMed] [Google Scholar]
- Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M.et al. (2003Mutations in PCSK9 cause autosomal dominant hypercholesterolemia Nat Genet 34154–156. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R., and, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert G, Charlton F, Rye KA., and, Piper DE. Molecular basis of PCSK9 function. Atherosclerosis. 2009;203:1–7. doi: 10.1016/j.atherosclerosis.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Maxwell KN, Soccio RE, Duncan EM, Sehayek E., and, Breslow JL. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res. 2003;44:2109–2119. doi: 10.1194/jlr.M300203-JLR200. [DOI] [PubMed] [Google Scholar]
- Attie AD., and, Seidah NG. Dual regulation of the LDL receptor–some clarity and new questions. Cell Metab. 2005;1:290–292. doi: 10.1016/j.cmet.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Cameron J, Ranheim T, Kulseth MA, Leren TP., and, Berge KE. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis. 2008;201:266–273. doi: 10.1016/j.atherosclerosis.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Kong W, Wei J, Abidi P, Lin M, Inaba S, Li C.et al. (2004Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins Nat Med 101344–1351. [DOI] [PubMed] [Google Scholar]
- Kong WJ, Wei J, Zuo ZY, Wang YM, Song DQ, You XF.et al. (2008Combination of simvastatin with berberine improves the lipid-lowering efficacy Metab Clin Exp 571029–1037. [DOI] [PubMed] [Google Scholar]
- Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W.et al. (2009A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates Proc Natl Acad Sci USA 1069820–9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A.et al. (2008Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates Proc Natl Acad Sci USA 10511915–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Fisker N, Asselin MC, Lindholm M, Rosenbohm C, Ørum H.et al. (2010A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo PLoS ONE 5e10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G.et al. (2007Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals Nucleic Acids Res 35687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Nakatani M, Narukawa K., and, Obika S. Antisense drug discovery and development. Future Med Chem. 2011;3:339–365. doi: 10.4155/fmc.11.2. [DOI] [PubMed] [Google Scholar]
- Rahman SM, Seki S, Obika S, Yoshikawa H, Miyashita K., and, Imanishi T. Design, synthesis, and properties of 2',4'-BNA(NC): a bridged nucleic acid analogue. J Am Chem Soc. 2008;130:4886–4896. doi: 10.1021/ja710342q. [DOI] [PubMed] [Google Scholar]
- Obika S, Rahman SMA, Fujisaka A, Kawada Y, Baba T., and, Imanishi T. Bridged nucleic acids: development, synthesis, and properties. Heterocycles. 2010;81:1347–1392. [Google Scholar]
- Miyashita K, Rahman SMA, Seki S, Obika S., and, Imanishi T. N-Methyl substituted 2′,4′-BNA(NC): a highly nuclease-resistant nucleic acid analogue with high-affinity RNA selective hybridization. Chem Commun. 2007. pp. 3765–3767. [DOI] [PubMed]
- Prakash TP, Siwkowski A, Allerson CR, Migawa MT, Lee S, Gaus HJ.et al. (2010Antisense oligonucleotides containing conformationally constrained 2',4'-(N-methoxy)aminomethylene and 2',4'-aminooxymethylene and 2'-O,4'-C-aminomethylene bridged nucleoside analogues show improved potency in animal models J Med Chem 531636–1650. [DOI] [PubMed] [Google Scholar]
- Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST.et al. (2007Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice J Lipid Res 48763–767. [DOI] [PubMed] [Google Scholar]
- Yu RZ, Baker B, Chappell A, Geary RS, Cheung E., and, Levin AA. Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem. 2002;304:19–25. doi: 10.1006/abio.2002.5576. [DOI] [PubMed] [Google Scholar]
- Straarup EM, Fisker N, Hedtjärn M, Lindholm MW, Rosenbohm C, Aarup V.et al. (2010Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates Nucleic Acids Res 387100–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S, Temsamani J., and, Tang JY. Pharmacokinetics, biodistribution, and stability of oligodeoxynucleotide phosphorothioates in mice. Proc Natl Acad Sci USA. 1991;88:7595–7599. doi: 10.1073/pnas.88.17.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JA, Craig SJ, Bayley D, Christian RA, Geary R., and, Nicklin PL. Pharmacokinetics, metabolism, and elimination of a 20-mer phosphorothioate oligodeoxynucleotide (CGP 69846A) after intravenous and subcutaneous administration. Biochem Pharmacol. 1997;54:657–668. doi: 10.1016/s0006-2952(97)00190-1. [DOI] [PubMed] [Google Scholar]
- Lendvai G, Velikyan I, Bergström M, Estrada S, Laryea D, Välilä M.et al. (2005Biodistribution of 68Ga-labelled phosphodiester, phosphorothioate, and 2'-O-methyl phosphodiester oligonucleotides in normal rats Eur J Pharm Sci 2626–38. [DOI] [PubMed] [Google Scholar]
- Akdim F, Visser ME, Tribble DL, Baker BF, Stroes ES, Yu R.et al. (2010Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia Am J Cardiol 1051413–1419. [DOI] [PubMed] [Google Scholar]
- Akdim F, Stroes ES, Sijbrands EJ, Tribble DL, Trip MD, Jukema JW.et al. (2010Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy J Am Coll Cardiol 551611–1618. [DOI] [PubMed] [Google Scholar]
- Heemskerk H, de Winter C, van Kuik P, Heuvelmans N, Sabatelli P, Rimessi P.et al. (2010Preclinical PK and PD studies on 2'-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model Mol Ther 181210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudling M. Hepatic mRNA levels for the LDL receptor and HMG-CoA reductase show coordinate regulation in vivo. J Lipid Res. 1992;33:493–501. [PubMed] [Google Scholar]
- Dueland S, Drisko J, Graf L, Machleder D, Lusis AJ., and, Davis RA. Effect of dietary cholesterol and taurocholate on cholesterol 7 alpha-hydroxylase and hepatic LDL receptors in inbred mice. J Lipid Res. 1993;34:923–931. [PubMed] [Google Scholar]
- Thomas C, Pellicciari R, Pruzanski M, Auwerx J., and, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK.et al. (2002Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype J Clin Invest 110109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machleder D, Ivandic B, Welch C, Castellani L, Reue K., and, Lusis AJ. Complex genetic control of HDL levels in mice in response to an atherogenic diet. Coordinate regulation of HDL levels and bile acid metabolism. J Clin Invest. 1997;99:1406–1419. doi: 10.1172/JCI119300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake JH, Duong-Polk XT, Taylor JM, Du EZ, Castellani LW, Lusis AJ.et al. (2002Transgenic expression of cholesterol-7-alpha-hydroxylase prevents atherosclerosis in C57BL/6J mice Arterioscler Thromb Vasc Biol 22121–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of intrahepatic cholesterol levels between control and 20 mg/kg/injection of P900SL-treated arms.
Relation between given dose and serum LDL-C levels of P901SL 6 weeks after treatment started.
Serum raw cholesterol levels in atherogenic diet-fed mice after 4 weeks of P900SL treatment.
Summary of histopathological findings.
Toxicological parameters.