

Abstract
Histone deacetylase (HDAC) enzymes posttranslationally modify lysines on histone and nonhistone proteins and play crucial roles in epigenetic regulation and other important cellular processes. HDAC inhibitors (e.g., suberoylanilide hydroxamic acid [SAHA; also known as vorinostat]) are used clinically to treat some cancers and are under investigation for use against many other diseases. Development of new HDAC inhibitors for noncancer indications has the potential to be accelerated by piggybacking onto cancer studies, as several HDAC inhibitors have undergone or are undergoing clinical trials. One such compound, SB939, is a new orally active hydroxamate-based HDAC inhibitor with an improved pharmacokinetic profile compared to that of SAHA. In this study, the in vitro and in vivo antiplasmodial activities of SB939 were investigated. SB939 was found to be a potent inhibitor of the growth of Plasmodium falciparum asexual-stage parasites in vitro (50% inhibitory concentration [IC50], 100 to 200 nM), causing hyperacetylation of parasite histone and nonhistone proteins. In combination with the aspartic protease inhibitor lopinavir, SB939 displayed additive activity. SB939 also potently inhibited the in vitro growth of exoerythrocytic-stage Plasmodium parasites in liver cells (IC50, ∼150 nM), suggesting that inhibitor targeting to multiple malaria parasite life cycle stages may be possible. In an experimental in vivo murine model of cerebral malaria, orally administered SB939 significantly inhibited P. berghei ANKA parasite growth, preventing development of cerebral malaria-like symptoms. These results identify SB939 as a potent new antimalarial HDAC inhibitor and underscore the potential of investigating next-generation anticancer HDAC inhibitors as prospective new drug leads for treatment of malaria.
INTRODUCTION
Widespread resistance of Plasmodium parasites to commonly available antimalarial drugs (23) and the lack of a licensed vaccine have necessitated increased efforts in the discovery and development of new antimalarial agents. Of particular interest are new antimalarial drug candidates which act on novel parasite targets or mechanisms associated with parasite survival or development. Transcriptional control in malaria parasites is complex, and regulatory mechanisms are the subject of intense investigation. Nevertheless, there is increasing evidence that targeting of transcriptional regulation represents a potential new therapeutic approach for malaria. Histone deacetylase (HDAC) enzymes, which are key regulators of transcription and validated therapeutic targets for some types of cancer, are promising new antimalarial drug targets (7).
Histone acetyltransferases (HATs) posttranslationally modify proteins by catalyzing the transfer of an acetyl group from acetyl-coenzyme A (acetyl-CoA) to the ε-nitrogen on the side chain of lysines, while HDACs catalyze the reverse reaction. These enzymes play crucial roles in modulating the acetylation state of histone proteins, contributing to alterations in chromatin structure and transcription (17). Many nonhistone proteins have also been identified as HDAC substrates (15, 22, 30). As a result, protein lysine acetylation is now considered common and is thought to play critical roles in regulating many important cellular processes, including protein stability, protein-protein interactions, protein localization, and DNA-binding properties of proteins (44).
Mammalian HDACs are classified into four classes (29, 31). Class I, II, and IV HDACs share a zinc cofactor catalytic core (27, 59), while class III HDACs use NAD+ to deacetylate their substrates. Five HDAC-encoding genes have been identified in the Plasmodium falciparum genome: one encoding a homologue of class I HDACs (PfHDAC1), two encoding homologues of class II HDACs (PfHDAC2 and -3), and two encoding class III HDAC homologues (PfSir2A and PfSir2B) (25, 28, 36, 61). PfSir2A and PfSir2B are not essential for asexual, blood-stage growth of P. falciparum in vitro. However, both play roles in regulating the expression of subsets of P. falciparum virulence genes, indicating potentially important roles in immune evasion in vivo (25, 28, 61). Although the functional roles of the class I and II PfHDAC homologues have not yet been elucidated, PfHDAC1 is a likely target of antimalarial HDAC inhibitors such as suberoylanilide hydroxamic acid (SAHA) (50).
In eukaryotic cells, interfering with HDAC action by using small-molecule inhibitors results in an accumulation of acetylated histones, alterations to transcription, and various cellular responses, such as apoptosis, differentiation, and changes in cell cycle progression. Three HDAC inhibitors are currently in clinical use for treatment of cancer, including two hydroxamate-based compounds (SAHA [Zolinza; Merck & Co.] and 4SC-201 [resminostat; also known as 4SC]) and a cyclic depsipeptide (FK228 [romidepsin; Gloucester Pharmaceuticals Inc.]). In addition, several HDAC inhibitors are now undergoing clinical trials (40, 42, 63). A number of HDAC inhibitors, particularly hydroxamates, are also potent (50% inhibitory concentrations [IC50s] of <200 nM) and selective (selectivity indexes of >100) inhibitors of P. falciparum parasites in vitro (reviewed in references 5 and 7). Some, including SAHA, also have good ex vivo activity (IC50s of ∼200 to 500 nM) against field isolates of P. falciparum and P. vivax, the second most important human-infecting malaria parasite (41). Limited data are available on HDAC inhibitor action in vivo in mouse malaria models, but one cyclic tetrapeptide (apicidin) and two hydroxamate compounds (suberic bishydroxamate [SBHA] and WR301801) have shown promising inhibitory activity profiles (1, 8, 21, 24). This validates further studies on next-generation HDAC inhibitors for treatment of malaria, especially novel versions with improved pharmacokinetic profiles. The extensive research being undertaken to develop HDAC inhibitors for treating human cancers provides a unique opportunity to piggyback onto these studies for noncancer indications such as malaria.
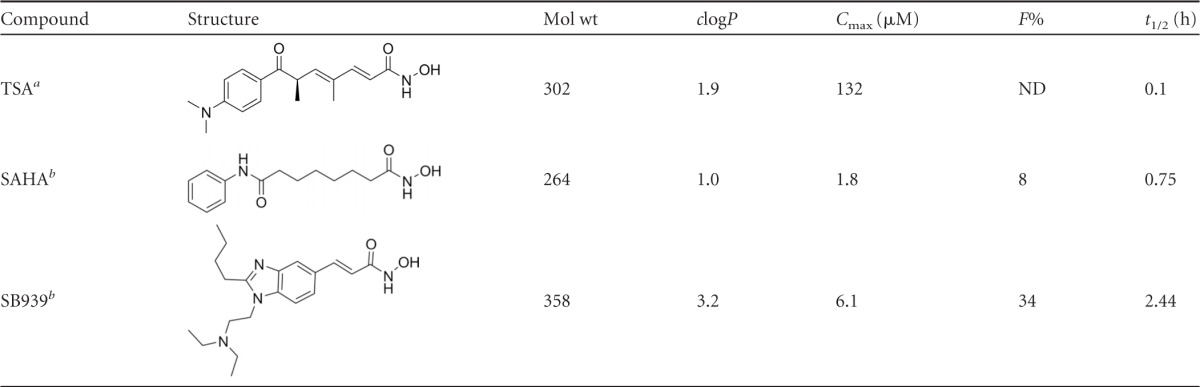
In this study, we tested the in vitro and in vivo antiplasmodial efficacies of a new, orally bioavailable, hydroxamate-based HDAC inhibitor undergoing clinical trials for cancer (35, 47, 52, 65, 67). SB939 (pracinostat; S*BIO), a pan-HDAC inhibitor acting on class I, II, and IV HDACs (47, 65), has a longer in vivo half-life (t1/2 of 2.4 h) than those of other hydroxamate-based HDAC inhibitors, such as SAHA (t1/2 of 0.75 h) (Table 1). Here we present data on the first reported noncancer application of this compound. We show that SB939 is a potent inhibitor of the in vitro growth of asexual P. falciparum parasites in human erythrocytes and of P. berghei exoerythrocytic-stage parasites in human hepatocytes. SB939 hyperacetylates P. falciparum histone and nonhistone proteins, demonstrating its mode of action as an HDAC inhibitor in P. falciparum. In vitro combination studies demonstrated an additive interaction with the antimalarial aspartic protease inhibitor lopinavir. In vivo, orally administered SB939 significantly inhibited parasite growth in a mouse model of infection, protecting against experimental cerebral malaria (ECM)-like symptoms.
Table 1.
Structures and properties of different HDAC inhibitors
MATERIALS AND METHODS
Compounds.
SAHA (Sigma) and (E)-3-{2-butyl-1-[2-(diethylamino)ethyl]benzimidazol-5-yl}prop-2-ene hydroxamic acid (SB939; Selleck Chemicals) were prepared as 10 mM stock solutions in 100% dimethyl sulfoxide (DMSO) for in vitro assays and in 50% DMSO–50% phosphate-buffered saline (PBS) immediately prior to use for in vivo mouse malaria studies. Chloroquine (chloroquine diphosphate salt; Sigma) was prepared in Milli-Q ultrapure water for in vitro and in vivo studies.
Culture of P. falciparum parasites.
P. falciparum 3D7 and Dd2 parasites were cultured in O+ human erythrocytes in RPMI 1640 medium (Life Technologies) supplemented with 10% heat-inactivated human serum and 50 mg/liter hypoxanthine as previously described (6).
P. falciparum growth inhibition assays.
Assessment of antimalarial activity against P. falciparum was carried out with a 72-h isotopic microtest as previously described (6, 24), using synchronous ring-stage-infected erythrocytes (0.5% parasitemia and 2.5% hematocrit). [3H]hypoxanthine incorporation was determined by harvesting cells onto MicroBeta 1450 filter mats (Wallac) and counting using a MicroBeta 1450 liquid scintillation counter. The percent inhibition of growth compared to that of matched DMSO (0.5%) controls was determined for at least three independent experiments, each carried out in triplicate. IC50s were calculated using linear interpolation of inhibition curves (33). A nonparametric two-tailed t test (Mann-Whitney) was used for statistical analysis.
Isobolograms.
Antimalarial drug combinations were assessed by isobologram analysis as previously described (58). All assays were performed in 96-well microtiter plates. Each well contained 100 μl of cell culture and 100 μl of each drug dilution or control. Plates were then labeled with [3H]hypoxanthine (0.5 μCi/well; Perkin Elmer). After 48 h, cells were harvested on MicroBeta 1450 filter mats (Wallac) and counted using a MicroBeta 1450 liquid scintillation counter. Three independent experiments were performed. Isobolograms were constructed using data from all experiments. Briefly, concentrations of each drug that alone or in combination resulted in 50% growth inhibition were plotted as fractional inhibitory concentrations (18). Using the SAAM II program (SAAM Institute, Seattle, WA), the following standard hyperbolic function (12) was fitted to the data: Yi = 1 − {XI/[XI + e−I × (1 − Xi)]}, where Yi is the IC50 of drug A combined with drug B, Xi is the IC50 of drug B combined with drug A, and I is the interaction value. Positive values for I indicate synergism, and negative values indicate antagonism; addition occurs when I is zero. The significance of the difference of I from zero (P values of >0.01 indicate an additive interaction) was assessed using Student's t test.
Mammalian cytotoxicity assays.
Human umbilical vein endothelial cells (HUVEC), proximal tubule epithelial cells (PTC), and neonatal foreskin fibroblast (NFF) cells were cultured in RPMI medium (Life Technologies) supplemented with 10% heat-inactivated fetal calf serum (CSL Biosciences) and 1% streptomycin (Life Technologies). Five thousand cells were seeded into wells of a 96-well tissue culture plate and cultured for 24 h at 37°C and 5% CO2. Following adhesion, HUVEC and PTC were starved in serum-free RPMI medium for a further 24 h. All three cell lines were treated with SB939, SAHA, and chloroquine in a 3-fold dilution series beginning at 100 μM. After 72 to 96 h, medium was removed from each, and 50 μl of staining solution was added, i.e., MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; 1 mg/ml) for HUVEC and PTC and sulfo-rhodamine B (0.4%) (Sigma) for NFF cells. After staining for 1 h, 100 μl isopropanol was added and plates were read at 570 nm in an enzyme-linked immunosorbent assay (ELISA) microplate reader.
Liver-stage assays.
P. berghei ANKA sporozoites dissected from the salivary glands of Anopheles stephensi mosquitoes were applied to HepG2 cells grown on coverslips (56). The medium was exchanged for fresh medium containing the test compound or the control antimalarial drug primaquine after 1 h and then replaced again at 24 and 48 h. Infected cells were incubated at 37°C and 8% CO2 for 53 h, fixed in 4% paraformaldehyde-PBS for 20 min, permeabilized using cold methanol, and labeled with a 1:200 dilution of mouse polyclonal antibody raised against whole P. berghei (46) and a 1:200 dilution of anti-P. falciparum ACP antibody (64) in 3% bovine serum albumin (BSA) for 1 h, followed by secondary labeling with Alexa Fluor antibodies. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). Parasites were imaged and counted using a Leica confocal microscope. Results are for three independent experiments and give the percent inhibition of growth compared to that of matched vehicle (0.01% DMSO)-treated parasites. IC50s were determined using the four-parameter model (Sigmaplot).
PfHDAC1 homology model and ligand docking.
The PfHADC1 amino acid sequence (http://www.uniprot.org; accession number Q7K6A1) was used in a BLAST (2) search of the Protein Data Bank (PDB) (14) to identify crystal structures of proteins with similar sequences, giving human HDAC2 (PDB accession no. 3max; resolution of 2.05 Å; 63% identity) (16) and HDAC8 (PDB accession no. 1t64; resolution of 1.90 Å; 41% identity). These template structures and PfHDAC1 sequences were aligned using ClustalW (60) and manually optimized by removing the N-terminal loop 1 of HDAC8 and PfHDAC1 loop 2. Residues Gly93 to Thr96 were not templated to either structure. Modeller (53) was used to build 15 all-atom homology models of PfHDAC1, including the active site Zn atom and the ligand trichostatin A (TSA). The homology model with the lowest DOPE score (57) was selected and structurally assessed via a Ramachandran plot (91% in favored regions) and via Verify_3D (95% of residues scored >0.2) (26). Sybyl-X 1.3 (Tripos International, St. Louis, MO) was used to superimpose crystal structures with the PfHDAC1 homology model; minor side chain geometries and protonation states near the active site Zn atom were corrected manually. This homology model was then used in GOLD for ligand docking studies.
The ligands SB939, TSA, and SAHA were built in Sybyl-X 1.3 and minimized using the MFF94s force field (MMFF94 charges; dielectric constant of 80; distance dependent) to a terminal gradient of 0.05 kcal/mol-A. GOLD was then used to perform 15 dockings of the ligands in the homology model, using the zinc-bound hydroxamate group of TSA (CONHOH atoms) as a scaffold constraint (10 kcal/mol). Side chains of Thr96, Asp97, Ser146, Arg268, and Leu269 were allowed to rotate freely during the docking experiment, and conformations were scored using Goldscore and rescored using Chemscore (48).
Protein hyperacetylation assays.
Protein hyperacetylation assays were carried out by incubating synchronous trophozoite-stage P. falciparum 3D7 parasites (3 to 5% parasitemia; 5% hematocrit) in six-well tissue culture plates for 3 h under standard in vitro culture conditions with different concentrations of compound or vehicle (0.05% DMSO). Infected erythrocytes were lysed with 0.15% saponin (Sigma), and parasite pellets were obtained by centrifugation. Pellets were washed extensively in cold PBS, pH 7.4, to remove hemoglobin and then resuspended in SDS-PAGE loading dye, heat denatured at 96°C for 3 min, and separated via SDS-PAGE. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Roche), and Western blotting was carried out using Odyssey blocking buffer (Li-Cor Biosciences) according to the manufacturer's instructions. Anti-(tetra)acetyl H4 histone primary antibody (Millipore) was used with goat anti-rabbit–infrared (IR) dye secondary antibody (Li-Cor Biosciences) to detect changes in histone H4 acetylation. Anti-pan-acetyl (K103) monoclonal antibody (Cell Signaling Technology) was used with goat anti-rabbit–IR dye secondary antiserum (Li-Cor Biosciences) to detect changes in nonhistone protein acetylation. Membranes were imaged using an Odyssey infrared imaging system (Li-Cor Biosciences).
In vivo antimalarial efficacy studies.
The in vivo antimalarial efficacy of compounds was examined in an ECM model using P. berghei ANKA parasites constitutively expressing a luciferase reporter gene (termed P. berghei ANKAluc) (3). Groups of five C57BL/6j mice were infected via intraperitoneal (i.p.) injection with 105 P. berghei ANKAluc-infected erythrocytes from an infected passage mouse. Two hours later, mice were treated with 100 μl of test compound or vehicle control (50% DMSO in PBS) via oral gavage. Mice were treated with compound or vehicle for a total of 3 days, either as a single dose or as a split dose (twice daily [BID]), with a 4-h interval between doses. A single daily dose of chloroquine (10 mg/kg of body weight in PBS) for 3 days, beginning at 2 h postinfection (p.i.), was used as a positive control. Peripheral parasitemia was monitored daily from day 4 p.i. by microscopic examination of Giemsa-stained thin blood smears prepared from tail bleeds. Parasite burden as a measure of bioluminescence (total flux; photons [p]/s) was measured from day 4 p.i. by anesthetizing mice with isoflurane (Bomac, New Zealand), injecting them with 100 μl luciferin (5 mg/ml in PBS; Caliper Life Sciences) substrate i.p., and then, after 4 min, imaging them using a Xenogen IVIS 100 imaging system (Caliper Life Sciences). Mice were housed in a ventilated cabinet (light from 8 p.m. to 8 a.m. and dark from 8 a.m. to 8 p.m.; Iffa Credo). All animal work was carried out according to protocols under the Animal Code of Practice and approved by the QIMR Animal Ethics Committee following guidelines mandated by the National Health and Medical Research Council of Australia.
RESULTS
In vitro activity of SB939 against P. falciparum asexual-stage parasites and mammalian cells.
In vitro inhibition studies against asexual-stage P. falciparum parasites growing in human erythrocytes showed that SB939 had potent activity (IC50s of ∼100 to 200 nM) against chloroquine-sensitive (3D7) and chloroquine-resistant (Dd2) P. falciparum parasites (Table 2). The IC50s of a reference HDAC inhibitor (SAHA) and the antimalarial drug chloroquine were similar to those previously reported (Table 2) (8, 24). The activity of SB939 against P. falciparum versus normal mammalian cell growth was assessed using HUVEC, PTC, and NFF cells (Table 2). These data were compared to previously published cytotoxicity data for normal human dermal fibroblast (NHDF) cells (47). Although low toxicity was reported against NHDF cells (IC50 of >100 μM) (47), SB939 had a greater inhibitory potency on the cells examined in this study, with IC50s ranging from 0.8 to 3.2 μM (Table 2).
Table 2.
In vitro antimalarial activity of SB939
| Compound |
P. falciparum IC50 (μM) |
Mammalian cell IC50 (μM)a |
Selectivity indexb | ||||
|---|---|---|---|---|---|---|---|
| 3D7 | Dd2 | NHDF cells | HUVEC | PTC | NFF cells | ||
| SAHA | 0.12 ± 0.04 | 0.19 ± 0.01 | 2.0 ± 0.04 | 1.8 ± 0.2 | 3.0 ± 0.5 | 4.90 ± 1.24 | 9–49 |
| SB939 | 0.08 ± 0.03 | 0.15 ± 0.03 | >100 | 0.8 ± 0.1 | 3.2 ± 0.5 | 1.48 ± 0.61 | 4–>1,250 |
| Chloroquine | 0.03 ± 0.09 | 0.15 ± 0.04 | >900 | ND | ND | 39.53 ± 1.32 | 258–>30,000 |
SAHA cytotoxicity was previously tested against NHDF cells (20); SB939 cytotoxicity was previously tested against NHDF cells (47); SB939 and SAHA cytotoxicity was tested in this study against HUVEC, PTC, and NFF cells; and chloroquine cytotoxicity was tested in this study against NFF cells and in previous studies against NHDF cells (55). ND, not determined.
Mammalian cell IC50/P. falciparum IC50. Larger values indicate greater parasite selectivity.
In vitro combination studies of SB939 with primaquine, piperaquine, and lopinavir.
Isobolograms describing the interactions of SB939 with primaquine and piperaquine against the chloroquine-sensitive P. falciparum line 3D7 demonstrated that the antiplasmodial activities of these compound combinations were antagonistic (see Fig. S1 in the supplemental material), with I values of <−5 and −1.4, respectively. However, SB939 and the protease inhibitor lopinavir behaved additively when combined (see Fig. S1).
In vitro activity of SB939 against P. berghei exoerythrocytic-stage parasites.
The activities of SB939 and the control HDAC inhibitor SAHA against the exoerythrocytic stage of the parasite life cycle were assayed using P. berghei ANKA sporozoites in cultured HepG2 cells. Following a 1-h invasion period, cells were cultured for 53 h in the presence of inhibitors and scored for successful parasite development. SB939 and SAHA displayed similar inhibitory profiles, with IC50s of 155.2 nM (±20.2 nM) and 164.8 nM (±18.3 nM), respectively (Fig. 1). At the concentrations examined (up to 1 μM), neither compound caused any toxicity to HepG2 cells compared to cells treated with vehicle (0.01% DMSO) or grown in normal medium (not shown). The antimalarial compound primaquine was used as a control and gave an IC50 of 3.48 μM (±0.42 μM) against exoerythrocytic-stage parasites, similar to the previously published IC50 of this compound (3.8 μM), which is less active than its metabolites (11).
Fig 1.
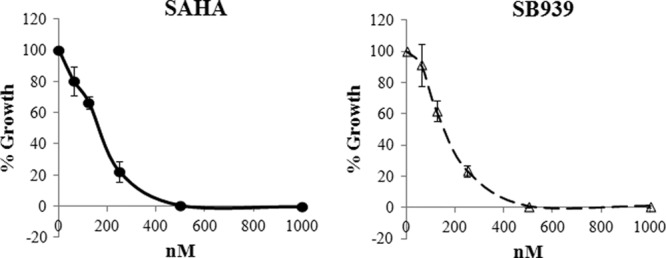
Inhibition of P. berghei exoerythrocytic-stage parasites. Dose-response curves are shown for exoerythrocytic-stage P. berghei parasites treated with HDAC inhibitors SAHA and SB939. Cells were treated with compounds for 53 h following invasion, fixed, stained, and scored by microscopy for successful development of parasites within HepG2 cells. The percent parasite growth compared to vehicle-only control parasite growth (mean ± standard error of the mean [SEM]) is shown.
In silico docking studies.
Binding of SB939 to an updated homology structure model of PfHDAC1 was also examined. This new PfHDAC1 homology model was generated with Modeller, using human HDAC8 and HDAC2 crystal structures and improving the loop 1 and loop 2 regions (an amino acid alignment is shown in Fig. S2 in the supplemental material). GOLD was used to dock TSA, SAHA, and SB939 (see Table 1 for structures) into the active site of PfHDAC1, using a hydroxamic acid scaffold constraint. Cross docking TSA back into PfHDAC1 resulted in the top-ranked conformation, with a heavy-atom root mean square deviation (RMSD) of 0.90 Å relative to the original TSA conformation (values of ≤2 Å are considered acceptable) (34). The top-ranked SAHA conformation docked with the phenyl head group docked into the pocket between loop 1 and loop 2 in a manner similar to that for the head group of TSA (59). SB939 docked into PfHDAC1 with the indole linker positioned between tunnel residues Phe148 and Phe203 and is shown in Fig. S3 in the supplemental material, with potential H bonds indicated. The SB939 hydroxamic acid carbonyl oxygen makes a putative interaction with Tyr301-OH, while the -NH group of the protonated and positively charged nitrogen of SB939 makes a potential interaction with Asp98. The linear butyl alkyl chain of SB939 was fitted into a surface pocket between loop 1 and loop 2 (see Fig. S3).
SB939's mode of action against P. falciparum asexual-stage parasites.
To assess the effect of SB939 on P. falciparum protein acetylation, 3D7 trophozoite-stage parasites were treated for 3 h with SB939, SAHA, or the antimalarial drug chloroquine, and Western blotting was conducted. Compared to vehicle and chloroquine control samples, SAHA and SB939 treatment resulted in hyperacetylation of proteins of ∼12 to 15 kDa, as viewed using antiserum specific for tetra-acetylated histone H4 (6) (Fig. 2). An increased signal was also observed for an ∼80- to 90-kDa protein in HDAC inhibitor-treated samples, using a commercial antiserum that recognizes acetylated lysine residues (pan-reactive). These data indicate that SB939 hyperacetylates both histone and nonhistone proteins.
Fig 2.

Hyperacetylation of P. falciparum proteins by SB939. Synchronous 3D7 trophozoite-stage P. falciparum parasites were treated with 50 or 500 nM chloroquine (CQ), SAHA, or SB939 or with vehicle only (control; 0.05% DMSO) for 3 h. Following saponin lysis, parasite protein lysates were prepared and SDS-PAGE and Western blotting carried out using anti-acetyl H4 or anti-pan-acetyl lysine (K103) antibodies. Coomassie blue staining was carried out as a loading control.
In vivo efficacy of oral SB939 in an experimental cerebral malaria model.
The in vivo efficacy of SB939 was examined in a murine ECM model using C57BL/6J mice and P. berghei ANKA parasites constitutively expressing a luciferase bioluminescence reporter protein. Oral administration of 25 mg/kg SB939 twice a day for 3 days resulted in a significant reduction in peripheral blood parasitemia (P < 0.05 for days 3 to 7 p.i.) and parasite biomass (P < 0.05 for days 3, 4, 6, and 7) compared to those of control mice (Fig. 3). Mice treated with the antimalarial drug chloroquine (10 mg/kg; single oral dose daily for 3 days) had subpatent peripheral blood parasitemias until day 8 to 9 days p.i. (e.g., parasitemias on day 9 p.i. ranged from 0 to 3.1% [mean parasitemia of 1.0%]), an effect previously reported with this treatment regimen. As expected, mice in the control group developed ECM-like symptoms on day 6 or 7 p.i. and were euthanized. None of the mice in the chloroquine- or SB939-treated groups had developed ECM-like symptoms by day 9 p.i., when the experiment was ended.
Fig 3.

Effect of twice-daily treatment with 25 mg/kg SB939 on P. berghei ANKA-infected mice. Groups of five P. berghei ANKAluc-infected C57BL/6j mice were treated orally with 25 mg/kg SAHA (dashed line) or vehicle only (50% DMSO in PBS) as a control (solid line) twice a day for 3 days, beginning at 2 h postinfection. (A) Mean (± SEM) peripheral blood parasitemia was determined by microscopic examination of Giemsa-stained thin blood smears. (B) Mean (± SEM) parasite-derived bioluminescence (flux) was determined using a Xenogen imager. Significant differences (*, P < 0.05; **, P < 0.01) in treated groups compared to controls are indicated. (C) Images of mice on day 6 postinfection show increasing parasite-derived bioluminescence intensities from purple to red (a luminescence scale is shown to the right).
Since cures were not achieved in mice treated with 2 doses of 25-mg/kg SB939 per day, the maximum reported tolerated daily dose of this compound (100 mg/kg) in mice (47, 65) was examined. Mice were treated orally with 50 mg/kg SB939 twice a day for three consecutive days to achieve a total daily dose of 100 mg/kg. Significant reductions in peripheral parasitemia and parasite burden were observed for SB939 versus the control group of mice on days 4 to 7 (P < 0.05) (Fig. 4); however, mice were not cured at this dose. Hyperparasitemias developed on days 12 to 16 p.i. in 4/5 mice treated with SB939. Mice were euthanized when their parasitemias reached ∼30%. One mouse had a peak peripheral parasitemia on day 12 p.i. of ∼12%, followed by a decrease in parasitemia (<1.5%) until day 22 p.i., when the experiment was ended. All mice in the control group succumbed to ECM-like symptoms and were euthanized on day 6 or 7. None of the mice in the SB939 group developed ECM-like symptoms.
Fig 4.

Effect of twice-daily treatment with 50 mg/kg SB939 in mice infected with P. berghei ANKA. P. berghei ANKAluc-infected C57BL/6j mice (n = 5) were treated orally with 50 mg/kg SB939 (B and D) or vehicle only (50% DMSO in PBS) (A and C) twice a day for 3 days, beginning at 2 h postinfection. Peripheral parasitemia (A and B) and parasite-derived bioluminescence (flux) (C and D) are shown for individual mice. Significant differences (*, P < 0.05; **, P < 0.01) between the SB939 and control groups are indicated.
DISCUSSION
Antimalarial drug resistance is driving the search for new agents able to either replace current drugs when they fail or complement existing antimalarials as partner drugs in new combination therapies. Here we investigated the antimalarial activity of a new hydroxamate-based HDAC inhibitor that is currently in phase I clinical trials for the treatment of cancer (52, 67). SB939 was developed from a medicinal chemistry program focused on addressing the general lack of metabolic stability and poor in vivo pharmacokinetics displayed by HDAC inhibitors (65). From a medicinal chemistry point of view, SB939 obeys Lipinski's rule of five (39) and has the appropriate size, hydrophobicity, and polar surface parameters (e.g., molecular weight = 358, octanol-water partition coefficient [clogP] = 3.2, number of H-bond donors = 2, number of H-bond acceptors = 6, and total polar surface area [TPSA] = 68%) required for oral bioavailability. It is therefore not surprising that SB939 has an improved pharmacokinetic profile compared to that of SAHA, the first HDAC inhibitor to be approved for human use against cutaneous T cell lymphoma (Table 1). Preclinical pharmacokinetic studies in mice have shown that orally administered SB939 (50 mg/kg; single dose) reaches a higher maximum concentration (Cmax), with higher oral bioavailability (F%) and a longer half-life (t1/2), than that of SAHA (Table 1). In vitro safety pharmacology data also show that SB939 (10 μM) does not interfere with the activity of non-HDAC enzymes, including other Zn2+-dependent enzymes, G-protein-coupled receptors, monoamine transporters, and ion channels (47, 65). SB939 thus has a promising profile as a new HDAC inhibitor.
This study is the first to report on noncancer activities of SB939. We found SB939 to be a potent inhibitor of the in vitro growth of P. falciparum malaria parasites, with the compound being active against both chloroquine-sensitive (3D7) and -resistant (Dd2) lines (Table 2). SB939 was less potent against Dd2 than against 3D7 (P < 0.05), suggesting that additional studies may be required to determine if cross-resistance is a potential concern. When the in vitro growth inhibition potency against P. falciparum parasites was compared to that of mammalian cell lines, a wide variation in selectivity indexes was observed. While SB939 was 1,000 times more active as an inhibitor against P. falciparum growth than against NHDF cells (47), much lower (1- to 5-fold) selectivity indexes were obtained for the other cell lines tested in this study (Table 2). This differential killing selectivity may be due to the different expression levels of the HDAC isoform(s) in different cell lines.
Malaria control and treatment currently rely on combination therapy to try to counteract the development of antimalarial drug resistance. As such, it is important that new antimalarial drugs be evaluated as therapeutic options in concert with complementary partner drugs. Only limited studies have examined the potential of HDAC inhibitors as partner drugs against malaria parasites. Antagonist effects have been reported for the antimalarial hydroxamate HDAC inhibitor WR301801 combined in vitro with mefloquine, chloroquine, artemisinin, or the antimicrobial agent azithromycin (24). In this study, we tested two clinically used antimalarial drugs that have not yet been combined with HDAC inhibitors, namely, piperaquine, which targets asexual-stage parasites, and primaquine, which is used primarily to target liver-stage parasites but also has modest activity against asexual-stage parasites (51, 62). We showed that SB939 antagonizes the antiplasmodial activity of both of these antimalarial drugs. We also tested a clinically utilized HIV protease inhibitor, lopinavir, that is currently used to treat HIV in HIV-Plasmodium-coinfected individuals and that we have previously shown to have antimalarial activity (4). We found that SB939 behaved in an additive fashion with lopinavir. This is the first positive in vitro interaction to be reported for an HDAC inhibitor and another antimalarial compound against P. falciparum. While this finding needs to be investigated further in vivo, it may also have important implications for the treatment of patients with malaria and HIV/AIDS, who may concurrently be receiving antiretroviral protease inhibitors. Moreover, given that recent data suggest that HIV protease inhibitor and HDAC inhibitor combination treatment regimens target latent virus (38, 43), additional antimalarial benefits of this combination may also be possible.
In this study, we conducted the first investigation of the in vitro growth inhibition activity of HDAC inhibitors against exoerythrocytic (liver)-stage Plasmodium parasites. Both SB939 and SAHA potently inhibited the development of P. berghei ANKA mouse malaria parasites within HepG2 liver cells. Although the target of these compounds in liver-stage parasites is not yet known, SAHA is known to inhibit recombinant PfHDAC1 activity (50), and the gene encoding this protein is transcribed in P. falciparum liver-stage parasites as well as in asexual- and gametocyte-stage parasites (9). Expression data for the P. berghei ANKA homologue (PbHDAC1) of PfHDAC1 in liver-stage parasites are not yet available, but considering that the two proteins share 94% amino acid identity (9), it is likely that HDAC inhibitors will target this protein in rodent malaria parasites. Our finding that HDAC inhibitors can target multiple Plasmodium life cycle stages in vitro has potentially important implications for the future development of this class of compounds as antimalarial drugs. Although most antimalarial drugs in clinical use target intraerythrocytic, asexual-stage parasites, very few are known to act on other life cycle stages, such as exoerythrocytic liver-stage parasites or gametocytes, the stage transmitted to the mosquito. Our data provide the first validation of HDAC inhibitors as liver-stage drug leads. Findings from this study suggest that SB939 acts as an HDAC inhibitor in malaria parasites.
Our in silico docking studies give an improved homology structural model of PfHDAC1 and also provide evidence for binding to SB939, in a fashion similar to that of TSA and SAHA. We also demonstrated that SB939 has the same mode of action as previously used antimalarial HDAC inhibitors (6, 21, 24), causing hyperacetylation of both histone and nonhistone proteins in P. falciparum asexual-stage parasites. The nonhistone protein hyperacetylated by SB939 treatment was not identified in this study, but its molecular mass (∼80 to 90 kDa) corresponds well to that of heat shock protein 90 (PfHSP90), which was recently identified as being hyperacetylated in TSA-treated P. falciparum parasites by use of the same pan-acetyl lysine antiserum as that used in this study (49). Interestingly, HSP90 is known to be a nonhistone target of HDAC action in mammalian cells (10, 37).
The in vivo efficacy of SB939 was examined using a murine ECM model. Cerebral malaria (CM) pathogenesis is poorly understood; however, cerebral pathology is associated with mature infected erythrocytes sequestering in the microvasculature of tissues, including the brain (13, 66). This results in immune evasion by mature P. falciparum-infected erythrocytes, preventing removal by the spleen and facilitating parasite survival (13). ECM caused by P. berghei ANKA parasites in mice shares some features with human CM, with susceptible mice developing neurological symptoms such as paralysis, ataxia, convulsions, and coma. This generally occurs 6 to 8 days after infection with P. berghei ANKA parasites, results in 80 to 100% mortality, and is associated with low peripheral blood parasitemia (3). Studies of ECM in mouse models of malaria have been facilitated greatly by the recent advent of a transgenic, luciferase-expressing P. berghei ANKA line that allows not only measurement of peripheral blood parasitemia but also measurement of parasite biomass in peripheral tissues (3). Orally administered SB939 was found to be effective in reducing both peripheral parasitemia and total parasite burden at doses of up to 100 mg/kg/day in the ECM model. Although mice were not cured, SB939 treatment prevented the development of ECM-like symptoms, and mice did not develop hyperparasitemia until day 15 to 22 p.i. In contrast to the result obtained with twice-daily dosing of SB939, when this compound was administered orally as a single daily dose of 50 or 100 mg/kg, a significant reduction in parasite biomass was observed on day 6 p.i., but there was no difference on day 7 p.i. (see Fig. S4 in the supplemental material). The poorer antiparasitic effect with a single dose of SB939 than with a twice-daily dose is likely due to the in vivo half-life of SB939 (2.4 h) being insufficient to attenuate parasite growth.
HDAC inhibitors are a promising new class of antimalarial compounds, with several compounds displaying potent in vitro and ex vivo activity against P. falciparum and P. vivax parasites. Recent studies on the effects of HDAC inhibitors on the growth and development of P. falciparum have also provided new insights into transcriptional regulation in malaria parasites (19, 32). Alterations in HDAC expression in malaria parasites may be associated with reduced clinical artemisinin efficacy (45), raising the possibility that HDAC inhibitors might contribute to future drug combinations aimed at addressing the issue of artemisinin resistance. Such findings underscore the potential for HDAC inhibitors to be used for malaria therapy. However, to progress HDAC inhibitors toward clinical trials for malaria, next-generation compounds must have potent antiplasmodial activity, highly selective killing action against parasites rather than normal host cells, and improved bioavailability and pharmacokinetic profiles after oral dosing. In this study, we demonstrated that SB939, a new orally bioavailable HDAC inhibitor with a superior pharmacokinetic profile over those of previous HDAC inhibitors such as SAHA, has potent activity against asexual- and sporozoite-stage Plasmodium parasites and promising in vivo activity in a mouse model of malaria infection. These findings support the development of HDAC inhibitors as a potential new antimalarial compound class.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Australian Research Council (grant FF0668705 to G.I.M., grant FF0668733 to D.P.F., and grant FT0991213 to K.T.A.) and the Australian National Health and Medical Research Council (grant 569735 to D.P.F. and grant 637406 to G.I.M.) for research and fellowship support.
We acknowledge the Australian Red Cross Blood Service for the provision of human blood and sera and the QIMR Animal Facility for animal handling support.
Footnotes
Published ahead of print 16 April 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Agbor-Enoh S, Seudieu C, Davidson E, Dritschilo A, Jung M. 2009. Novel inhibitor of Plasmodium histone deacetylase that cures P. berghei-infected mice. Antimicrob. Agents Chemother. 53:1727–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 3. Amante FH, et al. 2007. A role for natural regulatory T cells in the pathogenesis of experimental cerebral malaria. Am. J. Pathol. 171:548–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andrews KT, et al. 2006. Potencies of human immunodeficiency virus protease inhibitors in vitro against Plasmodium falciparum and in vivo against murine malaria. Antimicrob. Agents Chemother. 50:639–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andrews KT, Haque A, Jones MK. 2012. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 90:66–77 [DOI] [PubMed] [Google Scholar]
- 6. Andrews KT, et al. 2008. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 52:1454–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andrews KT, Tran TN, Wheatley NC, Fairlie DP. 2009. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 9:292–308 [DOI] [PubMed] [Google Scholar]
- 8. Andrews KT, et al. 2000. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int. J. Parasitol. 30:761–768 [DOI] [PubMed] [Google Scholar]
- 9. Aurrecoechea C, et al. 2009. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37:D539–D543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bali P, et al. 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 280:26729–26734 [DOI] [PubMed] [Google Scholar]
- 11. Bates MD, Meshnick SR, Sigler CI, Leland P, Hollingdale MR. 1990. In vitro effects of primaquine and primaquine metabolites on exoerythrocytic stages of Plasmodium berghei. Am. J. Trop. Med. Hyg. 42:532–537 [DOI] [PubMed] [Google Scholar]
- 12. Berenbaum M. 1978. A method for testing for synergy with any number of agents. J. Infect. Dis. 137:122–130 [DOI] [PubMed] [Google Scholar]
- 13. Berendt AR, Tumer GD, Newbold CI. 1994. Cerebral malaria: the sequestration hypothesis. Parasitol. Today 10:412–414 [DOI] [PubMed] [Google Scholar]
- 14. Berman HM, et al. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beumer JH, Tawbi H. 2010. Role of histone deacetylases and their inhibitors in cancer biology and treatment. Curr. Clin. Pharmacol. 5:196–208 [DOI] [PubMed] [Google Scholar]
- 16. Bressi JC, et al. 2010. Exploration of the HDAC2 foot pocket: synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 20:3142–3145 [DOI] [PubMed] [Google Scholar]
- 17. Cairns BR. 2001. Emerging roles for chromatin remodeling in cancer biology. Trends Cell Biol. 11:S15–S21 [DOI] [PubMed] [Google Scholar]
- 18. Canfield C, Pudney M, Gutteridge W. 1995. Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro. Exp. Parasitol. 80:373–381 [DOI] [PubMed] [Google Scholar]
- 19. Chaal BK, Gupta AP, Wastuwidyaningtyas BD, Luah YH, Bozdech Z. 2010. Histone deacetylases play a major role in the transcriptional regulation of the Plasmodium falciparum life cycle. PLoS Pathog. 6:e1000737 doi:10.1371/journal.ppat.1000737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crabb SJ, et al. 2008. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem. Pharmacol. 76:463–475 [DOI] [PubMed] [Google Scholar]
- 21. Darkin-Rattray SJ, et al. 1996. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. U. S. A. 93:13143–13147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dokmanovic M, Clarke C, Marks PA. 2007. Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 5:981–989 [DOI] [PubMed] [Google Scholar]
- 23. Dondorp AM, et al. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dow GS, et al. 2008. Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother. 52:3467–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duraisingh MT, et al. 2005. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell 121:13–24 [DOI] [PubMed] [Google Scholar]
- 26. Eisenberg D, Luthy R, Bowie JU. 1997. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 277:396–404 [DOI] [PubMed] [Google Scholar]
- 27. Finnin MS, et al. 1999. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401:188–193 [DOI] [PubMed] [Google Scholar]
- 28. Freitas-Junior LH, et al. 2005. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 121:25–36 [DOI] [PubMed] [Google Scholar]
- 29. Gao L, Cueto MA, Asselbergs F, Atadja P. 2002. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 277:25748–25755 [DOI] [PubMed] [Google Scholar]
- 30. Glozak MA, Sengupta N, Zhang X, Seto E. 2005. Acetylation and deacetylation of non-histone proteins. Gene 363:15–23 [DOI] [PubMed] [Google Scholar]
- 31. Grozinger CM, Schreiber SL. 2002. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 9:3–16 [DOI] [PubMed] [Google Scholar]
- 32. Hu G, et al. 2010. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 28:91–98 [DOI] [PubMed] [Google Scholar]
- 33. Huber W, Koella JC. 1993. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 55:257–261 [DOI] [PubMed] [Google Scholar]
- 34. Jain AN. 2008. Bias, reporting, and sharing: computational evaluations of docking methods. J. Comput. Aided Mol. Des. 22:201–212 [DOI] [PubMed] [Google Scholar]
- 35. Jayaraman R, et al. 2011. Preclinical metabolism and disposition of SB939 (pracinostat), an orally active histone deacetylase (HDAC) inhibitor, and prediction of human pharmacokinetics. Drug Metab. Dispos. 39:2219–2232 [DOI] [PubMed] [Google Scholar]
- 36. Joshi MB, et al. 1999. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol. Biochem. Parasitol. 99:11–19 [DOI] [PubMed] [Google Scholar]
- 37. Kekatpure VD, Dannenberg AJ, Subbaramaiah K. 2009. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem. 284:7436–7445 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38. Lehrman G, et al. 2005. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46:3–26 [DOI] [PubMed] [Google Scholar]
- 40. Ma X, Ezzeldin HH, Diasio RB. 2009. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs 69:1911–1934 [DOI] [PubMed] [Google Scholar]
- 41. Marfurt J, et al. 2011. Ex vivo activity of histone deacetylase inhibitors against multidrug-resistant clinical isolates of Plasmodium falciparum and P. vivax. Antimicrob. Agents Chemother. 55:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marks PA. 2010. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Invest. Drugs 19:1049–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matalon S, Rasmussen TA, Dinarello CA. 2011. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 17:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Minucci S, Pelicci PG. 2006. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 6:38–51 [DOI] [PubMed] [Google Scholar]
- 45. Mok S, et al. 2011. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics 12:391 doi:10.1186/1471-2164-12-391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nair SC, et al. 2011. Apicoplast isoprenoid precursor synthesis and the molecular basis of fosmidomycin resistance in Toxoplasma gondii. J. Exp. Med. 208:1547–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Novotny-Diermayr V, et al. 2010. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol. Cancer Ther. 9:642–652 [DOI] [PubMed] [Google Scholar]
- 48. Ortore G, Di Colo F, Martinelli A. 2009. Docking of hydroxamic acids into HDAC1 and HDAC8: a rationalization of activity trends and selectivities. J. Chem. Infect. Model. 49:2774–2785 [DOI] [PubMed] [Google Scholar]
- 49. Pallavi R, et al. 2010. Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of HSP90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J. Biol. Chem. 285:37964–37975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patel V, et al. 2009. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 52:2185–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pradines B, et al. 2006. In vitro activity of tafenoquine against the asexual blood stages of Plasmodium falciparum isolates from Gabon, Senegal, and Djibouti. Antimicrob. Agents Chemother. 50:3225–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Razak AR, et al. 2011. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer 104:756–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sali A, Blundell TL. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815 [DOI] [PubMed] [Google Scholar]
- 54. Sanderson L, et al. 2004. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin A after intraperitoneal administration to mice. Drug Metab. Dispos. 32:1132–1138 [DOI] [PubMed] [Google Scholar]
- 55. Sarr SO, et al. 2011. Icacina senegalensis (Icacinaceae), traditionally used for the treatment of malaria, inhibits in vitro Plasmodium falciparum growth without host cell toxicity. Malar. J. 10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmidt-Christensen A, Sturm A, Horstmann S, Heussler VT. 2008. Expression and processing of Plasmodium berghei SERA3 during liver stages. Cell. Microbiol. 10:1723–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shen MY, Sali A. 2006. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15:2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skinner-Adams TS, Andrews KT, Melville L, McCarthy J, Gardiner DL. 2007. Synergistic interactions of the antiretroviral protease inhibitors saquinavir and ritonavir with chloroquine and mefloquine against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 51:759–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Somoza JR, et al. 2004. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12:1325–1334 [DOI] [PubMed] [Google Scholar]
- 60. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tonkin CJ, et al. 2009. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 7:e84 doi:10.1371/journal.pbio.1000084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vennerstrom JL, et al. 1999. 8-Aminoquinolines active against blood-stage Plasmodium falciparum in vitro inhibit hematin polymerization. Antimicrob. Agents Chemother. 43:598–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wagner JM, Hackanson B, Lubbert M, Jung M. 2010. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenet. 1:117–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Waller RF, Reed MB, Cowman AF, McFadden GI. 2000. Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. EMBO J. 19:1794–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang H. 2011. Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. J. Med. Chem. 54:4694–4720 [DOI] [PubMed] [Google Scholar]
- 66. WHO Communicable Diseases Cluster 2000. Severe falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 94(Suppl 1):S1–S90 [PubMed] [Google Scholar]
- 67. Yong WP, et al. 2011. Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, SB939, in patients with refractory solid malignancies. Ann. Oncol. 22:2516–2522 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.