

Abstract
The mitochondrial bc1 complex is a multisubunit enzyme that catalyzes the transfer of electrons from ubiquinol to cytochrome c coupled to the vectorial translocation of protons across the inner mitochondrial membrane. The complex contains two distinct quinone-binding sites, the quinol oxidation site of the bc1 complex (Qo) and the quinone reduction site (Qi), located on opposite sides of the membrane within cytochrome b. Inhibitors of the Qo site such as atovaquone, active against the bc1 complex of Plasmodium falciparum, have been developed and formulated as antimalarial drugs. Unfortunately, single point mutations in the Qo site can rapidly render atovaquone ineffective. The development of drugs that could circumvent cross-resistance with atovaquone is needed. Here, we report on the mode of action of a potent inhibitor of P. falciparum proliferation, 1-hydroxy-2-dodecyl-4(1H)quinolone (HDQ). We show that the parasite bc1 complex—from both control and atovaquone-resistant strains—is inhibited by submicromolar concentrations of HDQ, indicating that the two drugs have different targets within the complex. The binding site of HDQ was then determined by using a yeast model. Introduction of point mutations into the Qi site, namely, G33A, H204Y, M221Q, and K228M, markedly decreased HDQ inhibition. In contrast, known inhibitor resistance mutations at the Qo site did not cause HDQ resistance. This study, using HDQ as a proof-of-principle inhibitor, indicates that the Qi site of the bc1 complex is a viable target for antimalarial drug development.
INTRODUCTION
Toxoplasma gondii and Plasmodium falciparum are apicomplexan parasites causing toxoplasmosis and malaria, respectively. The latter disease is among the most serious health problems in the world, leading to more than 1 million deaths per year. Over recent years, an increase in malaria mortality has been attributed to the development of parasite resistance to first line therapies, which has raised calls for the urgent development of new drugs with novel modes of action (see, for example, reference 6). The mitochondrial respiratory chain is an effective target for antimicrobial agents directed against these pathogens. Differences between respiratory chain enzymes of mammals and pathogenic organisms have been exploited to develop compounds used for drug therapy such as atovaquone. Atovaquone is a hydroxynapthoquinone active against different parasitic diseases (malaria, toxoplasmosis, and Pneumocystis pneumonia caused by the opportunistic pathogen fungus Pneumocystis jiroveci). Atovaquone inhibits the activity of the bc1 complex activity, a central enzyme of the respiratory chain (24). The mitochondrial bc1 complex is a multisubunit enzyme that catalyzes the transfer of electrons from ubiquinol to cytochrome c and couples this electron transfer to the vectorial translocation of protons across the inner mitochondrial membrane. The complex contains two distinct quinone-binding sites (the quinol oxidation site [Qo] and the quinone reduction site [Qi]), which are located on opposite sides of the membrane. Cytochrome b, the central membrane-embedded subunit, encoded by the mitochondrial genome, provides both Qo and Qi binding pockets. As a consequence of atovaquone-mediated bc1 complex inhibition, the electron transfer through the respiratory chain stops and the mitochondrial membrane potential of P. falciparum collapses. In addition, without a functioning bc1 complex to oxidize ubiquinol, the dihydro-ororate dehydrogenase oxidization (DHODH) comes to a halt as oxidized ubiquinone is required as an electron acceptor for the DHODH. Pyrimidine biosynthesis is thus inhibited which is lethal for the parasite (39, 44, 45). Unfortunately, resistance to atovaquone has been observed in both P. falciparum and T. gondii. This resistance is often associated with mutations in the target site, cytochrome b, the main subunit of the bc1 complex (reviewed in reference 21). Since atovaquone is effective against both circulating asexual stage parasites and liver stage parasites, it is a useful drug for both malaria treatment and prophylaxis. Therefore, new drugs that target the bc1 complex but that can circumvent atovaquone resistance and/or are more recalcitrant to resistance would be very welcomed. Currently, several different chemotypes targeting the bc1 complex have been developed, these include the hydroxynapthoquinones (atovaquone analogues), pyridones (clopidol analogues), and acridine-related compounds (acridinediones, acridones, and quinolones [reviewed in reference 1]).
1-Hydroxy-2-dodecyl-4(1H)quinolone (HDQ) was recently shown to inhibit parasite replication of T. gondii and P. falciparum in nanomolar concentrations (40). HDQ treatment in T. gondii causes a loss of the mitochondrial inner-membrane potential and a severe ATP depletion due to the block of the electron flow (34). Because of the structural similarity between HDQ and ubiquinol, it seems likely that the drug could target ubiquinol binding sites of respiratory enzymes. Consistent with this hypothesis, HDQ has been shown to inhibit the mitochondrial alternative NADH dehydrogenase (NDH2) and complex I of the yeast Yarrowia lipolytica, albeit with different efficiencies (the 50% inhibitory concentrations [IC50s] were 0.2 and 2 μM, respectively) (16). The T. gondii type-II NADH dehydrogenase expressed in Y. lipolytica has also been shown to be inhibited by HDQ with an IC50 of 0.3 μM (36). Furthermore, the HDQ-related compound HQNO (2-heptyl-4-hydroxyquinoline N-oxide) is a known inhibitor of mammalian and S. cerevisiae bc1 complex. Mutations causing resistance to HQNO have been reported in yeast; they are located at the Qi site of the complex (reviewed in reference 10). This led us to question whether the antimalarial activity of HDQ may be mediated via bc1 complex inhibition and, more specifically, by blocking the Qi site function.
We demonstrate here that in P. falciparum, HDQ, in addition to its inhibitory action toward the NADH:ubiquinone-oxidoreductase (PfNDH2), disrupts mitochondrial function through the potent inhibition of the bc1 complex. By studying yeast with specific cytochrome b mutations in the Qo and Qi sites, and using molecular modeling, we show that HDQ inhibition of the bc1 complex is mediated via Qi binding. HDQ therefore displays a novel inhibitory mode of action against an important antimalarial target.
MATERIALS AND METHODS
Chemical synthesis of HDQ.
The synthesis of HDQ was based on the method of Woscheck, et al. (49) (see Fig. S1 in the supplemental material for route of synthesis). Briefly, ethyl 3-oxopentadecanoate (prepared according to the method of Tietze and Ma [46]) was condensed with aniline using a catalytic amount of p-toluenesulfonic acid and azeotropic removal of water. The crude enamine was cyclized in Dowtherm to give 2-dodecylquinolin-4(1H)-one. Treatment of this 4(1H)-quinolone with potassium tert-butoxide, followed by esterification with ethyl chloroformate, yielded the 4-(ethoxycarbonyloxy)quinoline. Oxidation of this with mCPBA yielded the N-oxide. Hydrolysis of the N-oxide with potassium hydroxide in aqueous ethanol, followed by acidic workup and recrystallization gave HDQ. All spectroscopic data and CHN analyses were in accord with the proposed structure.
Parasites, culture, and drug sensitivity testing.
P. falciparum (3D7 strain) cultures consisted of a 2% suspension of O+ erythrocytes in RPMI 1640 medium (R8758, glutamine, and NaHCO3) supplemented with 10% pooled human AB+ serum, 25 mM HEPES (pH 7.4), and 20 μM gentamicin sulfate (47). Cultures were grown under a gaseous headspace of 4% O2 and 3% CO2 in N2 at 37°C. Parasite growth was synchronized by treatment with sorbitol (32). Drug susceptibilities were assessed by the measurement of fluorescence after the addition of SYBR green I as described previously (42). Drug IC50s were calculated from the log of the dose-response relationship, as fitted with Grafit software (Erithacus Software, Kent, United Kingdom). The results are expressed as the means of at least three separate experiments. The atovaquone-resistant isolate TM90C2B (Thailand) was generously provided by Dennis Kyle (College of Public Health, University of South Florida, Tampa, FL).
Transgenic parasites.
3D7-yDHOD·GFP, a transgenic derivative of P. falciparum 3D7 containing yeast dihydroorotate dehydrogenase (DHODH), was generated via the electroporation of purified pHHyDHOD-GFP plasmid into ring stages of P. falciparum using a Bio-Rad GenePulser as described previously (39). Purified pHHyDHOD-GFP plasmid was generously provided by Akhil Vaidya (Drexel University College of Medicine, Philadelphia, PA). This plasmid contains a human dihydrofolate reductase gene as a WR99210-selectable marker (39).
Preparation of P. falciparum cell extracts.
Free parasites were prepared from aliquots of infected erythrocytes (approximately 8 × 109 cells ml−1) by adding 5 volumes of 0.15% (wt/vol) saponin in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 1.76 mM K2HPO4, 8.0 mM Na2HPO4, 5.5 mM d-glucose [pH 7.4]) for 5 min, followed by three washes using centrifugation and resuspension in HEPES (25 mM)-buffered RPMI containing a protease inhibitor cocktail (Complete Mini; Roche). Cell extract was prepared by repeated freeze-thawing in liquid N2, followed by disruption with a sonicating probe.
Preparation of recombinant P. falciparum NADH dehydrogenase (PfNDH2).
Recombinant PfNDH2 was prepared from the E. coli heterologous expression strain F571 as described in reference 22 and used as a crude membrane preparation at a total protein concentration of 15 μg/ml for NADH:decylubiquinone oxidoreductase activity measurement.
Measurement of NADH:decylubiquinone oxidoreductase activity.
Recombinant PfNDH2 and yeast NDH2 activities (using samples prepared as described in reference 19) were assayed in a reaction medium consisting of 50 mM potassium phosphate (pH 7.5), 2 mM EDTA, 200 μM NADH, and 10 mM KCN. NADH:decylubiquinone oxidoreductase activity was initiated by the addition of 50 μM decylubiquinone. Decylubiquinone reduction was monitored at 283 nm (ε283 = 8.1 mM−1 cm−1) in a Cary 4000 spectrophotometer, with rate and IC50 data determined as described for the cytochrome c reductase assay.
Measurement of cytochrome c reductase activity in P. falciparum samples.
Cytochrome c reductase activity measurements were assayed in 50 mM potassium phosphate (pH 7.5), 2 mM EDTA, 10 mM KCN, and 30 μM equine cytochrome c (Sigma) at room temperature (4, 22). Cytochrome c reductase activity was initiated by the addition of decylubiquinol (50 μM). Reduction of cytochrome c was monitored in a Cary 4000 spectrophotometer at 550 versus 542 nm. Initial rates (computer-fitted as zero-order kinetics) were measured as a function of decylubiquinol concentration. Turnover rates of cytochrome c reduction were determined using ε550-542 = 18.1 mM−1 cm−1. Inhibitors of bc1 activity were added without prior incubation. dimethyl sulfoxide (DMSO) in the assays did not exceed 0.3% (vol/vol). The IC50s were calculated by using the four-parameter logistic method.
As previously shown by Fisher et al. (22), the spectrophotometric challenge presented by the presence of hemozoin is countered by adjusting the protein concentration within the cuvette such that the total absorbance is under 2.0 U (i.e., 1% transmitted light), which, coupled with the use of exogenous cytochrome c for the enzymatic assay (ΔA = 0.02/min), is well within the operating parameters of the Cary 4000 spectrophotometer used in the assays. It should be noted that, in addition to the above, hemozoin is chemically inert under our assay conditions and so will not cause a drift in the measured absorbance.
Real-time single-cell monitoring of membrane potential (Ψm).
The rhodamine derivative, TMRE (tetramethylrhodamine ethyl ester), was used to monitor the membrane potential (Ψm) of the plasma membrane and mitochondria of malaria-infected red blood cells, as described previously (4, 5).
Yeast mutant strains.
The mutations of the cytochrome b gene are listed in Table 1. The mutated and wild-type (WT) mitochondrial genomes were transferred by cytoduction into AD1-9 (α ura3 his1 yor1Δ::hisG snq2Δ::hisG pdr5Δ::hisG pdr10Δ::hisG pdr11Δ::hisG ycf1Δ::hisG pdr3Δ::hisG pdr15Δ::hisG pdr1Δ::hisG; kindly donated by M. Ghislain, UCL, Belgium) and into BY4742 Δcox7 (α ura3 his3 leu2 lys2 Δcox7::G418; purchased from Euroscarf). All of the strains analyzed in the present study of the AD1-9 or the BY4742 series were isogenic. The BY4742 Δcox7 series were used for the cytochrome c reductase assays. Since the strain lacks a functional cytochrome oxidase due to the nuclear Δcox7 mutation, the quinol cytochrome c reductase activity could be measured without added KCN. The AD1-9 series were used for all of the growth experiments, since the multiple deletions in the ABC transporter genes render the strains more sensitive to drugs than standard yeast strains (26). The yeast culture media were as described earlier (19).
Table 1.
Mutations in cytochrome b
| Mutation(s) | Source or referencea |
|---|---|
| G33A | This study |
| G37S | This study |
| T127I | 26 |
| G143A | 20 |
| I147V | 26 |
| S152A | 26 |
| H204Y | 11 |
| S206T/V | 11 |
| N208V | This study |
| R218K | This study |
| M221Q | 11 |
| F225L | This study |
| K228M | This study |
| K228I | This study |
| L275F | 26 |
| Y279C | 19 |
| Y279S | 20 |
| G143A+K228M | This study |
For mutations derived in this study, site-directed mutations were introduced in the mitochondrial cytochrome b gene by microprojectile bombardment-mediated mitochondrial transformation as described previously (26).
Measurement of quinol cytochrome c reductase activity in yeast mitochondria.
Mitochondria were prepared as described by Lemaire and Dujardin (33). Quinol cytochrome c reductase activity measurements were performed in 10 mM potassium phosphate (pH 7) and 20 μM equine cytochrome c at room temperature. Mitochondria were diluted to 5 to 30 nM bc1 complex. Concentrations were determined from reduced optical spectra, using ε = 28.5 mM−1 cm−1 at 562 to 575 nm. Activity was initiated by the addition of 40 μM decylubiquinol. Cytochrome c reduction was recorded at 550 nm versus 540 nm over a 3-min time course in a Beckmann DU 640 spectrophotometer. Initial rates were measured. The IC50s were determined by inhibitor titration over a 10 to 100% inhibition range. The measurements were repeated at least twice and averaged.
Molecular docking.
Equilibrium geometry was performed on HDQ using Spartan 08 (http://www.computational-chemistry.co.uk/spartan08.html) with molecular mechanics. Molecular docking of HDQ into the Qi site of yeast bc1 complex (Protein Data Bank [PDB] 3CX5) was performed using GOLD 5.0.1 (48). Hydrogen atoms were added to the protein, and all crystallographic water molecules were removed. The yeast protein was aligned with that of bovine bc1 complex (PDB 1SQX), which is highly conserved at the Qi site, and cocrystallized with ubiquinone. The location of ubiquinone was used in the modeling to define the search space for the docking algorithm. The site for docking was defined to be all residues within 5 Å of ubiquinone in the yeast protein. Docking was also performed including waters within 3 Å of the binding site but did not yield different results compared to no waters present. No water molecules were close enough to the various docking poses to form hydrogen bonds. Analysis is based on results with no water molecules present. GoldScore fitness function was used to perform the docking. GoldScore is a molecular mechanics-based scoring function that uses a protein-ligand hydrogen bond and Van der Waals terms to optimize for the prediction of ligand-binding poses. The docking was repeated 10 times, with early termination criteria disabled and default GA settings applied.
RESULTS
HDQ disrupts P. falciparum mitochondrial function and inhibits PfNDH2 and the bc1 complex.
HDQ displayed potent inhibitory activity of P. falciparum growth proliferation with an IC50 of 86.5 ± 2.6 nM against the control strain 3D7 (all values are derived from three or more independent experiments performed as described in Materials and Methods). The drug was markedly less effective against the transgenic strain containing yeast dihydroorotate dehydrogenase (3D7-yDHODH·GFP) with an IC50 of 6.1 ± 0.9 μM, as previously reported (15). The decreased sensitivity clearly indicates that HDQ inhibits mitochondrial function, since the expression of the yeast dihydroorotate dehydrogenase has been shown to bypass the need for an efficient quinol oxidation by the bc1 complex (39). The addition of proguanil (1 μM final concentration) was observed to reverse the resistance of the transgenic 3D7-yDHODH·GFP strain to HDQ, as has been shown for atovaquone (39). Interestingly, the isolate TM90C2B that carries the cytochrome b mutation Y268S (yeast Y279S) and shows a high level of atovaquone resistance (IC50 mutant/control > 1,000), is highly sensitive to HDQ (IC50 = 64 ± 7.2 nM), which suggests that these inhibitors have a different mode/site of action. As described earlier, HDQ has previously been reported to act as an inhibitor of the type II NADH dehydrogenase (NDH2) in Y. lipolytica (16) and of NDH2 from T. gondii expressed in Y. lipolytica (36). In agreement with these published data, we found that the activity of the recombinant malaria parasite PfNDH2 was sensitive to submicromolar concentrations of HDQ with a measured IC50 of 77 ± 4.2 nM (Fig. 1a). A previous study reported that PfNDH2 was not sensitive to HDQ (15). The discrepancy is likely to originate from the significant differences in the heterologous expression strategy used for this enzyme, as well as differences in the assay conditions, these issues have been discussed previously (22).
Fig 1.

Effect of HDQ on P. falciparum PfNDH2, bc1 complex and mitochondrial membrane potential. (a) The PfNDH2 activity (●) was determined by monitoring NADH oxidation and concomitant decylquinone reduction; the bc1 activity (○) was determined by monitoring cytochrome c reduction using decylubiquinol as an electron donor (see Materials and Methods). All data have been acquired from multiple observations from at least three separate preparations. DMSO in the assays did not exceed 0.3%. The IC50s were calculated by using the four-parameter logistic method. (b) Effect of HDQ on the mitochondrial membrane potential (Ψm) of P. falciparum. The time course of TMRE-dependent fluorescence after the addition of HDQ (100 nM) to P. falciparum-infected erythrocytes is shown. The data were normalized to 100% in untreated and to 0% in CCCP (10 μM)-treated cells. Graph shows the mean data derived from experiments performed independently ± the standard errors (n ≥ 3).
Interestingly, the parasite bc1 complex activity was also found to be inhibited by nanomolar concentration of HDQ with a IC50 = 19 ± 1.3 nM (Fig. 1a), which is in the same range as the IC50 for the Qo site inhibitors atovaquone and the acridinedione WR249685 (IC50 = 3 to 5 nM) (4). Thus, the quinol analog appears to have a dual action, targeting two respiratory enzymes, the PfNDH2 and the bc1 complex. Consistent with HDQ inhibiting these respiratory enzymes, perfusion of HDQ to trophozoites-stage parasites resulted in a rapid depolarization of mitochondrial membrane potential (Fig. 1b).
Furthermore, we observed that the bc1 complex activity of the atovaquone-resistant isolate TM90C2B was as sensitive to HDQ as the wild-type (atovaquone sensitive) strain 3D7, with 200 nM HDQ reducing QH2-cytochrome c reductase activity by 74 and 77%, respectively. The TM902CB and 3D7 strains were similarly sensitive to the Qi site inhibitor antimycin (5 μM), displaying 80 and 73% inhibition, respectively, whereas 50 nM atovaquone reduced 3D7 bc1 activity by 75%, while only affecting TM902CB bc1 activity by 10% (all experiments performed in triplicate as described in Materials and Methods). These data confirm that the atovaquone resistance mutation Y268S found in the TM902CB strain does not confer cross-resistance to HDQ, indicating that the two drugs have distinct binding sites in the bc1 complex. In order to further probe the specific binding site of HDQ to the bc1 complex, we used yeast as a model system, owing to its tractability in being genetically/biochemically manipulated. We then generated a number of yeast mutants with specific point mutations in the Qo and Qi sites and monitored their impact on HDQ sensitivity.
HDQ blocks the respiratory growth of S. cerevisiae through inhibition of the bc1 complex.
We first monitored the effect of HDQ on the growth of yeast cells cultured in respiratory medium, using ethanol as a substrate, and in fermentable medium, using glucose as substrate (10% glucose without aeration) (data not shown). HDQ inhibited the respiratory growth with an IC50 of ∼1 μM. Yeast growth in fermentable medium was not affected by 20 μM HDQ, indicating that HDQ has no effect on cell growth supported by fermentation. For comparison, the IC50s for atovaquone and HQNO were approximately 20 and 5 μM, respectively. The sensitivity of the yeast NDH2 activity to HDQ was then assayed. We found that HDQ was a weak inhibitor of the S. cerevisiae NDH2 (<10% inhibition of NADH:decylubiquinone oxidoreductase activity with 1 μM HDQ, measured as described in Materials and Methods). Yeast bc1 complex activity, on the contrary, was sensitive to HDQ (Fig. 2), with a ratio of IC50 per bc1 complex of 4 to 5, which is similar to the inhibitory effect of azoxystrobin, a bc1 complex inhibitor acting at the Qo site and used to control plant pathogenic fungi.
Fig 2.
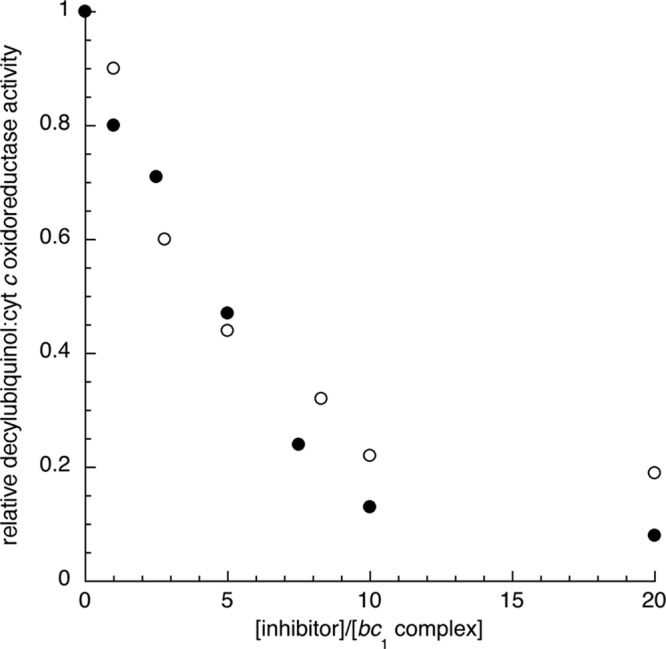
Sensitivity of the yeast bc1 complex activity to HDQ and to the Qo site inhibitor azoxystobin. The quinol cytochrome c reductase activity was measured using mitochondria prepared from WT yeast cells (see Materials and Methods). The measures were repeated at least three times and averaged. The errors did not exceed 10% of the presented values. The data are presented as the ratio of the inhibitor concentration (○, azoxystrobin; ●, HDQ) to the bc1 complex concentration. An IC50 ratio of 4 to 5 (inhibitor molecules per bc1 complex) was estimated from the plots.
HDQ acts at the Qi site of the bc1 complex.
In order to determine the site of binding of HDQ, we monitored the impact of mutations introduced in the Qo and Qi sites on the sensitivity to HDQ. First, we measured the quinol-cytochrome c reductase activity as described in Fig. 2 and determined the IC50 for HDQ. For comparison, we estimated the IC50 for two other bc1 complex inhibitors: the Qi site binding compound, antimycin and the Qo site binding compound, azoxystrobin (Table 2). Two Qi site mutations G33A and K228M, previously reported as causing resistance to HQNO and antimycin, respectively (8, 14), decreased the sensitivity of the bc1 complex toward HDQ (Table 2): the IC50s increased approximately 7- and 10-fold for G33A and K228M, respectively. K228M had a moderate effect on antimycin sensitivity while the sensitivity toward the Qo site inhibitor (azoxystrobin) was unchanged. The Qo site mutation G143A that causes a high level of resistance toward azoxystrobin, myxothiazol, and atovaquone (20) had no effect on HDQ sensitivity. When G143A and K228M mutations were combined, the resulting mutant showed resistance toward both Qo and Qi site inhibitors. We tested a third Qi site change, G37S, reported as causing weak resistance to ilicicolin H (13). This mutation had no effect on HDQ sensitivity. For all of the mutations located in the active sites (Qo or Qi), we observed a 1.5- to 2-fold decrease in bc1 complex activity.
Table 2.
Inhibitory effect of HDQ on the quinol cytochrome c reductase of WT and mutant mitochondria
| Cytochrome b WT and mutation(s) | Turnover no.a (s−1) | IC50/bc1 complexb |
||
|---|---|---|---|---|
| HDQ | Antimycin | Azoxystrobin | ||
| WT | 33 | 4 | 0.5 | 5 |
| Qi site | ||||
| G33A | 20 | 30 | 0.4 | ND |
| G37S | 20 | 4 | 0.4 | ND |
| K228M | 16 | 30–50 | 0.8 | 4 |
| Qo site | ||||
| G143A | 15 | 4 | 0.5 | >20 |
| Qi and Qo | ||||
| G143A+K228M | 18 | 30–40 | 0.9 | >20 |
The turnover number is the amount of cytochrome c reduced per bc1 complex per s using 40 μM decylubiquinol (see Materials and Methods). The measurements were repeated at least twice. The errors did not exceed 10% of the presented values.
Values are presented as the ratio of the IC50 to the concentration of the monomeric bc1 complex (estimated using the cytochrome optical signal as described in Materials and Methods). For example, 0.5 molecules of the tight binding inhibitor antimycin were added per monomeric bc1 complex to inhibit the quinol cytochrome c reductase activity by 50%. ND, not determined.
We then monitored the inhibitory effect of HDQ on the respiratory growth of a collection of Qi mutants chosen on the basis of the structure (28, 29) and/or on previous report of their effect on HQNO sensitivity (10) (Table 3). In line with the enzymatic data presented in Table 2, G33A and K228M (and the double-change G143A+K228M) conferred a high level of resistance (IC50 of >10 μM compared to 1 μM for the WT). These mutants also showed a marked (>10-fold) resistance to HQNO (data not shown). Two other Qi mutations (causing HQNO resistance [10]) resulted in an increased resistance toward HDQ, H204Y and M221Q, whereas mutations in positions 208, 218, and 225 had little effect. Interestingly, the HDQ sensitivity of mutant K228I was similar to the WT, in direct contrast to K228M. S206T/V and G37S appeared to be more sensitive to HDQ. The increased sensitivity of the respiratory growth of G37S might seem surprising, since the sensitivity of the mutated bc1 complex was unchanged compared to the WT. However, this behavior can be explained by the decreased activity of the bc1 complex in the mutant (Table 2). Since the level of bc1 complex activity is lower in the mutant cells, its titration by HDQ in the culture gave a lower value. A similar behavior explains the increased sensitivity of S206V/T. Note that the respiratory growth of the mutants with lower bc1 activity was not decreased in the absence of inhibitor. The bc1 complex activity is in excess in WT yeast cell, and only a severe decrease in the activity or level of the complex results in a defective respiratory growth.
Table 3.
Differential effects of cytochrome b mutations on the sensitivity of the respiratory growth toward HDQ
| WT and site mutation(s) | IC50 (μM)a |
|---|---|
| WT | 1 |
| Qi site | |
| G33A | >10 |
| G37S | <0.05 |
| H204Y | >10 |
| S206T/V | <1 |
| N208V | 1–2 |
| R218K | 1–2 |
| M221Q | >10 |
| F225L | 1–2 |
| K228M | >10 |
| K228I | 1 |
| G143A+K228M | >10 |
| Qo site | |
| T127I | 1 |
| G143A | 0.05–0.1 |
| I147V | 0.5 |
| S152A | 0.05 |
| L275F | 0.5–1 |
| Y279C | <0.05 |
| Y279S | <0.05 |
The IC50 values were estimated as described in Materials and Methods. Cells were cultured in respiratory medium (ethanol) in the presence of increasing concentrations of HDQ. The cell density was measured at the early stationary phase. The cell density of the mutants in the absence of inhibitors was similar to that of the WT. The measurements were repeated at least twice.
For comparison, we monitored the inhibitory effect of HDQ on respiratory growth of Qo site mutants (Table 3). T127I, I147V, S152A, and L275F are mutations found in the human pathogenic fungus P. jiroveci, after atovaquone treatment (26); Y279C and Y279S are atovaquone resistance mutations reported in P. falciparum (Y268C/S in P. falciparum). G143A causes a high level of resistance toward Qo site fungicides in plant pathogenic fungi and was shown to confer atovaquone resistance when introduced in yeast (20; for a review of cytochrome b mutations, see reference 21). None of the atovaquone resistance mutations conferred cross-resistance to HDQ. An increased sensitivity toward HDQ was observed for several mutants. The increased sensitivity correlates with a decreased activity of the mutated bc1 complex caused by the Qo site mutation. T127I and I147V had no or mild effect on the bc1 complex activity (26) and on the sensitivity toward HDQ, whereas S152A caused a 5-fold decrease in activity (26) and a marked sensitivity toward HDQ. The G143A mutation resulted in a 2-fold decrease in quinol cytochrome c reductase activity and a >10-fold decrease in the sensitivity of the respiratory growth to HDQ (Tables 2 and 3). L275F had no effect on the respiratory growth sensitivity and the bc1 complex activity (not shown). Y279C/S has previously been shown to severely impair bc1 complex activity (31) and, accordingly, the IC50 on respiratory growth was markedly more sensitive to HDQ (<0.05 μM). Consistent with these data, Qi site mutants with reduced bc1 activity showed an increased sensitivity toward the Qo site inhibitor atovaquone (data not shown). The double mutant that combines Qo and Qi site resistance mutations G143A+K228M expressed the combined resistance toward Qo and Qi inhibitors.
In summary, four Qi site HQNO-resistance mutations caused a marked cross-resistance to HDQ, whereas none of the atovaquone-resistant Qo site mutations tested conferred cross-resistance to HDQ. It is interesting that, as described above for P. falciparum TM902CB, the mutation Y279S (Y268S in P. falciparum), when introduced in yeast, caused a marked decreased in bc1 complex activity, a high level of resistance to atovaquone (20, 31) but no cross-resistance toward HDQ. These data are consistent with the hypothesis that HDQ targets the Qi site of the bc1 complex. Molecular modeling was then performed to further explore the molecular interactions of HDQ binding at the Qi site of the bc1 complex.
Molecular modeling of HDQ binding at the Qi site of the S. cerevisiae bc1 complex.
The analysis was performed using GOLD 5.0.1 with PDB 3CX5 as a template (see Materials and Methods). The best scoring pose had a GoldScore of 59.4, indicative of a good binding orientation within the Qi site. Subsequent poses supported the predicted orientation. A detailed description of the model of docking and associated data are presented in the supplementary data (see Fig. S2 and Table S1 in the supplemental material). For comparison purposes, in silico docking of ubiquinone-6 within the Qi site of yeast bc1 resulted in a “best solution” binding pose with a GoldScore of 50.4. Docking of stigmatellin at the Qo and Qi sites of yeast bc1 resulted in GoldScores of 111.6 and 56.3, respectively, in agreement with the expected binding site for this inhibitor. Interestingly, the favored docking pose for HDQ within the Qo site had a GoldScore value of 65.4, which is suggestive of an apparently more favorable interaction than that observed at Qi. However, it should be noted that hydrogen bonds are more highly weighted than hydrophobic and van der Waals interactions as determined by the GOLD docking algorithm, and the more hydrophilic nature of the Qo site compared to Qi may artificially favor HDQ docking in silico at Qo. The mobility of the ectodomain of the Rieske protein and the presence of protein-bound water within Qo also presents challenges for in silico modeling methods.
The protein-HDQ interactions were similar to that of Qi-bound substrate ubiquinone-6 in PDB entry 1EZV and to the binding interactions of the quinolone headgroup of NQNO (2-nonyl-4-hydroxyquinoline N-oxide), the C9-alkyl variant of HDQ, in the bovine structure (25). The Qi site mutations analyzed above were reexamined in light of this model (Fig. 3). G33A (a conserved residue in mammalian, yeast, and P. falciparum cytochrome b and located in transmembrane helix A) has been previously shown to cause resistance to HQNO but not to antimycin (8). In our binding model, the αC atom of G33 has a closest approach of 4.4 Å to the quinolone headgroup of HDQ, so it is reasonable to suggest that introduction of a methyl group at this position may result in steric hindrance with Qi-bound molecules. H204, conserved in yeast and Plasmodium bc1 complex, is replaced by a threonine in mammals and has a closest approach of 10 Å to the quinolone headgroup of NQNO in the bovine atomic structure (25). A similar separation is observed in our HDQ-docked yeast model. It is thus not immediately apparent why this mutation should affect quinolone binding and yet not be deleterious for bc1 complex activity (9), although it may be postulated that introduction of the bulkier tyrosyl side chain results in local reorganization in the packing of side chains or conformation in the DE loop. M221Q (transmembrane helix E) causes resistance to HQNO but not antimycin and has a nearly WT level of bc1 complex activity (8). The εC methyl group of M221 is predicted to be in a stabilizing hydrophobic interaction with the quinolone ring of HDQ in our yeast structural model (3.6 Å closest approach). This residue is replaced by phenylalanine in mammalian and Plasmodium cytochrome b. In the NQNO-inhibited bovine enzyme, this phenylalanine side chain participates in a stabilizing aromatic-aromatic interaction with the bound quinolone. Mutation to the more polar glutamine side chain removes this interaction. K228M (transmembrane helix E) confers weak resistance to antimycin but marked resistance to HDQ (Table 2). The terminal amino group of this residue participates in a water-mediated H-bond to the formyl amino oxygen atom of bound antimycin in bovine bc1 complex (28). A similar bridged hydrogen-bonding association has been suggested for this residue and substrate ubiquinone in the yeast bc1 structure, where it was proposed to form part of a proton-uptake pathway for quinone redox chemistry at Qi (30). Interestingly, the side chain appears to demonstrate considerably mobility and can be modeled in two different conformations (the terminal NE atoms of K228 point in different directions in the yeast and bovine crystal structures [25]). This residue has a closest approach of 5.5 Å to NQNO in the bovine structure, although it is unclear whether it is participating in a water-bridged H-bond with the quinolone N-oxide moiety. In our structural model, which was based on the yeast coordinates (3CX5 [43]), the side chain of K228 is oriented “distal” to the Qi pocket, and so we observe a separation of 13 Å to the quinone headgroup of bound HDQ. Rotation of this residue into the Qi “proximal” position would facilitate a bridged hydrogen bond between the terminal amino group and the HDQ quinolone headgroup. It should be noted, however, that the mutation of this residue to isoleucine did not confer resistance to HDQ (and K228 is replaced by leucine in P. falciparum cytochrome b), and so it is unlikely that hydrogen bonds to this position are a significant factor in quinolone binding. It is therefore unclear why the K228M mutation should confer HDQ resistance, it may arise from distortion of the nearby aA loop, a region of the Qi site predicted to contain several residues in close association with bound HDQ.
Fig 3.

Model of HDQ binding in the Qi site and localization of Qi mutations. (a) Structural model (obtained as described in Materials and Methods) showing mutations causing resistance to HDQ (red) and mutations without effect on HDQ resistance (green). (b) Comparison of the sequences of Qi site region between bovine (Bt), yeast S. cerevisiae (Sc), and P. falciparum (Pf) cytochromes b. Residues in close contact (>4 Å) with the antimycin bound at the Qi site in the bovine enzyme are indicated in boldface (28). The mutated residues analyzed in the present study are marked with an asterisk (*).
G37S (transmembrane helix A) has been reported previously as causing resistance to ilicicolin H (13); no resistance to HDQ was observed here. This residue has a closest approach of 8.3 Å to HDQ in our docking model. S206 (DE loop) is conserved in eukaryotic cytochrome b and is predicted to be a H-bond donor via its hydroxyl moiety to the methoxy oxygen atom of Qi-bound ubiquinone in the bovine and yeast bc1 complex crystal structures (28, 29). Mutation to threonine or valine, however, has no effect on bc1 complex catalytic activity (9) and confers no resistance to HDQ. F225L (transmembrane helix E) has previously reported as causing resistance to diuron (14). The phenyl ring of this residue has a closest contact of 3.1 Å with the quinolone group of HDQ in our docking model, with the rings oriented at 120 degrees with respect to each other and a van der Waals contact surface area of 19 Å2. The isobutyl group of leucine is likely to be able to form a similar stabilizing hydrophobic interaction with HDQ in the F225L mutant and, indeed, leucine is found in this position in the sequence of P. falciparum cytochrome b. N208V and R218K, both located in the DE loop (proposed as a proton input pathway for quinone redox chemistry at Qi [30]), are ∼10 Å from HDQ in our binding model.
DISCUSSION
HDQ has been shown previously to display potent antimalarial activity (40). Here, we confirm this observation and additionally show that this compound is active against the atovaquone resistance parasite TM90C2B (carrying the Y268S mutation), suggesting a different target site of action to atovaquone. The lack of activity of HDQ against the transgenic 3D7-yDHODH·GFP strain (39), indicated that this compound targets mitochondrial function. Consistent with this, assessment of mitochondrial function using single-cell imaging of parasite mitochondria revealed that addition of HDQ rapidly depolarized the mitochondrial membrane potential. Assessment of the electron transport chain enzymes NADH:ubiquinone oxidoreductase (PfNDH2 [18]) and bc1 complex revealed that HDQ is a potent inhibitor of both enzymes. HDQ therefore displays characteristics of a privileged pharmacophore able to inhibit more than one enzyme. It is not clear, however, from our data alone whether parasite kill is afforded via the inhibition of bc1 alone or via a combination of the inhibition of bc1 and PfNDH2. A recent study, performed in the rodent malaria P. berghei, indicates that deletion of the NDH2 gene is not lethal to erythrocytic stages of the parasite (7). Our observation that proguanil reverses the resistance of the transgenic 3D7-yDHODH·GFP to HDQ, is consistent with similar observations using bc1-acting inhibitors (39). However, this same experiment has not been performed with a known PfNDH2-selective drug and is therefore difficult to interpret. Clearly further, definitive investigations are required to determine the essentiality of PfNDH2 in P. falciparum. It is worth noting, however, that, historically, anti-infectives displaying polypharmacology show greater efficacy over single-targeting inhibitors (27). In Toxoplasma gondii, the deletion of type II NADH:dehydrogenase genes is not lethal but is required for optimal tachyzoite growth (35). Interestingly, HDQ has been shown to be synergistic with atovaquone for growth inhibition (36), which may be attributed to the polypharmacological effect of HDQ against the type II NADH: dehydrogenases as well as the Qo and Qi sites of the bc1 complex.
In order to determine the mode of action of HDQ against the bc1 complex, yeast mutants were generated carrying specific amino acid substitutions in the two catalytic sites, Qo and Qi (Table 1). The specific Qi point mutations tested were chosen because of their involvement in the binding of antimycin and ubiquinol in the Qi site of the bovine enzyme, as revealed by crystal structures (28, 29) and/or by a previous report of the effect on HQNO sensitivity (10): S206T/V, H204Y, K228M/I, G33A, G37S, M221Q, and F225L. Two residues located in a possible proton pathway toward the Qi site (30), R218K and N208V, were also tested. The Qo site mutations were atovaquone-resistant mutations reported in human and plant pathogens. None of the Qo site mutations conferred cross-resistance to HDQ, while four Qi site mutations caused a cross-resistance toward HDQ and HQNO, namely, G33A, H204Y, M221Q, and K228M (Tables 2 and 3). Thus, it could be suggested that these residues are involved in the stabilization of HDQ in the Qi site. As described above, molecular modeling of HDQ bound to the Qi site supports the importance of residues G33, H204, and M221 for the binding interaction; however, it is not clear at this stage why the K228M mutation affects this process.
The mutational study cannot exclude that HDQ could also bind at the Qo site with a lower affinity. Inhibitors have been described that bind at both Qo and Qi sites. For instance, spectroscopic studies have shown that Ascochlorin acted at both sites of the bacterial and vertebrate bc1 complex and crystallographic analyses has revealed its precise binding sites in the two quinone pockets of the chicken enzyme (3). Crystallographic analysis of the bovine bc1 complex showed that 2-nonyl-4-hydroxyquinoline N-oxide (NQNO) binds at both sites in the crystal structure (25). It is likely that HQNO could also bind at the Qo site. However, amino acid substitutions causing resistance to HQNO have only been found in the Qi site (10). This suggests that the affinity of NQNO/HQNO for the Qo site would be lower; thus, resistance could not develop by mutation of the Qo site.
Comparison of sequences (Fig. 3B) shows that Qi site residues involved in HDQ stabilization (as judged by mutational analysis) are well conserved between yeast and P. falciparum enzyme. It is likely that HDQ would also target the Qi site of P. falciparum, which is in agreement with the observation that the atovaquone resistance P. falciparum isolate TM90C2B (with the Qo site mutation Y279S [Y268S in P. falciparum]) is sensitive to HDQ.
The bc1 complex of the malaria parasite is a proven drug target and is an essential component for various stages of the parasite life cycle, including the liver stages and the circulating asexual stages. This enzyme is therefore the only validated malarial drug target that has utility for both curative and prophylaxis treatment. Unfortunately, atovaquone-resistant parasites have been observed in the field following atovaquone or Malarone treatment failures (see, for example, references 2, 23, 38, and 41). Atovaquone, as do the fungicides and pesticides targeting the bc1 complex, binds at the Qo site (the only exception is the fungicide cyazofamid that targets the Qi site and is only active against oomycetes [37]). Mutations in the Qo site have been reported that compromise the pathogen control (reviewed in reference 21). In addition, the Qo inhibitors have similar modes of binding, and thus cross-resistance toward the drugs is observed. Thus, inhibitors acting at the Qi site would be invaluable tools against pathogens.
With the exception of cyazofamid, most bc1 inhibitors that have been developed into drugs or pesticides do not target the Qi. The Qi site is less conserved among species than the Qo site. From an alignment of 16 diverse cytochrome b sequences, including vertebrates, invertebrates, fungi, plants, and protozoa, the estimated sequence similarity at the Qo site is 48% for the ef loop and 60% for the C terminus of the helix C and the cd1 loop; the sequence similarity at the Qi site is 30% for the aA loop and N terminus of helix A, 36% for the C terminus of helix D and the DE loop and 44% for the N terminus of helix E (data not shown). Thus, from the drug development point of view, the relative sequence diversity of the Qi pocket should have favored selectivity. On the other hand, Qi appears to be more structurally rigid (25, 28), with no evidence of movement on antimycin binding, whereas the Qo site shows more mechanical flexibility on inhibitor binding (17). It was previously reported that in the Qo pocket, H-bonds between side chains of a few conserved residues hold together the components of the site that are regions of the cytochrome b distant in the sequence. Crofts et al. described the feature as a “loose stitching” that would allow the expansion of the site upon binding of inhibitors (12). That property presumably allows the site to accommodate more diverse molecules and could explain the development of specific Qo inhibitors active against pathogens.
We showed here that HDQ, active against P. falciparum, is a proof-of-concept molecule that targets the Qi site and that can therefore circumvent atovaquone resistance. HDQ is not active in vivo using the P. berghei rodent model (7), but given its potent activity against bc1, this is most probably due to poor drug exposure. Drugs developed on the HDQ scaffold targeting the Qi site, but with improved pharmacokinetic features, may therefore be a valuable tool in combating parasite drug resistance as we would predict that if used in combination, Qi and Qo site inhibitors would be more recalcitrant to the emergence of resistance. Double mutations at both active sites, if they arose, would result in a severe loss of function and thus of cell fitness and are unlikely to be tolerated. Further synthetic studies are now under way to develop more drug-like inhibitors of the Qi site of the bc1 complex.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dennis Kyle (College of Public Health, University of South Florida) for supplying the atovaquone-resistant isolate TM90C2B (Thailand) and Akhil Vaidya (Drexel University College of Medicine, Philadelphia) for supplying purified pHHyDHOD-GFP plasmid. We also thank the staff and patients of Ward 7Y and the Gastroenterology Unit, Royal Liverpool Hospital, for their generous donation of blood.
This study was supported by grants from the Leverhulme Trust, the Wellcome Trust, the National Institute of Health Research (BRC Liverpool), and the Agence National de la Recherche.
Footnotes
Published ahead of print 30 April 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Barton V, Fisher N, Biagini GA, Ward SA, O'Neill P. 2010. Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr. Opin. Chem. Biol. 14:1–7 [DOI] [PubMed] [Google Scholar]
- 2. Berry A, et al. 2006. Prevalence of Plasmodium falciparum cytochrome b gene mutations in isolates imported from Africa, and implications for atovaquone resistance. Trans. R. Soc. Trop. Med. Hyg. 100:986–988 [DOI] [PubMed] [Google Scholar]
- 3. Berry EA, et al. 2010. Ascochlorin is a novel, specific inhibitor of the mitochondrial cytochrome bc1 complex. Biochim. Biophys. Acta 1797:360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biagini G, et al. 2008. Acridinediones: selective and potent inhibitors of the malaria parasite mitochondrial bc1 complex. Mol. Pharmacol. 73:1347–1355 [DOI] [PubMed] [Google Scholar]
- 5. Biagini GA, Viriyavejakul P, O'Neill PM, Bray PG, Ward SA. 2006. Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrob. Agents Chemother. 50:1841–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boulton IC, et al. 2010. CRIMALDDI: a coordinated, rational, and integrated effort to set logical priorities in anti-malarial drug discovery initiatives. Malar. J. 9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boysen KE, Matuschewski K. 2011. Arrested oocyst maturation in Plasmodium parasites lacking type II NADH:ubiquinone dehydrogenase. J. Biol. Chem. 286:32661–32671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brasseur G, Brivet-Chevillotte P. 1995. Characterization of mutations in the mitochondrial cytochrome b gene of Saccharomyces cerevisiae affecting the quinone reductase site (Qn). Eur. J. Biochem. 230:1118–1124 [DOI] [PubMed] [Google Scholar]
- 9. Brasseur G, Coppee J-Y, Colson A-M, Brivet-Chevillotte P. 1995. Structure-function relationships of the mitochondrial bc1 complex in temperature-sensitive mutants of the cytochrome b gene, impaired in the catalytic center N. J. Biol. Chem. 270:29356–29364 [DOI] [PubMed] [Google Scholar]
- 10. Brasseur G, Saribas AS, Daldal F. 1996. A compilation of mutations located in the cytochrome b subunit of the bacterial and mitochondrial bc1 complex. Biochim. Biophys. Acta 1275:61–69 [DOI] [PubMed] [Google Scholar]
- 11. Coppee J-Y, Brasseur G, Brivet-Chevillotte P, Colson A-M. 1994. Non-native intragenic reversions selected from Saccharomyces cerevisiae cytochrome b-deficient mutants. J. Biol. Chem. 269:4221–4226 [PubMed] [Google Scholar]
- 12. Crofts AR, et al. 1999. Mechanism of ubiquinol oxidation by the bc1 complex: different domains of the quinol binding pocket and their role in the mechanism and binding of inhibitors. Biochemistry 38:15807–15826 [DOI] [PubMed] [Google Scholar]
- 13. Ding MG, di Rago J-P, Trumpower BL. 2006. Investigating the Qn site of the cytochrome bc1 complex in Saccharomyces cerevisiae with mutants resistant to ilicicolin H, a novel Qn site inhibitor. J. Biol. Chem. 281:36036–36043 [DOI] [PubMed] [Google Scholar]
- 14. di Rago J-P, Colson A-M. 1988. Molecular basis for resistance to antimycin and diruon, Q-cycle inhibitors acting at the Qi site in the mitochondrial ubiquinol-cytochrome c reductase in Saccharomyces cerevisiae. J. Biol. Chem. 263:12564–12570 [PubMed] [Google Scholar]
- 15. Dong CK, et al. 2009. Type II NADH dehydrogenase of the respiratory chain of Plasmodium falciparum and its inhibitors. Bioorg. Med. Chem. Lett. 19:972–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eschemann A, Galkin AG, Oettmeier W, Brandt U, Kerscher S. 2005. HDQ (1-hydroxy-2-dodecyl-4(1H)quinolone), a high-affinity inhibitor for mitochondrial alternative NADH dehydrogenase. J. Biol. Chem. 280:3138–3142 [DOI] [PubMed] [Google Scholar]
- 17. Esser L, et al. 2004. Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for cytochrome bc1 complex. J. Mol. Biol. 341:281–302 [DOI] [PubMed] [Google Scholar]
- 18. Fisher N, Bray PG, Ward SA, Biagini GA. 2007. The malaria parasite type II NADH:quinone oxidoreductase: and alternative enzyme for an alternative lifestyle. Trends Parasitol. 23:305–310 [DOI] [PubMed] [Google Scholar]
- 19. Fisher N, et al. 2004. Human disease-related mutations in cytochrome b studied in yeast. J. Biol. Chem. 279:12951–12958 [DOI] [PubMed] [Google Scholar]
- 20. Fisher N, Meunier B. 2005. Re-examination of inhibitor resistance conferred by Qo site mutations in cytochrome b using yeast as a model system. Pest. Manag. Sci. 61:973–978 [DOI] [PubMed] [Google Scholar]
- 21. Fisher N, Meunier B. 2008. Molecular basis of resistance to cytochrome bc1 inhibitors. FEMS Yeast Res. 8:183–192 [DOI] [PubMed] [Google Scholar]
- 22. Fisher N, Warman AJ, Ward SA, Biagini GA. 2009. Chapter 17 type II NADH: quinone oxidoreductases of Plasmodium falciparum and Mycobacterium tuberculosis kinetic and high-throughput assays. Methods Enzymol. 456:303–320 [DOI] [PubMed] [Google Scholar]
- 23. Fivelman QL, Butcher GA, Adagu IS, Warhurst DC, Pasvol G. 2002. Malarone treatment failure and in vitro confirmation of resistance of Plasmodium falciparum isolate from Lagos, Nigeria. Malar. J. 1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fry M, Pudney M. 1992. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43:1545–1553 [DOI] [PubMed] [Google Scholar]
- 25. Gao X, et al. 2003. Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry 42:9067–9080 [DOI] [PubMed] [Google Scholar]
- 26. Hill P, et al. 2003. Recapitulation in Saccharomyces cerevisiae of cytochrome b mutations conferring resistance to atovaquone in Pneumocystis jiroveci. Antimicrob. Agents Chemother. 47:2725–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hopkins AL. 2008. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 4:682–690 [DOI] [PubMed] [Google Scholar]
- 28. Huang L-S, Cobessi D, Tung E, Berry EA. 2005. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol. 351:573–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hunte C, Koepke J, Lange C, Rossmanith T, Michel H. 2000. Structure at 2.3 angstrom resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Struct. Fold. Des. 8:669–684 [DOI] [PubMed] [Google Scholar]
- 30. Hunte C, Palsdottir H, Trumpower BL. 2003. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 545:39–46 [DOI] [PubMed] [Google Scholar]
- 31. Kessl J, et al. 2005. Cytochrome b mutations that modify the ubiquinol-binding pocket if the cytochrome bc1 and confer anti-malarial drug resistance in Saccharomyces cerevisiae. J. Biol. Chem. 280:17142–17148 [DOI] [PubMed] [Google Scholar]
- 32. Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418–420 [PubMed] [Google Scholar]
- 33. Lemaire C, Dujardin G. 2008. Preparation of respiratory chain complexes from Saccharomyces cerevisiae wild-type and mutant mitochondria: activity measurement and subunit composition analysis. Methods Mol. Biol. 432:65–81 [DOI] [PubMed] [Google Scholar]
- 34. Lin SS, Gross U, Bohne W. 2009. Type II NADH dehydrogenase inhibitor 1-hydroxy-2-dodecyl-4(1H)quinolone leads to collapse of mitochondrial inner-membrane potential and ATP depletion in Toxoplasma gondii. Eukaryot. Cell 8:877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin SS, Gross U, Bohne W. 2011. Two internal type II NADH dehydrogenases of Toxoplasma gondii are both required for optimal tachyzoite growth. Mol. Microbiol. 82:209–221 [DOI] [PubMed] [Google Scholar]
- 36. Lin SS, et al. 2008. The Toxoplasma gondii type-II NADH dehydrogenase TgNDH2-I is inhibited by 1-hydroxy-2-alkyl-4(1H)quinolones. Biochim. Biophys. Acta 1777:1455–1462 [DOI] [PubMed] [Google Scholar]
- 37. Mitani S, et al. 2001. The biochemical mode of action of the novel selective fungicide Cyazofamid: specific inhibition of mitochondrial complex III in Phythium spinosum. Pestic. Biochem. Physiol. 71:107–115 [Google Scholar]
- 38. Musset L, Bouchaud O, Matheron S, Massias L, Le Bras J. 2006. Clinical atovaquone-proguanil resistance of Plasmodium falciparum associated with cytochrome b codon 268 mutations. Microbes Infect. 8:2599–2604 [DOI] [PubMed] [Google Scholar]
- 39. Painter HJ, Morrisey JM, Mather MW, Vaidya AB. 2007. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446:88–91 [DOI] [PubMed] [Google Scholar]
- 40. Saleh A, Friesen J, Baumeister S, Gross U, Bohne W. 2007. Growth inhibition of Toxoplasma gondii and Plasmodium falciparum by nanomolar concentrations of 1-hydroxy-2-dodecyl-4(1H)quinolone, a high-affinity inhibitor of alternative (type II) NADH dehydrogenases. Antimicrob. Agents Chemother. 51:1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwartz E, Bujanover S, Kain KC. 2003. Genetic confirmation of atovaquone-proguanil-resistant Plasmodium falciparum malaria acquired by a nonimmune traveler to East Africa. Clin. Infect. Dis. 37:450–451 [DOI] [PubMed] [Google Scholar]
- 42. Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48:1803–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solmaz SR, Hunte C. 2008. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J. Biol. Chem. 283:17542–17549 [DOI] [PubMed] [Google Scholar]
- 44. Srivastava IK, Morrisey JM, Darrouzet E, Daldal F, Vaidya AB. 1999. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 33:704–711 [DOI] [PubMed] [Google Scholar]
- 45. Srivastava IK, Rottenburg Vaidya AB. 1997. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 272:3961–3966 [DOI] [PubMed] [Google Scholar]
- 46. Tietze LF, Ma L. 2010. Synthesis of novel 1-hydroxyquinolones with high anti-toxoplasma activity. Heterocycles 82:377–396 [Google Scholar]
- 47. Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675 [DOI] [PubMed] [Google Scholar]
- 48. Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. 2003. Improved protein-ligand docking using GOLD. Proteins 52:609–623 [DOI] [PubMed] [Google Scholar]
- 49. Woschek A, Mahout M, Mereiter K, Hammerschmidt F. 2007. Synthesis of 2-heptyl-1-hydroxy-4(1H)-quinolone: unexpected rearrangement of 4-(alkoxycarbonyloxy)quinoline N-oxides to 1-(alkoxycarbonyl-oxy)-4(1H)-quinolones. Synthesis Stuttgart 10:1517–1522 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.