

Abstract
The WalK (histidine kinase)/WalR (response regulator) two-component signal transduction system is a master regulatory system for cell wall metabolism and growth. This system is conserved in low G+C Gram-positive bacteria, including Bacillus subtilis, Staphylococcus aureus, Enterococcus faecalis, and Streptococcus mutans. In this study, we found the first antibiotic that functions as a WalK inhibitor (signermycin B) by screening 10,000 Streptomyces extracts. The chemical structure (C23H35NO4; molecular weight, 389.5) comprises a tetramic acid moiety and a decalin ring. Signermycin B exhibited antimicrobial activity, with MIC values ranging from 3.13 μg/ml (8 μM) to 6.25 μg/ml (16 μM) against Gram-positive bacteria that possess the WalK/WalR two-component signal transduction system, including the drug-resistant bacteria methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecalis. The half-maximal inhibitory concentrations of signermycin B against WalK in these organisms ranged from 37 to 61 μM. To determine the mechanism of action of signermycin B, surface plasmon resonance response analysis with the two WalK domains of Bacillus subtilis and competition assay with ATP were performed. The results showed that signermycin B binds to the dimerization domain but not the ATP-binding domain of WalK. In the presence of the cross-linker glutaraldehyde, signermycin B did not cause protein aggregation but interfered with the cross-linking of WalK dimers. These results suggest that signermycin B targets the conserved dimerization domain of WalK to inhibit autophosphorylation. In Bacillus subtilis and Staphylococcus aureus, signermycin B preferentially controlled the WalR regulon, thereby inhibiting cell division. These phenotypes are consistent with those of cells starved for the WalK/WalR system.
INTRODUCTION
Two-component signal transduction systems (TCSs) allow bacteria to rapidly adapt to physical, chemical, and biological stresses originating from outside the cell. The bacterial TCS consists of a membrane-bound sensor histidine kinase (HK) and a cytosolic response regulator (RR) (12). In response to an environmental signal, the sensor HK autophosphorylates at a conserved histidine residue in the dimerization domain and transfers the phosphoryl group to the conserved residue in the regulatory domain of its cognate RR. The phosphorylated RR then binds to upstream promoter regions of target genes to regulate their expression. Some TCSs control gene clusters essential for cell viability, whereas others control genes involved in virulence, biofilm formation, and quorum sensing in pathogenic bacteria (14). Thus, TCS inhibitors are expected to exert a range of effects (2, 14).
The QseC/QseB system is involved in the virulence of the human pathogen Escherichia coli O157 (i.e., enterohemorrhagic E. coli [EHEC]). When activated by autoinducer 3, the QseC/QseB system rapidly induces locus of enterocyte effacement virulence genes of EHEC when cell densities are high (i.e., a quorum is achieved) (21, 25). Inhibitors that target QseC/QseB could suppress the virulence factors of EHEC without killing cells.
In contrast, the WalK/WalR (i.e., YycG/YycF) TCS, which is specific to low G+C Gram-positive bacteria (e.g., Bacillus subtilis, Staphylococcus aureus, Enterococcus faecalis, and Streptococcus mutans), is essential for bacterial growth rather than virulence (9, 10). WalK/WalR is a novel target for antibacterial agents against multidrug-resistant bacteria, including methicillin-resistant S. aureus and vancomycin-resistant E. faecalis (9, 14).
WalK is a membrane-linked HK that possesses a catalytic domain and a dimerization domain conserved at the cytoplasmic C-terminal region. WalK cytoplasmic domains have been purified, and results of in vitro studies show that they are autophosphorylated and phosphorylate their cognate RRs (6, 7, 17, 31, 32). Cytoplasmic or truncated forms of HKs have been reported to dimerize in vitro (15). In addition, a solution-state nuclear magnetic resonance (NMR) study (30) described dimerization of the homodimeric core domain of EnvZ, which belongs to the same Pho subfamily of HKs as WalK. Dimerization appears to be essential for HK autophosphorylation (18).
WalR is a typical RR with an N-terminal receiver domain and a C-terminal DNA-binding domain. The receiver domain contains the invariant active-site residues, including a conserved aspartic acid residue that serves as the phosphorylation site. The WalR C-terminal domains of B. subtilis and S. aureus are similar to that of the RR PhoB from E. coli and contain a winged helix-turn-helix motif for DNA binding (3, 24). Both WalRs form a dimer to bind to a DNA sequence consisting of two hexanucleotide direct repeats separated by five nucleotides (5′-TGTWAN-N5-TGTWAH-3′) (4).
In B. subtilis, WalK/WalR activate expression of yocH, yvcE, and lytE, which encode autolysins, and ydjM, which is predicted to encode a cell wall-associated protein (1, 17, 27). In addition, WalK/WalR negatively regulate the expression of yoeB and yjeA, which encode proteins that modulate autolysin activity (1, 26). In S. aureus, the WalK/WalR-dependent regulation of nine cell wall metabolism genes was reported (5). These genes encode two major S. aureus autolysins (AtlA and LytM) and lytic transglycosylases (IsaA and SceD) (5, 6, 27) and five proteins that contain a CHAP amidase domain (SsaA, SA0620, SA2097, SA2353, and SA0710) (4, 5). These studies have revealed that the WalK/WalR TCS functions as a master regulatory system for cell wall metabolism (4, 5, 6).
Taken together, these results suggest the WalK/WalR TCS to be a potential new antibiotic target. We previously developed a high-throughput genetic system using a synthetic chemical library to isolate antibacterial agents able to inhibit the homodimerization of WalK and WalR. The results showed that WalK and WalR inhibitors have antibacterial activity (11, 13). The WalK dimerization domain is also expected to be an effective target for inhibition. In this study, we screened natural products to find an HK inhibitor that targets the dimerization domain of WalK and discovered a new antibiotic, signermycin B.
MATERIALS AND METHODS
Strains, plasmids, and chemicals.
Bacterial strains and plasmids used in this study are listed in Table 1. All chemicals were obtained from Sigma-Aldrich (Milwaukee, WI), Nacalai Tesque (Kyoto, Japan), or Wako (Osaka, Japan).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli BL21(DE3) | F− ompT hsdSB(rB− mB−) gal(λcI857 ind1, Sam7 nin5 lacUV5-T7 gene1) dcm(DE3) | Novagen |
| B. subtilis 168 | trpC2 | Laboratory stock |
| S. aureus | ||
| FDA209P | Wild type | MCRFa |
| N315 | Methicillin sensitive | 13 |
| MS16526 | Methicillin resistant | MCRF |
| Streptomyces sp. strain MK851-mF8 | Signermycin B producing | This study |
| E. faecalis | ||
| JCM5803 | Wild type | MCRF |
| NCTC12201 | Vancomycin resistant | MCRF |
| S. mutans UA159 | Wild type | ATCC |
| Plasmids | ||
| pHT10 | Expression vector for E. coli and B. subtilis; Apr Cmr | MoBi Tec |
| pET-21a(+) | Expression vector, Apr, C-terminal His tag | Novagen |
| pET-22b(+) | Expression vector, Apr, C-terminal His tag | Novagen |
| pBsG-full | WalK(Bs) (B. subtilis 168 aa 1 to 611) cloned into pHT10 | This study |
| pETYycG-tru | WalK(Bs) (B. subtilis 168 aa 207 to 611) cloned into pET-21a(+) | 23 |
| pBsGsubA | WalK(Bs) (B. subtilis 168 aa 366 to 611) cloned into pET-21a(+) | This study |
| pBsGsubB | WalK(Bs) (B. subtilis 168 aa 437 to 611) cloned into pET-21a(+) | This study |
| pABF | B. subtilis 168 WalR under control of Pspac | 13 |
| pYycG Sa | WalK(Sa) (S. aureus N315 aa 204 to 608) cloned into pET-21a (+) | 31 |
| pETEfWalK | WalK(Efa) (E. faecalis JCM5803 aa 203 to 611) cloned into pET-21a(+) | This study |
| pETSMVicK31-450 | WalK(Sm) (VicK of S. mutans UA159, aa 31 to 450) cloned into pET22b(+) | 8 |
MCRF, Microbial Chemistry Research Foundation, Tokyo, Japan.
Plasmid construction.
A DNA fragment containing full-length WalK of B. subtilis was amplified by PCR using pBY33 as the template (23) and primers HT10-BsGfull-F and HT10-BsGstop-R (see Table S1 in the supplemental material). The amplicon was ligated into the BamHI and XbaI sites of pHT10 to obtain pBSG-full. To construct pBsGsubA and pBsGsubB, the WalK dimerization domain (amino acid residues [aa] 366 to 436) and ATP-binding domain (aa 437 to 611) were amplified using pBY33 and specific primers (see Table S1 in the supplemental material). These fragments were ligated into the NdeI and XhoI sites of pET21a(+).
Physicochemical measurement.
Optical rotation of signermycin B was measured using a P1030 spectropolarimeter (JASCO, Tokyo, Japan). UV spectra were recorded with a U2800 spectrophotometer (Hitachi, Tokyo, Japan). Infrared (IR) spectra were recorded with an FT-210 Fourier transform infrared spectrometer (Horiba, Kyoto, Japan). Proton and carbon NMR spectra were recorded using a JNM ECA-600 (600-MHz) spectrometer (JEOL, Tokyo, Japan). High-resolution mass spectra were obtained using an LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, MA). The stereostructure of signermycin B was determined by X-ray crystallography with an R-Axis Rapid imaging plate area detector (Rigaku, Tokyo, Japan) with filtered Cu-Ka radiation.
Isolation of signermycin B.
A slant culture of signermycin-producing Streptomyces sp. strain MK851-mF8 was inoculated into a 500-ml baffled Erlenmeyer flask containing 110 ml seed medium consisting of 2.0% galactose, 2.0% dextrin, 1.0% glycerin, 0.5% Bacto Soytone (Difco), 0.5% corn steep liquor, and 0.2% CaCO3 in deionized water. The inoculated medium was cultured on a rotary shaker (180 rpm) at 30°C for 3 days, and then 2.5 ml of the seed culture was transferred to a 500-ml baffled Erlenmeyer flask containing 110 ml of producing medium consisting of 2.0% glycerin, 2.0% dextrin, 1.0% Bacto Soytone, 0.3% yeast extract, 0.2% (NH4)2SO4, and 0.2% CaCO3 in deionized water (pH 7.4 before sterilization). Fermentation was carried out on a rotary shaker (180 rpm) at 27°C for 5 days. The fermentation broth (3 liters) was centrifuged to separate the mycelial cake from the supernatant. The mycelial cake was then extracted with methanol (MeOH) (1 liter). After addition of an equivalent volume of water, the solution was applied to a Diaion HP-20 column (60 by 220 mm; Mitsubishi Chemical Co.). The column was then eluted with 50% aqueous MeOH (450 ml) and 80% aqueous MeOH (450 ml). The active principle was eluted with 80% MeOH and concentrated in vacuo to yield a brown oil (842 mg). The brown oil, including the active compound, was chromatographed on a Sephadex LH-20 column (GE Healthcare) with MeOH. The active fractions were collected and concentrated in vacuo to give a pale brown oil (660 mg). The active material was further chromatographed on a reversed-phase high-performance liquid chromatography column developed with acetonitrile-H2O-trifluoroacetic acid (TFA) at 60:40:0.001 (Capcell Pak C18 UG120, 30 by 250 mm; Shiseido Co., Ltd., Japan) at a flow rate of 15 ml/min. The active fractions were collected and concentrated in vacuo to yield pure signermycin B (206 mg) as a colorless powder. Because signermycin B dissolves poorly in dimethyl sulfoxide (DMSO), its sodium salt was prepared and used in this study.
Autophosphorylation activity.
The cytoplasmic WalK domains (with C-terminal His tag) of Gram-positive B. subtilis, S. aureus, E. faecalis, and S. mutans were expressed and purified as described previously using E. coli BL21(DE3) containing pETYycG-tru (23), pYycGSa (31), pETEfwalK (Table 1), and pETSMVicK31-450 (8), respectively. The WalK cytoplasmic domains were preincubated in kinase buffer (50 mM Tris-HCl [pH 8.5], 100 mM KCl, 100 mM NH4Cl, 5 mM MgCl2) at 30°C, as previously reported (8, 22, 23, 31). Signermycin B was then added. After a 5-min incubation, ATP solution (2.5 μM ATP and 16.7 nM [γ-32P]ATP) was added, and the reaction mixture was incubated for an additional 10 min. The reaction was stopped by adding sodium dodecyl sulfate (SDS) sample buffer (300 mM Tris-HCl [pH 8.0], 10% SDS, 25% β-mercaptoethanol, 50% glycerol, 0.25% bromophenol blue). The samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE). The gel was then dried, exposed to an imaging plate, and analyzed using an FLA-7000 imaging analyzer (Fuji Film, Tokyo, Japan) and Multi Gauge version 3.0 software (Fuji Film, Tokyo, Japan). Prism 4.1 (GraphPad Software, CA) was used to determine the 50% inhibitory concentration (IC50) and in the kinetic analysis.
Antimicrobial activity.
MICs were determined by the standard agar dilution method recommended by the Japan Society of Chemotherapy (23).
SPR.
Surface plasmon resonance (SPR) analysis was carried out with a Biacore X100 system (GE Healthcare, Japan) at 25°C. The immobilization buffer for the system consisted of 10 mM HEPES (pH 7.5), 150 mM NaCl, and 0.05% Tween 20. WalK(A) and WalK(B) of B. subtilis (see Fig. 4) were immobilized on the surface of a CM5 sensor chip according to the manufacturer's instructions. WalK(A) and WalK(B) were expressed and purified as described previously using E. coli BL21(DE3) containing pBsGsubA and pBsGsubB, respectively (23). Briefly, the surface of flow cell 2 on the chip was activated by injecting a mixture of 0.4 M N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride and 0.1 M N-hydroxysuccinimide for 7 min at a flow rate of 5 ml/min. Next, solutions of WalK(A) and WalK(B) (800 μg/ml) in a 10 mM acetate buffer (pH 5.5) were injected for 14 min over flow cell 2. To cap the remaining N-hydroxysuccinimide esters, 1 M ethanolamine (pH 8.5) was injected for 7 min. Flow cell 1 was left unmodified to serve as a reference. To obtain a dose-dependent response curve set for signermycin B, a 1.5-fold dilution series was prepared with running buffer (10 mM HEPES [pH 7.5], 65 mM NaCl, 0.1% Tween 20), ranging in concentration from 33.4 to 385 μM signermycin B. Each sample was injected in duplicate for 3 min at a flow rate of 30 μl/min. Dissociation of the compound was monitored for 20 min to ensure full removal from the surface. Running buffer blanks were included to subtract systematic artifacts. With ATP as a ligand, conditions included a running buffer of 50 mM Tris HCl (pH 8.5), 100 mM KCl, 100 mM NH4Cl, and 10 mM MgCl2, a concentration of 15.6 to 500 μM, a dissociation time of 800 s, a contact time of 180 s, and a flow rate of 10 μl/min.
Fig 4.

WalK(Bs) domain structures (4).
Cross-linking experiment.
WalK(Sm)31-450 (final concentration, 0.5 μM) was first autophosphorylated with [γ-32P]ATP in 50 mM Tris-HCl (pH 7.5), 50 mM KCl, and 10 mM MgCl2 for 20 min. DMSO or serially diluted signermycin B was added to the autophosphorylated kinase, incubated for 5 min, and then cross-linked by adding glutaraldehyde (0.03%) and incubating for 30 min. The reaction was terminated by adding SDS sample buffer, and the reaction mixture was subjected to SDS-PAGE. Phosphorylated proteins were detected and analyzed with the FLA-7000 imaging analyzer.
Inhibition of autophosphorylation was measured in parallel with the cross-linking study using the same protein sample, reaction buffer, and [γ-32P]ATP. WalK was first incubated with DMSO or signermycin B for 5 min. Autophosphorylation was then assessed by adding [γ-32P]ATP and incubating for 20 min at 25°C.
RNA preparation and cDNA synthesis.
For quantitative reverse-transcription PCR (qRT-PCR) analysis, B. subtilis 168 and S. aureus N315 were grown with aeration to an optical density at 660 nm (OD660) of 0.3 at 37°C. After addition of signermycin B, the culture was incubated for another 5 min. Cells were then harvested by centrifugation and immediately suspended in RNAlater (Ambion, Austin, TX). RNA was then purified with the SV Total RNA Isolation System (Promega, Madison, WI), followed by a DNase I treatment (Turbo DNA-free kit; Ambion) to eliminate genomic DNA contamination. Reverse transcription was carried out with the high-capacity cDNA reverse transcription kit (ABI, Foster City, CA).
qRT-PCR.
PCR primers (see Table S1 in the supplemental material) were designed with Primer Express software version 3.0 (ABI). Amplification of cDNA (1.0 ng) was carried out with 250 nM gene-specific primers and PowerSYBR Green master mix (ABI). PCR amplification, detection, and analysis were performed using the StepOne real-time PCR System (ABI). PCR conditions included an initial denaturation step at 95°C for 10 min followed by 40-cycle amplification (95°C for 15 s and 60°C for 60 s). The specificity of the amplified product and absence of primer dimer formation were verified by generating a melting curve with an initial denaturation step of 95°C for 15 s followed by 60°C for 1 min and 95°C for 15 s. Fluorescence was measured continually during the melting curve cycle (stepwise 0.3°C) beginning at 60°C. The absence of contaminating genomic DNA was verified by testing each sample in control reactions without a previous reverse transcription step. The critical threshold cycle was defined for each sample, and expression levels were normalized using the 16S rRNA gene as an internal standard; 16S rRNA levels did not vary under our experimental conditions. Each assay was performed in triplicate.
Stereostructure accession number.
The absolute stereostructure of signermycin B was deposited at the Cambridge Crystallographic Data Centre with accession number CCDC 815220.
RESULTS AND DISCUSSION
Isolation and structure determination.
We previously developed a sensitive differential growth assay to screen for WalK inhibitors from natural sources (22, 31). Using this screening method, significant bactericidal activity was detected in broth cultures of Streptomyces sp. strain MK851-mF8. Isolation and characterization of the active compound revealed that the inhibitory activity was derived from a novel antibiotic, which we named signermycin B (Fig. 1). The physicochemical properties are as follows: [α]D20°+66.4°; high-resolution mass spectrometry (electrospray ionization [ESI], positive), m/z 412.2456 (M + Na)+ (calculated for C23H35NO4, 412.2458); UVmax MeOH (ε)0.005 M HCl 222 (sh), 285 (11,700), 243 (9500), 284 (13,000); IRνmax (KBr, cm−1), 3,500 to 3,200, 2,956, 2,871, 1,697, 1,655, 1,603, 1,458, 1,377, 1,338, 1,292, 1,232, 1,209, and 1034). The characteristic UV signals (acidic, 285 nm; basic, 243 and 284 nm) and 13C NMR signals (d 102.5, 174.9, and 195.2) suggested the presence of a tetramic acid moiety. The structure of signermycin B consisting of a tetramic acid moiety and decalin ring was determined by mass spectrometry, NMR, X-ray crystallography, and the advanced Marfey method (20) (see Fig. S1 to S3 in the supplemental material). The absolute stereostructure of signermycin B was determined (Fig. 2).
Fig 1.
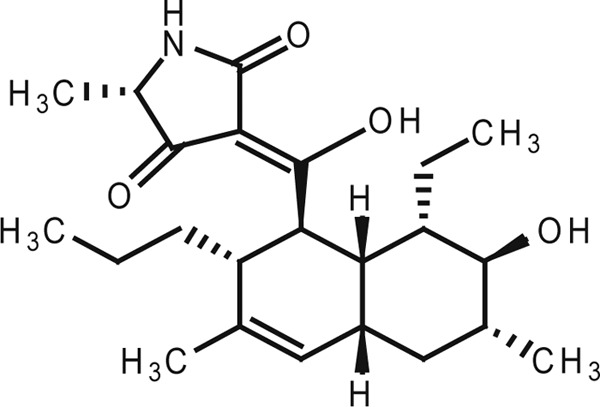
Structure of signermycin B.
Fig 2.
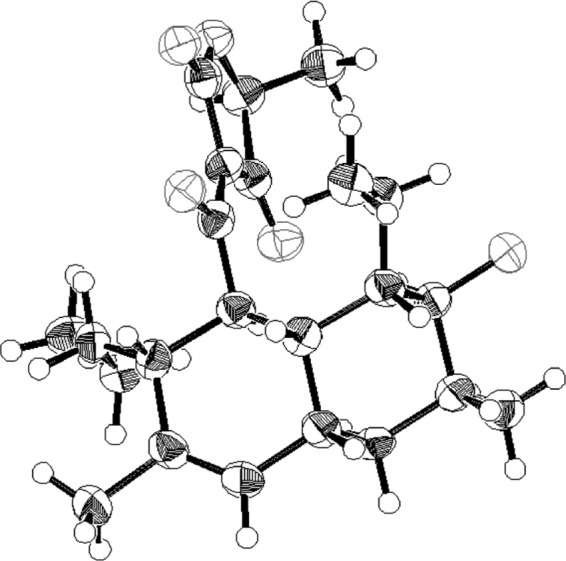
Stereostructure (ORTEP drawing) of signermycin B.
IC50 and MIC values.
Signermycin B exhibited antimicrobial activity against Gram-positive bacteria that possess the WalK/WalR TCS, including the drug-resistant bacteria methicillin-resistant S. aureus, and vancomycin-resistant E. faecalis, with MIC values ranging from 3.13 μg/ml (8 μM) to 6.25 μg/ml (16 μM). The IC50s of signermycin B against WalK in these organisms were 37 μM to 62 μM (Table 2). However, signermycin B was ineffective against all Gram-negative bacteria tested that do not possess TCSs controlling essential genes: Escherichia coli K-12, Salmonella enterica serovar Enteritidis 1891, Pseudomonas aeruginosa A3, Klebsiella pneumoniae PCI602, and Shigella dysenteriae JS11910. To confirm that signermycin B targets WalK, the effects of WalK induction on cell growth were analyzed in B. subtilis (Fig. 3). B. subtilis 168 containing pBSG-full and pHT10 was grown in the presence of isopropyl β-d-1-thiogalactopyranoside (IPTG) until the OD660 reached 0.3 and then was incubated in the presence or absence of signermycin B at the MIC of 3.13 μg/ml. The result showed that WalK-induced cells were more resistant against signermycin B than noninduced cells (Fig. 3a). However, when WalR was induced in the presence of IPTG, signermycin B resistance was not observed (Fig. 3b). These results show that signermycin B inhibited bacterial growth by targeting WalK.
Table 2.
IC50s and MICs of signermycin B
| Histidine kinase (WalK) | IC50 against WalK (μM) | MIC (μg/ml) |
|---|---|---|
| B. subtilis 168 | 43 | 3.13 |
| S. aureus FDA 209P | 37 | 3.13 |
| E. faecalis JCM5803 | 61 | 6.25 |
| S. mutans UA159 | 62 | 6.25 |
| S. aureus MS16526 (methicillin resistant) | 3.13 | |
| E. faecalis NCTC12201 (vancomycin resistant) | 6.25 |
Fig 3.
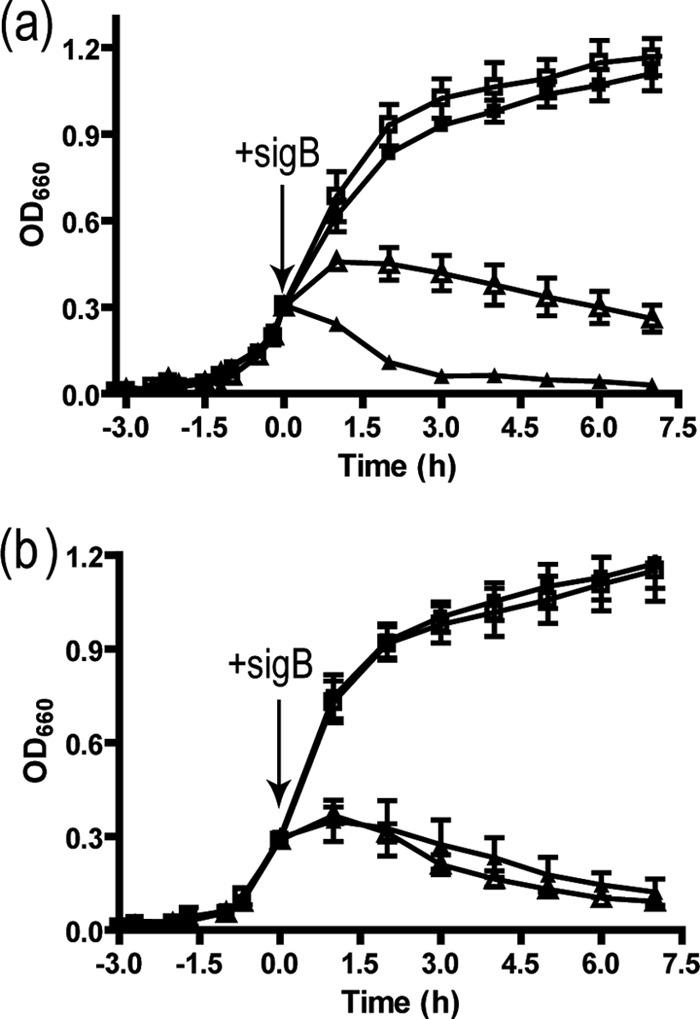
Effect of signermycin B on cell growth after induction of WalK or WalR. (a) B. subtilis 168 was transformed with pHT10 (■, ▲) and pBsG-full (□, △) and cultured in Luria broth containing 1 mM IPTG at 37°C until the OD660 reached 0.3. Cultures were incubated in the presence (▲, △) or absence (■, □) of signermycin B (final concentration, 3.13 μg/ml), and the OD660 was determined at the indicated times. The arrow shows the time at which signermycin B was added to the culture. Error bars represent standard errors of the means from three independent assays. (b) B. subtilis 168 was transformed with pAN18 (■, ▲) and pABF (□, △) and cultured until the OD660 reached 0.3. Cultures were incubated in the presence (▲, △) or absence (■, □) of signermycin B (final concentration, 3.13 μg/ml) to measure OD660 at the indicated times.
SPR analysis.
The HK cytoplasmic region contains several functional domains, as shown in Fig. 4 using WalK of B. subtilis as an example. Although dimerization and ATP-binding domains are common to most HKs, the other regions are less conserved and show less similarity in amino acid sequences. We therefore examined the binding affinity of signermycin B for the dimerization (A) and ATP-binding (B) domains of B. subtilis WalK. SPR analysis was used to identify the potential signermycin B binding domain on the target protein. WalK(A) and WalK(B) were immobilized on CM5 sensor chips, and signermycin B binding was determined using ATP as a control. SPR sensorgrams clearly showed that signermycin B bound to the dimerization domain but not the ATP-binding domain of WalK (Fig. 5a and b). In contrast, ATP bound to the ATP-binding domain but not the dimerization domain (Fig. 5c and d).
Fig 5.
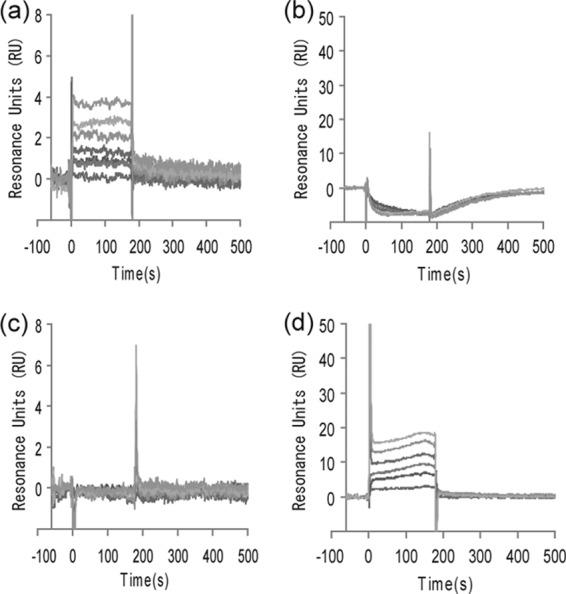
Surface plasmon resonance sensorgrams. The dimerization domain (a and c) and ATP-binding domain (b and d) of B. subtilis WalK (Fig. 3) were immobilized on a sensor chip. Signermycin B (a and b) and ATP (c and d) were used as analytes. Normalized response units (RUs) were plotted against time (s). Sensorgrams with the following analyte concentrations were plotted in order of increasing normalized RUs: signermycin B, 0, 33.4, 50.2, 75.3, 113, 170, 256, and 385 μM; ATP, 15.6, 31.25, 62.5, 125, 250, and 500 μM.
Competition assay.
Signermycin B is the first example of an antibiotic that binds to the WalK dimerization domain. To confirm that signermycin B does not target the ATP-binding domain of WalK, the IC50s of signermycin B were determined in the presence of different concentrations of ATP and compared with those of AMP-PNP, a nonhydrolyzable ATP analog. We found that at concentrations of up to 100 μM, ATP had no effect on the IC50s of signermycin B, whereas the IC50 of AMP-PNP exceeded 500 μM in the presence of high ATP concentrations (25 μM and 100 μM) (Table 3). Signermycin B did not compete with ATP to inhibit the autophosphorylation of WalK.
Table 3.
Competition assaya
| ATP concn (μM) | IC50 (μM) |
|
|---|---|---|
| Signermycin B | AMP-PNP | |
| 2.5 | 38 | 69 |
| 25 | 37 | >500 |
| 100 | 31 | >500 |
Signermycin B and AMP-PNP were tested against WalK(Bs) with ATP in an autophosphorylation assay.
WalR regulon gene expression.
To establish that signermycin B acts on the WalK/WalR system, the effect of signermycin B on in vivo expression of WalR regulon genes (1, 5, 6, 10, 17) was evaluated by qRT-PCR. In B. subtilis, WalR positively regulates the expression of yocH and yvcE (cwlO), while negatively regulating the expression of yoeB and yjeA. In S. aureus, WalR positively regulates the expression of isaA and ssaA. As shown in Fig. 6a, adding signermycin B to B. subtilis reduced the expression of yocH and yvcE 0.4-fold and increased the expression of yoeB 4-fold and that of yjeA 4.5-fold within 5 min. Likewise, transcription of isaA and ssaA was also inhibited 0.2-fold within 5 min of adding signermycin B to the S. aureus culture (Fig. 6b). These results suggest that signermycin B preferentially controls the WalR regulon genes.
Fig 6.

Transcriptional regulation of the WalR regulon by signermycin B. Signermycin B was added to exponentially growing cultures of B. subtilis 168 (a) and S. aureus N315 (b). Bar colors indicate signermycin B concentrations: white, 0 μM; gray, 1.61 μM; and black, 3.21 μM. After 5 min, total RNA was isolated and quantified by qRT-PCR. Relative results are expressed as the mean and standard deviation from triplicate experiments.
The WalR regulon (including autolysins) plays a crucial role in cell wall metabolism (4, 10). As autolysins are involved in the separation of daughter cells, one would expect WalK inhibition to result in filamentous cells in B. subtilis and cell aggregation in S. aureus (13). In fact, untreated B. subtilis 168 exhibited the typical short-rod morphology, whereas signermycin B-treated B. subtilis 168 cells formed extremely long aseptate filaments. S. aureus cells aggregated in the presence of signermycin B (data not shown). These results are consistent with previously reported phenotypes of WalK- or WalR-depleted mutants (5, 9, 10).
Signermycin B mechanism of action.
Several other HK inhibitors have been identified, but most caused protein aggregation and disrupted the membrane integrity of bacteria and equine erythrocytes. The precise mechanism of action is not known in most cases (16, 19, 28, 29). Stephenson et al. reported that the HK inhibitors RWO-49815 and closantel acted on the autokinase domains of unrelated sensor kinases and caused protein aggregation in the presence of the cross-linker glutaraldehyde (29). To clarify the mechanism of action of signermycin B, protein cross-linking was performed in the presence of glutaraldehyde. Both glutaraldehyde aldehyde groups covalently linked the ε-amino groups of WalK lysine residues, located within the spacer arm of glutaraldehyde. Based on the four-helix bundle structure of the WalK(Sm) dimerization domain resolved by T. Okajima et al. (unpublished data), ε-amino groups of K227 and K248 in the WalK(Sm) dimerization domain were expected to be bridged by glutaraldehyde. Therefore, we carried out a cross-linking experiment using WalK(Sm) and glutaraldehyde (Fig. 7). After glutaraldehyde treatment, the autophosphorylated protein bands of WalK migrated from a monomer to a dimer position on SDS-PAGE (Fig. 7a, lanes 1 and 2). Treating WalK with signermycin before adding glutaraldehyde inhibited covalent dimer formation (Fig. 7a, lanes 5 to 8), with an inhibitory concentration almost the same as the IC50 of WalK (Fig. 7b).
Fig 7.
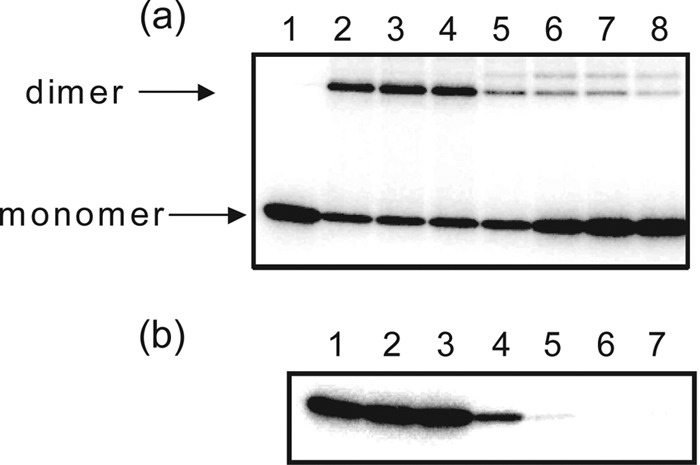
Effects of signermycin B on WalK cross-linking and autophosphorylation. (a) Autophosphorylated WalK(Sm)31-450 was treated with increasing concentrations of signermycin B (lane 2, 0 μM; lane 3, 20 μM; lane 4, 40 μM; lane 5, 80 μM; lane 6, 160 μM; lane 7, 320 μM; lane 8, 640 μM) and cross-linked with glutaraldehyde. Lane 1, autophosphorylated HK in the absence of glutaraldehyde and signermycin B (control). (b) WalK(Sm)31-450 was autophosphorylated in the presence of increasing concentrations of signermycin B (lane 1, 0 μM; lane 2, 20 μM; lane 3, 40 μM; lane 4, 80 μM; lane 5, 160 μM; lane 6, 320 μM; lane 7, 640 μM).
In this study, we discovered a novel WalK inhibitor, signermycin B, which targets the WalK dimerization domain without causing protein aggregation in the presence of a cross-linker (Fig. 8). Its mode of action differed considerably from that of HK inhibitors described previously (29). Signermycin B binds to the WalK dimerization domain to inhibit autophosphorylation activity, thereby hindering WalK/WalR signal transduction involved in cell growth and division. Our findings suggest that the dimerization domain as well as the ATP-binding domain of WalK should be targeted. This finding opens up possibilities for the development of HK inhibitors that are effective against various drug-resistant bacteria.
Fig 8.

Proposed action of signermycin B.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Ho for valuable comments on the manuscript.
This work was supported by the Research and Development Program for New Bio-Industry Initiatives (2006 to 2010) from the Bio-Oriented Technology Research Advancement Institution (BRAIN), a Grant-in-Aid for Scientific Research (A, 20248012) from the Japan Society for the Promotion of Science (JSPS), the Adaptable and Seamless Technology Transfer Program through Target-Driven R&D, Japan Science and Technology Agency, the Strategic Project to Support the Formation of Research Bases at Private Universities: Matching Fund Subsidy from MEXT (Ministry of Education, Culture, Sports, Science and Technology), 2011-2015 (S1101035), and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices” from MEXT.
Footnotes
Published ahead of print 23 April 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Bisicchia P, et al. 2007. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol. Microbiol. 65:180–200 [DOI] [PubMed] [Google Scholar]
- 2. Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. 2008. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 6:17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doi A, Okajima T, Gotoh Y, Tanizawa K, Utsumi R. 2010. X-ray crystal structure of the DNA-binding domain of response regulator WalR essential to the cell viability of Staphylococcus aureus and interaction with target DNA. Biosci. Biotechnol. Biochem. 74:1901–1907 [DOI] [PubMed] [Google Scholar]
- 4. Dubrac S, Bisicchia P, Devine KM, Msadek T. 2008. A matter of life and death: cell wall homeostasis and the WalK/WalR (YycGF) essential signal transduction pathway. Mol. Microbiol. 70:1307–1322 [DOI] [PubMed] [Google Scholar]
- 5. Dubrac S, Boneca IG, Poupel O, Msadek T. 2007. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J. Bacteriol. 189:8257–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dubrac S, Msadek T. 2004. Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J. Bacteriol. 186:1175–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Echenique JR, Trombe MC. 2001. Competence repression under oxygen limitation through the two-component MicAB signal-transducing system in Streptococcus pneumoniae and involvement of the PAS domain of MicB. J. Bacteriol. 183:4599–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eguchi Y, Kubo N, Matsunaga H, Igarashi M, Utsumi R. 2011. Development of an antivirulence drug against Streptococcus mutans: repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, walkmycin C. Antimicrob. Agents Chemother. 55:1475–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fabret C, Hoch JA. 1998. A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J. Bacteriol. 180:6375–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fukuchi K, et al. 2000. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiology 146:1573–1583 [DOI] [PubMed] [Google Scholar]
- 11. Furuta E, et al. 2005. Targeting protein homodimerization: a novel drug discovery system. FEBS Lett. 579:2065–2070 [DOI] [PubMed] [Google Scholar]
- 12. Gao R, Stock AM. 2009. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63:133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gotoh Y, et al. 2010. Novel antibacterial compounds specifically targeting the essential WalR response regulator. J. Antibiot. (Tokyo) 63:127–134 [DOI] [PubMed] [Google Scholar]
- 14. Gotoh Y, et al. 2010. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 13:232–239 [DOI] [PubMed] [Google Scholar]
- 15. Hidaka Y, Park H, Inouye M. 1997. Demonstration of dimer formation of the cytoplasmic domain of a transmembrane osmosensor protein, EnvZ, of Escherichia coli using Ni-histidine tag affinity chromatography. FEBS Lett. 400:238–242 [DOI] [PubMed] [Google Scholar]
- 16. Hilliard JJ, Goldschmidt RM, Licata L, Baum EZ, Bush K. 1999. Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob. Agents Chemother. 43:1693–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Howell A, et al. 2003. Genes controlled by the essential YycG/YycF two-component system of Bacillus subtilis revealed through a novel hybrid regulator approach. Mol. Microbiol. 49:1639–1655 [DOI] [PubMed] [Google Scholar]
- 18. Inouye M, Dutta R. 2003. Regulation of porins in Escherichia coli by the osmosensing histidine kinase/phosphatase EnvZ, p25–46 Academic Press, San Diego, CA [Google Scholar]
- 19. Macielag MJ, Goldschmidt R. 2000. Inhibitors of bacterial two-component signalling systems. Expert Opin. Invest. Drugs 9:2351–2369 [DOI] [PubMed] [Google Scholar]
- 20. Marfey P. 1984. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 49:591–596 [Google Scholar]
- 21. Njoroge J, Sperandio V. 2009. Jamming bacterial communication: new approaches for the treatment of infectious diseases. EMBO Mol. Med. 1:201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Okada A, et al. 2007. Targeting two-component signal transduction: a novel drug discovery system. Methods Enzymol. 422:386–395 [DOI] [PubMed] [Google Scholar]
- 23. Okada A, et al. 2010. Walkmycin B targets WalK (YycG), a histidine kinase essential for bacterial cell growth. J. Antibiot. (Tokyo) 63:89–94 [DOI] [PubMed] [Google Scholar]
- 24. Okajima T, et al. 2008. Response regulator YycF essential for bacterial growth: X-ray crystal structure of the DNA-binding domain and its PhoB-like DNA recognition motif. FEBS Lett. 582:3434–3438 [DOI] [PubMed] [Google Scholar]
- 25. Rasko DA, et al. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salzberg LI, Helmann JD. 2007. An antibiotic-inducible cell wall-associated protein that protects Bacillus subtilis from autolysis. J. Bacteriol. 189:4671–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stapleton MR, et al. 2007. Characterization of IsaA and SceD, two putative lytic transglycosylases of Staphylococcus aureus. J. Bacteriol. 189:7316–7325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stephenson K, Hoch JA. 2004. Developing inhibitors to selectively target two-component and phosphorelay signal transduction systems of pathogenic microorganisms. Curr. Med. Chem. 11:765–773 [DOI] [PubMed] [Google Scholar]
- 29. Stephenson K, Yamaguchi Y, Hoch JA. 2000. The mechanism of action of inhibitors of bacterial two-component signal transduction systems. J. Biol. Chem. 275:38900–38904 [DOI] [PubMed] [Google Scholar]
- 30. Tomomori C, et al. 1999. Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Nat. Struct. Biol. 6:729–734 [DOI] [PubMed] [Google Scholar]
- 31. Watanabe T, et al. 2003. Isolation and characterization of inhibitors of the essential histidine kinase YycG in Bacillus subtilis and Staphylococcus aureus. J. Antibiot. (Tokyo) 56:1045–1052 [DOI] [PubMed] [Google Scholar]
- 32. Yamamoto K, et al. 2001. Antibacterial agents that inhibit histidine protein kinase YycG of Bacillus subtilis. Biosci. Biotechnol. Biochem. 65:2306–2310 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.