

Abstract
Doxycycline was found to act synergistically with the antifungal fluconazole against Candida albicans. Combination with doxycycline converts fluconazole from fungistatic to fungicidal, prevents the onset of drug resistance, and is also effective against a clinical isolate characterized by elevated resistance to fluconazole. Investigation of the interactions between the two drugs by way of checkerboard assays indicated that doxycycline had an influence on the MIC for fluconazole, as defined by CLSI standards, only at high concentrations (200 μg/ml). However, lower concentrations were effective at eliminating residual cell growth at supra-MICs of fluconazole. Using MIC-0, defined as a drug combination resulting in optically clear wells, as an endpoint, doxycycline was found to be synergistic with fluconazole at a concentration as low as 25 μg/ml, with a fractional inhibitory concentration index of <0.5. Doxycycline-mediated growth inhibition can be reversed by externally added iron, indicating that iron depletion may account for the synergism. Consistently, we confirmed old literature data about iron-chelating activity of doxycycline. Synergism of fluconazole with doxycycline does not appear to be mediated by calcineurin, since doxycycline further aggravates the susceptibility to fluconazole of mutants lacking the catalytic or the regulatory subunits of calcineurin. Growth in the presence of fluconazole and doxycycline is restored by an elevated dosage of ERG11 in Saccharomyces cerevisiae but not in C. albicans, despite the full competence of the pathogen's protein to act as a suppressor in baker's yeast.
INTRODUCTION
Infections by Candida albicans have been on the rise in the past decades, mostly due to changes in the clinical practice, such as the increased use of immunosuppressants and broad-spectrum antibacterial agents (26, 29, 47). Fungal infections represent a challenge for clinicians, because of the scarcity and sometimes the limited efficacy of antifungal drugs (1, 44). Fluconazole and other azole antifungals, targeting ergosterol biosynthesis (23), have been extensively used in the recent clinical practice thanks to their reduced toxicity and lower cost, compared to amphotericin B in its conventional and lipidic forms, respectively (44). The major disadvantage of azole antifungals, however, is their fungistatic nature, an aspect that favors the onset of drug resistance. The development of isogenic strains of C. albicans characterized by stepwise increased tolerance to fluconazole in patients undergoing continued treatment with this drug has been documented in several cases (34, 56, 57). For these reasons, the conversion of fluconazole from fungistatic to fungicidal via combinations with other drugs is highly desirable.
In a different perspective, tools for the detailed study of the molecular genetics of C. albicans have been greatly improved in recent times (6, 7, 40). Regulatable promoters are available to the research community, to induce or repress gene expression in C. albicans. Most of them, such as the PCK1 and the MAL2 promoters, allow conditional expression/repression of the gene of interest upon incubation of cells in growth media containing the repressing or the activating carbon source (3, 28). The tetracycline direct and reverse systems, which allow gene induction or repression by addition of doxycycline to the growth medium, have also been successfully used to regulate gene expression in C. albicans (38, 45). However, it was found that, compared to other organisms, significantly higher concentrations of doxycycline are necessary for full induction of tetracycline-inducible promoters in C. albicans (45).
During the course of unpublished experiments with C. albicans strains carrying tetracycline-regulated promoters, we noticed that doxycycline interfered heavily with cell growth in the presence of fluconazole and decided to investigate further this unexpected interaction. We believe that researchers in the field of C. albicans molecular genetics may benefit from the conclusions of our studies.
MATERIALS AND METHODS
Strains and media.
The strains used in this study are listed in Table 1. Most experiments were conducted with the Candida albicans reference strain SC5314 (18). Yeast (Saccharomyces cerevisiae) and C. albicans strains were routinely refreshed from frozen stocks at −80°C and maintained on YPD (1% yeast extract, 2% peptone, 2% dextrose, 2% agar) plates. Experiments were carried out using minimal medium (1.7 g/liter Difco yeast nitrogen base without ammonium sulfate, 5 g/liter ammonium sulfate) supplemented with 2% glucose (synthetic defined [SD] medium), 3% glycerol, and 3% ethanol (SEG), 2% galactose (SGal), or 2% Casamino Acids (SCAA). Minimal medium was supplemented with uridine when ura3Δ/ura3Δ strains of C. albicans were used. RPMI 1640 medium with l-glutamine without sodium bicarbonate (Sigma) was buffered with 0.165 M morpholinepropanesulfonic acid (MOPS). Glucose at the final concentration of 2% was added only to RPMI agar medium. Fetal bovine serum (FBS) medium consisted of 10% fetal bovine serum in sterile Milli-Q water (Millipore). Media were solidified with 2% agar. Sabouraud dextrose agar medium was bought from Fluka. Minimal medium for yeast experiments was supplemented with histidine, lysine, leucine, and uracil at appropriate concentrations (59). Leucine and uracyl were omitted from media for the experiments shown in Fig. 5A and D, respectively. All plate tests for C. albicans were carried out at 37°C; plate tests for S. cerevisiae were carried out at 30°C. The standard concentration of fluconazole on plates was 10 μg/ml, whereas that of doxycycline was 50 μg/ml. Indications are given in the text for experiments conducted with different drug concentrations.
Table 1.
Fungal strains used in this study
| Strain | Genotype | Comment | Source or Reference |
|---|---|---|---|
| Candida albicans | |||
| SC5314 | Wild type | 18 | |
| CAI4 | ura3Δ::imm434/ura3Δ::imm434 | 16a | |
| AFA59b | ura3Δ::imm434/ura3Δ::imm434 RPS10::pAFC89b | CAI4 carrying extra copy of CaERG11 under ACT1 promoter | This study |
| AFA60a | ura3Δ::imm434/ura3Δ::imm434 RPS10::CIp10 | CAI4 transformed with URA3 vector | This study |
| AFA63a | ura3Δ::imm434/ura3Δ::imm434 RPS10::pAFC92a | CAI4 carrying extra copy of CaERG11 under PCK1 promoter | This study |
| AFA21 | ura3Δ::imm434/ura3Δ::imm434 RPS10::pPCK1-GFP | CAI4 transformed with URA3-PCK1 vector | This study |
| FH5 | Clinical isolate, highly resistant to fluconazole | 34, 57 | |
| DSY2091 | ura3Δ::imm434/ura3Δ::imm434 cna1Δ::hisG-URA3-hisG/cna1Δ::hisG | cna1Δ/cna1Δ mutant | 55 |
| DSY2115 | ura3Δ::imm434/ura3Δ::imm434 cna1Δ::hisG/cna1Δ::hisG LEU2::CNA1::URA3 | CNA1 reintegrant | 55 |
| DSY2146 | ura3Δ::imm434/ura3Δ::imm434 cna1Δ::hisG/cna1Δ::hisG LEU2::CNA1tr::URA3 | CNA1tr reintegrant | 55 |
| Saccharomyces cerevisiae BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Reference strain | Euroscarf |
Fig 5.
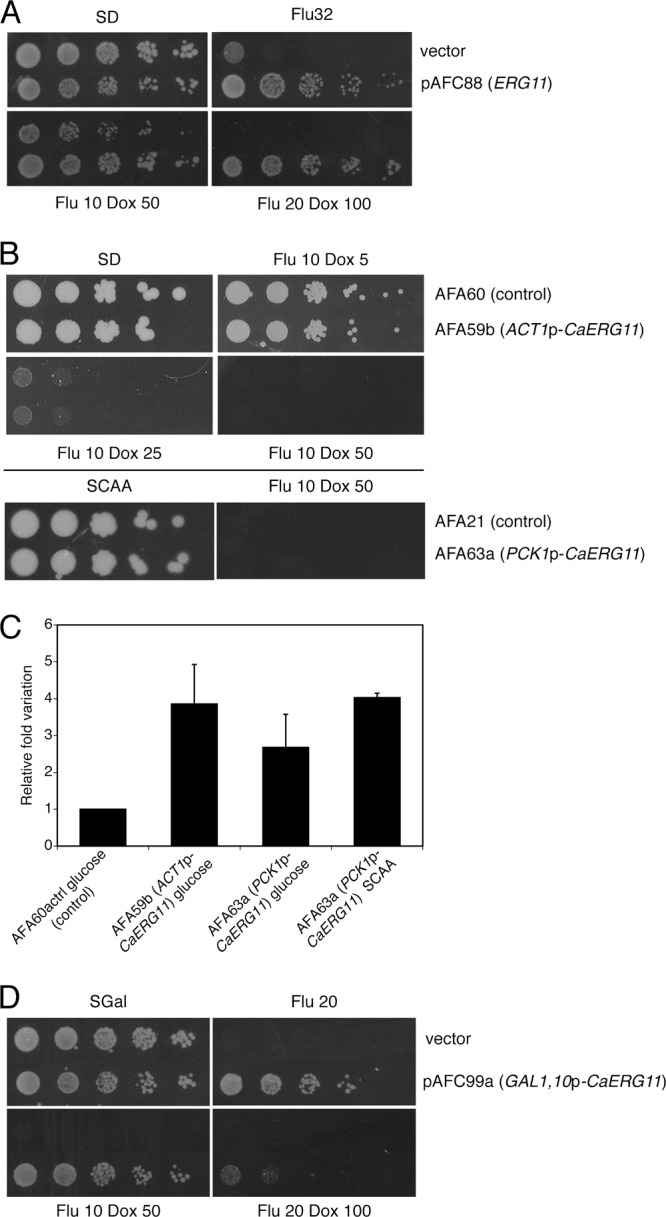
Overexpression of ERG11 suppresses fluconazole-doxycycline susceptibility in S. cerevisiae but not in C. albicans. (A) Transformants of the S. cerevisiae reference strain BY4742 with plasmid pAFC88 (ERG11) or with an empty plasmid (control) were spotted onto SD plates containing combinations of fluconazole and doxycycline, at the indicated concentrations (μg/ml). Plates were incubated for 4 days before being scanned. (B, top panel) Cells of the C. albicans strains AFA60 (control) and AFA59b (ACT1p-CaERG11) were spotted onto SD plates containing the indicated concentrations of fluconazole plus doxycycline. (Bottom panel) Cells of strains AFA21 (control) and AFA63a (PCK1p-CaERG11) were spotted onto SCAA plates with or without fluconazole plus doxycycline. Plates were incubated for 4 days before being scanned. (C) Quantitative real-time PCR assessment of expression of CaERG11 in strains carrying an extra copy of the gene under the control of the constitutive ACT1 gene or of the inducible PCK1 promoter. The graph shows mean values with standard deviations from two independent experiments. (D) Transformants of the S. cerevisiae reference strain BY4742 with plasmid pAFC99a (GAL1,10p-CaERG11) or with an empty plasmid (control) were spotted onto SGal plates containing combinations of fluconazole and doxycycline at the indicated concentrations (μg/ml). Plates were incubated for 5 days before being scanned.
Fluconazole, doxycycline, tetracycline, gentamicin, neomycin, potassium cyanide, ferric chloride hexahydrate, ferric citrate, zinc sulfate, and bathophenanthroline disulfonic acid (BSP) were added, starting from concentrated solutions, to autoclave-sterilized, precooled agar media. With the exception of doxycycline, which was dissolved in 50% ethanol, all of the chemicals mentioned above were dissolved in Milli-Q water.
MIC testing in broth microdilution assays.
The fluconazole MIC was determined according to the approved CLSI standard reference method for broth dilution antifungal susceptibility testing of yeasts M27-A3 (10). Inocula of C. albicans SC5314 and FH5 were prepared from cultures grown overnight on Sabouraud medium, adjusted to obtain final cell suspensions of 0.5 × 103 to 2.5 × 103 CFU/ml. Viable counts of the inocula were verified by plating serial dilutions on YPD plates. MIC plates were incubated at 35°C for 48 h for the reference strain SC5314 and for 24 h for strain FH5. MIC endpoints were defined as the lowest concentration of fluconazole causing a >50% decrease in optical density (prominent decrease in turbidity, or score of 2, according to CLSI guidelines) and a >90% decrease in viability compared to the drug-free control. Optical densities were recorded at 540 nm using a Molecular Devices SpectraMax Plus 384 absorbance microplate reader. Viable counts were measured by plating 100 μl of 10−1 to 10−5 serial dilutions on YPD plates. For viable count checks of optically clear wells, 100 μl of the undiluted wells' content was also plated (8, 33). Where appropriate, comparison of results obtained from plating 10−1/10−2 dilutions and from 100 μl of undiluted wells' content did not evidence a significant influence of the drugs' carryover in our experimental conditions. Experiments were repeated a minimum of three times.
Checkerboard assays.
The interaction of doxycycline with fluconazole was investigated by way of checkerboard tests on 96-well plates, performed according to the CLSI approved standard M27-A3 (10), with 48-h incubations for strain SC5314 and 24-h incubations for strain FH5. For SC5314, the fluconazole and doxycycline concentrations tested ranged from 0.03 to 32 μg/ml and from 3.125 to 200 μg/ml, respectively. For FH5, fluconazole concentrations ranged from 0.125 to 128 μg/ml and doxycycline concentrations ranged from 25 to 800 μg/ml. Concentrations of doxycycline higher than 200 μg/ml were repeatedly found to cause cloudiness of the RPMI medium, altering spectrophotometric readings and the general outcome of experiments (our unpublished observations). Results obtained with such concentrations of doxycycline were therefore not considered further. Viable count determinations were performed as described above. Elimination of fluconazole tolerance, defined as incomplete growth inhibition at supra-MICs of fluconazole, by doxycycline, allowed the use of MIC-0 as an endpoint. MIC-0 was defined as the lowest drug combination resulting in optically clear wells. The fractional inhibitory concentration (FIC) index was calculated as the sum of the FICs of either drugs (MICfluconazole + doxycycline/MICfluconazole + MICdoxycycline + fluconazole/MICdoxycycline). Drug interactions were classified as synergistic when the FIC value was ≤0.5 and antagonistic when the FIC value was >4 (22, 41). FIC values that were >0.5 but ≤4 were not considered informative for the establishment of interactions (22, 41). The MIC for doxycycline, which was >200 μg/ml experimentally, was considered to be 400 μg/ml for calculation purposes. Similarly, the MIC-0 values for fluconazole and doxycycline, which were >32 μg/ml and >200 μg/ml, respectively, were considered to be 64 and 400 μg/ml. Experiments were repeated a minimum of three times.
MIC testing using Etest strips.
Determination of fluconazole MICs using Etest strips (AB Biodisk) was carried out on RPMI-glucose plates. Overnight cultures in YPD were diluted in sterile saline to an optical density at 600 nm (OD600) of 0.1 and plated using sterile swabs, following the directions from the manufacturer. Plates were incubated at 37°C for 48 h after the overlay of strips. Results of Etest measurement are normally considered in agreement with those of broth microdilution tests when discrepancies between MIC values are not higher than 2 dilutions (48).
Spot tests and resistance-to-fluconazole assays.
Cultures were grown overnight in minimal glucose medium, diluted to an OD600 of 1, and serially diluted in presterilized, flat-bottom 96-well plates. Ten-fold (C. albicans) or 5-fold (S. cerevisiae) dilutions were spotted onto the indicated media using a spotter (Sigma). The spot test presented in Fig. 2A was performed by spotting 5 μl of each microtiter well using a multichannel pipette. Plates were incubated for the time indicated in the figure legends at 37°C (C. albicans) or 30°C (S. cerevisiae). Plates containing fluconazole and/or doxycycline were used within 24 h from preparation. All spot tests presented in this work were repeated a minimum of three times. For experiments on resistance to fluconazole, 104 cells of C. albicans SC5314 were spread on SD medium containing 128 μg/ml fluconazole, with or without 50 μg/ml doxycycline. Plates were incubated at 37°C for 4 days.
Fig 2.
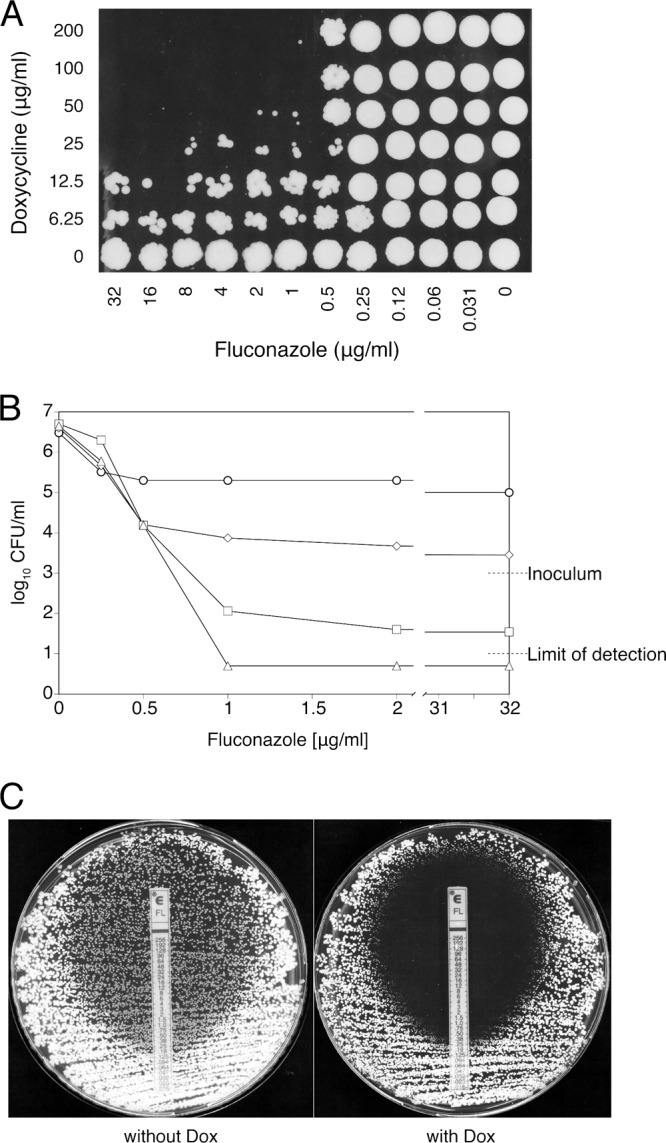
Doxycycline eliminates fluconazole tolerance. (A) A 5-μl concentration of each well from one of the checkerboard assays was spotted onto a YPD plate after 48 h of incubation, to monitor the viability of drug-treated cells. The recovery plate was incubated for 2 days before being scanned. (B) Viable cell counts after 48 h of incubation in the presence of fluconazole alone or with 25, 50, or 100 μg/ml doxycycline added (corresponding to rows 7, 4, 3, and 2 in panel A, respectively). Circles, fluconazole alone; diamonds, fluconazole plus 25 μg/ml doxycycline; squares, fluconazole plus 50 μg/ml doxycycline; triangles, fluconazole plus 100 μg/ml doxycycline. Values falling below the detection limit of 101 CFU/ml were approximated to 0.5 × 101 CFU/ml. Mean values from three independent experiments are shown. (C) Etest measurement of the fluconazole MIC of strain SC5314 on RPMI-glucose plates. The left panel shows medium without doxycycline, and the right panel shows medium with 50 μg/ml doxycycline.
Determination of fungicidal activity.
Determination of fungicidal activity was performed in microtiter plates using the standard CLSI M27-A3 procedure for MIC determination (10), with the exception that larger inocula of 104 CFU/ml were used (8, 49). Plates were incubated for either 48 or 96 h. Viable count checks were carried out by plating 100 μl of the appropriate dilutions or 100 μl of undiluted wells' content (for optically clear wells) on YPD plates. Both kinds of experiments were repeated three times.
Iron chelation assay.
Iron chelation was assayed using the colorimetric SideroTec assay kit from Emergen Bio (Maynooth, Ireland), following the manufacturer's instructions. Briefly, fluconazole, tetracycline, and doxycycline at concentrations of 25, 50, 100, 200, and 400 μg/ml were mixed with the provided reagents in flat-bottom 96-well plates and incubated at room temperature for 15 min. Plates were read at 630 nm on the above-mentioned plate reader. Decreases in absorbance are inversely proportional to iron chelation activity. Milli-Q water was substituted for the chemicals in the negative control.
Genomic screen for multicopy suppressors of susceptibility to fluconazole plus doxycycline.
We transformed the S. cerevisiae genomic library in vector pFL44 constructed in the laboratory of F. Lacroute (60) in the yeast strain BY4742 by the lithium acetate procedure. One aliquot of each transformation mixture was plated on SD medium containing histidine, leucine, and lysine for estimation of the transformation efficiency. The remaining transformation mixture was outgrown for 4 h in liquid YPD before being spread on plates the same as those described above containing 10 μg/ml fluconazole and 50 μg/ml doxycycline. Truly resistant transformants were separated from false positives by further restreaking them under selective conditions and by cosegregation analysis of resistance to fluconazole plus doxycycline with the presence of a transforming plasmid. Interesting plasmids were extracted from yeast, transformed in Escherichia coli, and retransformed into BY4742 for confirmation of their suppressor activity. Genomic inserts of suppressor plasmids were sequenced using oligonucleotides M13F (5′-GTAAAACGACGGCCAG-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′), identifying an ∼5.4-kb fragment containing incomplete SOD2, YHR007, ERG11, and incomplete STP2. Reintroduction of pAFC88 (described further below) into BY4742 and subsequent suppression of the susceptibility to fluconazole plus doxycycline of the resulting transformants confirmed ERG11 as the suppressor gene.
Plasmids used in this study.
A HindIII-SmaI fragment, containing ERG11 surrounded by ∼730 nucleotides at its 5′ end and ∼670 nucleotides at its 3′ end, was excised from the Lacroute library suppressor plasmid and cloned into YEplac181 digested with HindIII and SmaI (17), creating plasmid pAFC88. For overexpression studies in C. albicans, CaERG11 was amplified from genomic DNA of strain SC5314 using primers CaERG11s (5′-CCCAAGCTTATGGCTATTGTTGAAACTGTCA-3′) and CaERG11as (5′-GCGGCTAGCTGAATCGAAAGAAAGTTGCCG-3′). The amplified fragment was subcloned with HindIII-NheI, replacing the luciferase gene, in plasmid Cip10::ACT1p-gLUC59 (15), and in plasmid pPCK1-GFP (5), replacing the green fluorescent protein (GFP) gene. The resulting plasmids are named pAFC89b (ACT1p-CaERG11) and pAFC92a (PCK1p-CaERG11). Cloned CaERG11 was sequenced to verify the absence of mutations.
For cross-complementation experiments, CaERG11 was extracted with HindIII-NheI from pAFC89b, blunted with the Klenow enzyme, and subcloned into SmaI pBEVY-GL (35), creating plasmid pAFC99a.
Quantitative real-time PCR.
Cells of the AFA59b, AFA60a, and AFA63a strains were grown to mid-log phase in minimal glucose medium before their RNA was extracted. For AFA63a, cells were collected and split, and each half was resuspended in either SD or SCAA medium and then outgrown for 4 h before RNA extraction. cDNA was prepared from DNase-treated RNA samples with the Promega A3500 reverse transcription kit. Quantitative PCR was performed on a StepOne Plus real-time PCR system (Applied Biosystems) using the Kapa SYBR Fast kit (Kapa Biosystems). The fold regulation of each target gene was calculated using the threshold cycle (ΔΔCT) method, using expression of the TEF1 to normalize data. The CT value of TEF1 was subtracted from that of CaERG11 to calculate the ΔCT. The ΔCT value of AFA60a was used as a reference and was therefore subtracted from the ΔCT value of the other samples to obtain ΔΔCT values. Gene expression levels relative to the reference were expressed as 2−ΔΔCT. The primers used were CaERG11up (5′-TTACCTCATTATTGGAGACGTGATG-3′), CaERG11down (5′-CACGTTCTCTTCTCAGTTTAATTTCTTTC-3′), TEF1a-fw (5′-CCACTGAAGTCAAGTCCGTTGA-3′), and TEF1a-rv (5′-CACCTTCAGCCAATTGTTCGT-3′).
RESULTS
Doxycycline acts synergistically with fluconazole in a dosage-dependent manner.
Growth of wild-type Candida albicans SC5314 cells is prevented in minimal medium containing a combination of fluconazole (10 μg/ml) and doxycycline (50 μg/ml) at concentrations at which these chemicals cause either no growth defects (doxycycline) or only have a mild one (fluconazole) when used separately (Fig. 1). Growth inhibition exerted by doxycycline in the presence of fluconazole is dosage dependent (Fig. 1A), suggesting that the action of doxycycline is specific. No growth of any kind was observed on fluconazole-doxycycline plates containing 50 μg/ml doxycycline, even when these were incubated for up to 10 days (our unpublished observations). Tests on different media, such as RPMI-glucose, YPD, and fetal bovine serum-containing medium, produced identical results (Fig. 1B), demonstrating that growth inhibition is not dependent on the composition of the medium. Similar results were also obtained when glucose was replaced with glycerol and ethanol in the medium (Fig. 1B), indicating that growth inhibition is not dependent on the carbon source. Finally, tests in liquid media produced the same results as those on solid media (our unpublished observations).
Fig 1.

Doxycycline synergizes with fluconazole in a dosage-dependent and medium- and carbon source-independent manner. Cells of the C. albicans reference strain SC5314 were spotted onto plates containing different drug combinations. Where indicated, fluconazole (Flu) is present at 10 μg/ml, whereas the concentration of doxycycline (Dox) is expressed in μg/ml. (A) SD plates with 3 days of incubation. (B) YPD plates with 1 day of incubation, RPMI-glucose plates and FBS plates with 2 days of incubation, and SEG plates with 5 days of incubation. (C) SC5314 cells were spotted onto SD plates containing fluconazole and either tetracycline (Tetr), gentamicin, or neomycin. The concentration of the antibiotics is indicated in μg/ml. Plates were incubated for 2 days before being scanned.
Tetracycline acts synergistically with fluconazole with minor efficiency.
Tetracyclines are antibacterial drugs that prevent the association of acylated tRNAs with the ribosome, therefore inhibiting translation (9). Since doxycycline is a derivative of tetracycline with improved pharmacokinetic properties, we decided to test whether amelioration of the antifungal properties of fluconazole was an exclusive feature of doxycycline or whether it could be extended to tetracycline.
Tetracycline proved to be also synergistic with fluconazole, albeit with reduced potency (Fig. 1C). In fact, even though an inhibitory effect of fluconazole plus tetracycline could be observed against C. albicans, this was less robust than that observed with doxycycline, given that a higher concentration of tetracycline was required to prevent fungal growth. Notably, the 200-μg/ml tetracycline concentration that appeared to completely prevent growth of C. albicans on solid medium was considerably less efficient in liquid medium (our unpublished observations). Gentamicin and neomycin, two unrelated antibiotics that also prevent bacterial translation, showed no effect in combination with fluconazole against C. albicans (Fig. 1C).
Doxycycline eliminates fluconazole tolerance.
Using the CLSI standard reference method (M27-A3) for broth dilution antifungal susceptibility testing of yeasts (10), the MIC for fluconazole of standard wild-type strain SC5314 was 0.5 μg/ml, whereas the MIC of doxycycline alone was >200 μg/ml. In order to verify the nature of interactions between the two drugs, fluconazole and doxycycline at concentrations ranging from 0.03 to 32 μg/ml and from 6.25 to 200 μg/ml, respectively, were tested in all possible concentration permutations by using checkerboard microtiter assays.
Doxycycline reduced the MIC for fluconazole 1-fold (0.25 μg/ml) when added at 200 μg/ml (not shown) and led to no detectable changes at lower concentrations (described further below). Hence, calculation of the fractional inhibitory concentration index (see Materials and Methods) produced values of >0.5 for concentrations of doxycycline that were ≤200 μg/ml, failing to indicate synergism with fluconazole (22, 41).
Growth of strain SC5314 at supra-MICs of fluconazole, a phenomenon referred to as “tolerance” (55), is commonly observed, fluconazole being only mildly fungistatic against this strain (33). These observations were confirmed in our experiments: Figure 2A shows robust recovery of cells incubated for 48 h with fluconazole alone at all concentrations tested (bottom row of cells) and decreases in viability in the presence of doxycycline. Starting from inocula of 103 CFU/ml, viable counts indicated increases of 3 and 2 logs, respectively, for cells incubated in the absence or presence of 0.5 μg/ml fluconazole (Fig. 2B). In contrast, doxycycline further reduced growth in the presence of fluconazole by 1 log, and resulted in optically clear wells (Fig. 2B). Addition of 25 μg/ml doxycycline stably reduced the viable counts by 1 log and was associated with a strong potentiation of the fungistatic power of fluconazole (Fig. 2B). Higher concentrations of doxycycline caused further reductions in viable counts at supra-MICs of fluconazole, to levels just above the detection limit of 101 CFU/ml (50 μg/ml doxycycline) (Fig. 2B) or below that (100 μg/ml doxycycline) (Fig. 2B).
These observations indicate that tolerance to fluconazole is lost in the presence of doxycycline and that killing of C. albicans at supra-MICs of fluconazole occurs in the presence of >25 μg/ml doxycycline. Since in these experiments, determination of the 99.9% killing activity that classically defines the minimal fungicidal concentration (MFC) (16) was not possible, the MFC could not be used as an endpoint. Nevertheless, the ∼3-log decrease in cell viability, compared to that of the drug-free control, observed at supra-MICs of fluconazole in the presence of 25 μg/ml doxycycline (Fig. 2B) met the requirements that define optically clear wells according to the CLSI guidelines (10), making possible the use of MIC-0 as an endpoint. Given the MIC-0 value of fluconazole alone as >32 μg/ml and the MIC-0 value when tested in combination with doxycycline as 2 μg/ml, and with the MIC-0 values of doxycycline as >200 μg/ml alone and as 25 μg/ml in combination with fluconazole, recalculation of the FICs resulted in values of 0.031 for fluconazole and 0.063 for doxycycline. The recalculated FIC index of 0.094 indicates synergism between fluconazole and doxycycline.
Fluconazole MIC testing using Etest strips has been reported to deliver results comparable to those obtained using the broth microdilution method, within a 2-dilution margin of discrepancy (48). Consistent with the results reported above for broth microdilution MIC determinations, 50 μg/ml doxycycline did not affect the fluconazole MIC of strain SC5314 in Etest measurements (Fig. 2C). However, the robust growth of smaller colonies within the fluconazole inhibition ellipse that is normally observed in these assays (48) was completely eliminated in the presence of the antibacterial (Fig. 2C). Clearing of the subpopulation of cells growing inside the fluconazole inhibition ellipse by doxycycline represents another piece of evidence of the interference of doxycycline with cell growth at supra-MIC fluconazole concentrations.
Taken together, results obtained by broth microdilution tests and Etests indicate that doxycycline potentiates the activity of fluconazole abrogating tolerance to the antifungal.
Doxycycline converts the action of fluconazole from fungistatic to fungicidal.
Given its fungistatic nature against C. albicans, fluconazole is unable to eradicate fungal infections, and the presence of elevated levels of the drug tends to favor the onset of drug resistance. Experiments shown in Fig. 2 indicated that killing of C. albicans cells occurred upon treatment with supra-MICs of fluconazole in the presence of 50 and 100 μg/ml doxycycline (96.5% and >99% killing, respectively, with an initial inoculum of 103 CFU/ml). In order to test whether killing of C. albicans by the fluconazole-doxycycline combination could reach the minimal fungicidal concentration (MFC) defined by 99.9% killing (16), cell survival was tested using the CLSI procedure for MIC determinations, with the modification proposed by Canton et al. (8). This consisted of using initial inocula of 104 CFU/ml. Sampling of 100 μl of the well's volume under these conditions allows the detection of survival rates up to 101 CFU/ml, corresponding to a decrease in viability of 3 logs (99.9% killing) (49). Using this procedure, no CFU could be recovered from wells containing fluconazole at ≥1 μg/ml in the presence of 100 μg/ml doxycycline (Fig. 3A), indicating a killing activity of >99.9% of initial cells. Under similar fluconazole conditions, 50 μg/ml doxycycline led to an average detection of 2 × 101 to 4 × 101 CFU/ml (Fig. 3A), indicating 99.6 to 99.8% killing activity. However, it has to be noted that a long incubation period (96 h) was necessary to obtain such a high percentage of killing. At standard 48-h incubations, a strong fungistatic effect was observed for 50 μg/ml doxycycline (Fig. 3A), whereas for 100 μg/ml doxycycline, cell viability was already below the initial inoculum (Fig. 3A). These results corroborate those presented in Fig. 2 and demonstrate that addition of doxycycline renders fluconazole fungicidal.
Fig 3.
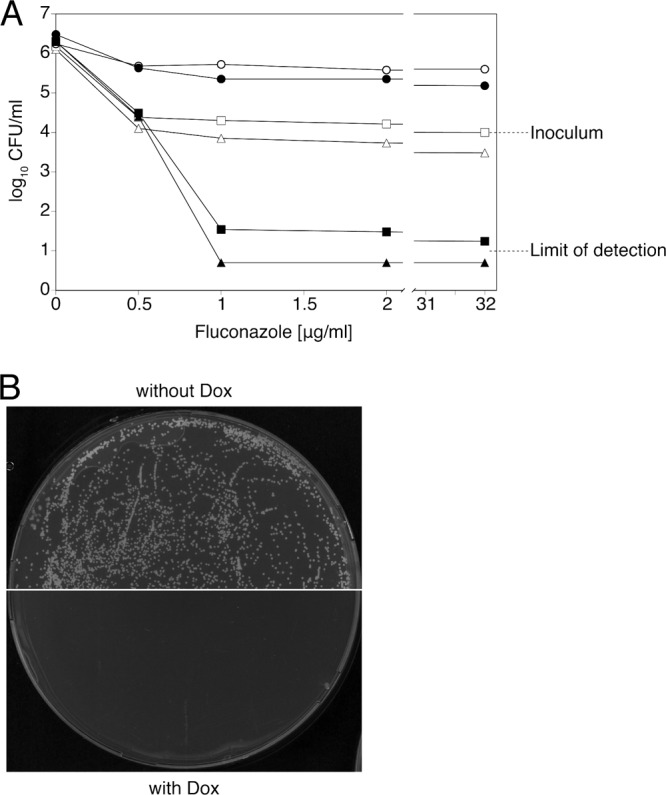
Doxycycline converts fluconazole from fungistatic to fungicidal and prevents the onset of drug resistance. (A) Viable cell counts after 48 h or 96 h of incubation in the presence of fluconazole alone or with 50 or 100 μg/ml doxycycline added. Open symbols, viable cell counts after 48 h of incubation; filled symbols, viable cell counts after 96 h of incubation. Circles, fluconazole alone; squares, fluconazole plus 50 μg/ml doxycycline; triangles, fluconazole plus 100 μg/ml doxycycline. Values falling below the detection limit of 101 CFU/ml were approximated to 0.5 × 101 CFU/ml. Mean values from three independent experiments are shown. (B) Emergence of fluconazole-resistant strains on plates containing 128 μg/ml fluconazole without (top) or with (bottom) 50 μg/ml doxycycline. Plates were incubated for 4 days before being scanned.
Doxycycline prevents the onset of resistance to fluconazole.
In Candida albicans, resistance to fluconazole has been documented to arise via different mechanisms, including mutations in ERG11 (encoding the drug target protein lanosterol 14α-demethylase) or increased expression of multidrug transporters and efflux pump-encoding genes such as CDR1, CDR2, and MDR1 (reviewed in references 1, 36, and 58). In recent years, it has become clear that in patients, resistance of C. albicans to fluconazole develops as the result of multiple genetic alterations (30, 57).
To test whether the presence of doxycycline could also influence the onset of resistance to fluconazole, we plated large numbers of C. albicans cells on media containing a high concentration of fluconazole (128 μg/ml), with and without doxycycline. As observed in Fig. 3B, a large number of colonies became visible on plates containing fluconazole only. However, no resistant colonies were present on plates that also contained doxycycline after 4 days (Fig. 3B) nor after 10 days of incubation (our unpublished observations).
Thus, the fluconazole-doxycycline combination is instrumental in the prevention of cellular resistance to fluconazole, a result that is likely to arise from the conversion of fluconazole to a fungicidal drug. In other words, cells of C. albicans may be killed before resistance-conferring mutations can be fixed by cell proliferation.
The fluconazole-doxycycline combination is also effective on a clinical isolate of Candida albicans highly resistant to fluconazole.
Clinical isolate FH5, isolated from a patient who had undergone marrow transplantation (34), was described as extremely resistant to fluconazole (57). Resistance in this strain is considered to be the result of isochromosome 5L duplication and loss of heterozygosity at the TAC1 gene locus, events that generated four copies of a nonhyperactive ERG11 allele and four copies of the hyperactive TAC1-7 allele (57). When FH5 was tested for growth in fluconazole-doxycycline-containing medium, it proved able to grow in the presence of the standard concentrations of 10 μg/ml fluconazole and 50 μg/ml doxycycline (Fig. 4A). However, growth inhibition was observed when the concentration of both chemicals was raised 4-fold (Fig. 4A), although it has to be noted that, under these conditions, growth was severely retarded but not completely abolished (data not shown). Interestingly, while stepwise increases in the concentration of doxycycline alone did not affect the fitness of strain FH5, they imparted detectable growth retardation on the reference strain SC5314 (Fig. 4A) (see Discussion).
Fig 4.

The fluconazole-doxycycline combination is also effective on a clinical isolate of C. albicans characterized by elevated resistance to fluconazole. (A) Cells of the C. albicans reference strain SC5314 and of the clinical isolate FH5 were spotted onto SD plates containing doxycycline, without (middle panels) or with (right panels) fluconazole. Concentrations are expressed in μg/ml. Plates were incubated for 2 days before being scanned. (B) Etest measurement of fluconazole MIC of strain FH5 on RPMI-glucose plates. The left panel shows medium without doxycycline, the middle panel shows medium with 50 μg/ml doxycycline, and the right panel shows medium with 200 μg/ml doxycycline.
Interactions between fluconazole and doxycycline were tested by way of checkerboard tests also with strain FH5. In broth microdilution assays, 50 to 100 μg/ml doxycycline had little effect on the MIC for fluconazole of this strain, whereas a reduction from 64 to 16 μg/ml was observed in the presence of 200 μg/ml doxycycline (data not shown). Similar results were obtained by measuring the MIC for fluconazole by way of Etest strips on RPMI-glucose (Fig. 4B). The effect of doxycycline on growth at supra-MICs of fluconazole could not be established, since this phenomenon is only minimally present in FH5 (our unpublished observations).
These observations indicate that the fluconazole-doxycycline combination can also be effective against strains with increased resistance to fluconazole.
Genomewide screen for suppressors of fluconazole-doxycycline susceptibility in Saccharomyces cerevisiae reveals suppression by increased dosage of ERG11.
Saccharomyces cerevisiae and Candida albicans are estimated to have diverged ∼800 million years ago (21). Baker's yeast offers a number of tools for genetic studies that cannot be performed with C. albicans, because of the diploid nature of the latter and the absence of a known sexual cycle. For this reason, a number of studies of pathogenic fungi, including susceptibility/resistance to azole antifungals, have been conducted using S. cerevisiae as a tool, with the aim of translating the obtained results to pathogenic species (36, 58). In order to verify whether we could use baker's yeast for genetic studies on the susceptibility to fluconazole-doxycycline, we checked whether yeast was also susceptible to this combination. Addition of doxycycline to fluconazole plates inhibited growth of S. cerevisiae wild-type strain BY4742, with some differences with respect to C. albicans. First, a dosage of chemicals higher than the standard 10 μg/ml fluconazole and 50 μg/ml doxycycline was necessary to obtain more sensitized experimental conditions (i.e., to eliminate background growth) (Fig. 5A); second, growth in fluconazole-doxycycline was severely retarded but not completely abolished, as in the case of the pathogen (our unpublished observations). Furthermore, while we never observed the onset of stable clones of C. albicans resistant to fluconazole plus doxycycline, colonies of S. cerevisiae appeared at high frequency on plates containing this drug combination. These colonies proved to be true resistant mutants at a further analysis, since their fluconazole-doxycycline-resistant phenotype was maintained after repeated passages under nonselective conditions (absence of fluconazole plus doxycycline) (our unpublished observations).
In order to identify multicopy suppressors of fluconazole-doxycycline susceptibility, BY4742 was transformed with a yeast genomic library in a 2μm plasmid (60), and transformations were plated on minimal medium containing fluconazole plus doxycycline. Putative suppressors were separated from false positives by restreaking them on the same medium and by allowing them to lose the transforming plasmids upon growth under nonselective conditions. Strains for which uracil prototrophy cosegregated with resistance to fluconazole plus doxycycline were analyzed further: their plasmids were extracted, amplified in E. coli, and retransformed into yeast to confirm plasmid-linked suppression. Two plasmids that reproducibly conferred resistance to fluconazole plus doxycycline were isolated, sequenced, and found to contain the same insert (see Materials and Methods). The insert DNA was subcloned so that the second-generation plasmid contained a yeast genomic fragment carrying a single gene. By use of this procedure, the suppressor gene was identified as ERG11 (Fig. 5A), encoding the target of fluconazole. Overexpressed ERG11 is already known to confer resistance to fluconazole and itraconazole in S. cerevisiae (24, 32), a result that was confirmed here (Fig. 5A). Hence, these results suggest that, compatibly with the synergistic hypothesis, doxycycline enhances the toxicity of fluconazole rather than adding toxicity via a different mechanism.
Overexpression of CaERG11 is not sufficient for resistance to fluconazole and doxycycline in C. albicans.
Isolation of ERG11 as a multicopy suppressor in S. cerevisiae prompted us to test whether the corresponding gene of C. albicans was also capable of suppression. The Candida Genome database (http://www.candidagenome.org) indicated ORF19.922 and ORF19.8538 as the C. albicans ERG11 alleles (hereafter referred to as CaERG11), with the corresponding predicted proteins differing at two amino acids only. We amplified ORF19.922 and cloned it in two CIp10-derived plasmids (37), so as to obtain expression of CaERG11 under the control of the strong constitutive ACT1 promoter (15) or the strong, gluconeogenic carbon source-inducible PCK1 promoter PCK1p (5). Both plasmids were integrated at the RPS10 locus of the wild-type CAI4 strain of C. albicans and tested for suppression. The corresponding strains failed to grow in the presence of the fluconazole-doxycycline combination (Fig. 5B). Furthermore, no difference between growth of these strains and their parental ones was evidenced, even when smaller amounts of doxycycline were used (Fig. 5B) (our unpublished observations), nor was any difference in the MIC for fluconazole observed (our unpublished data). Overexpression of ERG11 in these transformants was verified via quantitative real-time PCR and shown to be ∼3-fold higher in overexpression strains than that in the controls (Fig. 5C).
Finally, since overexpression of CaERG11 failed to suppress susceptibility to the fluconazole-doxycycline combination in C. albicans, we decided to investigate whether this failure was to be attributed to a functional divergence of the CaErg11 versus Erg11 protein or to a wider difference in the physiology of C. albicans versus S. cerevisiae. For this purpose, the CaERG11 gene was cloned in a yeast vector under the control of a galactose-inducible promoter, and the corresponding transformants were tested for suppression of fluconazole and fluconazole-doxycycline susceptibility. As shown in Fig. 5D, yeast cells overexpressing CaERG11 were less susceptible to fluconazole and fluconazole plus doxycycline than control cells, demonstrating that increased dosage of CaErg11 can suppress fluconazole susceptibility in yeast but not in C. albicans, and, as a consequence, that the suppression activity is retained by the CaERG11 gene. Taken together, the results on overexpression of ERG11 in the two yeast species underline the difference existing between them in terms of their responses to the antifungal fluconazole.
Doxycycline potentiates the action of fluconazole via iron chelation.
Metal chelation by tetracycline antibiotics in vitro, with a high affinity for Fe3+ ions, has been described in the past (reviewed in reference 39). Grenier and collaborators (19) proposed that the activity of tetracyclines against Actinobacillus actinomycetemcomitans may be due not only to their antibacterial properties, but also to their iron-chelating activity. In addition, doxycycline is also known to be an inhibitor of mammalian matrix metalloproteases, an action mediated by chelation of the catalytic Zn2+ ions of these enzymes (20, 53).
Recovery of Erg11, a hemoprotein, as a multicopy suppressor of hypersusceptibility of baker's yeast to fluconazole plus doxycycline led us to hypothesize that perturbation of iron homeostasis in the presence of doxycycline may play a role in establishing the hypersusceptibility of C. albicans to fluconazole. To this aim, we tested whether addition of ferric iron could reverse the doxycycline effect. As can be observed in Fig. 6A and B, addition of ferric chloride or ferric citrate restored growth in the presence of fluconazole plus doxycycline, rescuing C. albicans as well as baker's yeast from hypersusceptibility. Importantly, growth in the presence of fluconazole plus doxycycline was not restored by the addition of zinc, nor did addition of Fe3+ alter growth of C. albicans in the presence of fluconazole alone (our unpublished observations).
Fig 6.

Doxycycline synergizes with fluconazole via iron sequestration. (A) Cells of C. albicans SC5314 were spotted onto SD plates containing combinations of fluconazole and doxycycline at standard concentrations, with and without ferric citrate or ferric chloride added at the indicated concentrations. Plates were incubated for 2 days before being scanned. (B) Cells of S. cerevisiae BY4742 were similarly spotted onto SD plates. Plate scans were taken after 2 days. (C) Colorimetric assessment of iron-chelating activity of doxycycline, tetracycline, and fluconazole. Iron chelation is expressed as a reduction of absorbance at 630 nm. Filled circles, doxycycline; open circles, tetracycline; filled squares, fluconazole; open squares, water. Mean values with standard deviations from three independent experiments are shown. (D) Cells of C. albicans SC5314 were spotted onto SD plates containing combinations of fluconazole (10 μg/ml) and BPS (20 μg/ml). Plates were incubated for 4 days before being scanned.
Iron-chelating activity of tetracyclines was confirmed using a colorimetric assay. We could demonstrate that both tetracycline and doxycycline chelate iron, with doxycycline showing higher affinity than tetracycline (Fig. 6C), whereas fluconazole showed no intrinsic iron-binding activity. Notably, the stronger iron-chelating activity of doxycycline correlated with its stronger synergism with fluconazole.
Potentiation of the action of fluconazole against C. albicans by iron depletion has been demonstrated by Prasad et al. (50), via the addition of the iron chelator bathophenanthroline disulfonic acid (BPS) to the growth medium. While we confirmed this observation (Fig. 6D), we also noticed that a concentration of BPS of as little as 20 μg/ml had a negative effect on cell growth even when used alone (Fig. 6D), as also observed in S. cerevisiae (12).
Taken together, the observations described here indicate that iron depletion by doxycycline may underlie its synergism with fluconazole.
Synergism between fluconazole and doxycycline is not mediated by calcineurin.
Despite showing no intrinsic antifungal activity of their own, the immunosuppressants cyclosporine (CsA) and FK506 were reported to be synergistic with fluconazole against Candida albicans (31, 33), converting fluconazole to a fungicidal drug and eliminating trailing growth (33). The synergistic action of fluconazole with CsA and FK506 is mediated by inhibition of the phosphatase activity of calcineurin by the immunosuppressants (13, 55).
Given the similarities between the combined action of CsA/FK506 and doxycycline with fluconazole, we decided to investigate on the possibility that a decrease in or abrogation of calcineurin activity may also result from the presence of doxycycline. For this purpose, we made use of cna1Δ/cna1Δ mutants lacking subunit A of calcineurin (55). If doxycycline exerts its action via inhibition of calcineurin, one would expect the presence of doxycycline to have no effect on the hypersusceptibility of these mutants to fluconazole. Hypersusceptibility of cna1Δ/cna1Δ mutants to fluconazole (55) was confirmed in our tests (Fig. 7). However, growth of these mutants in the presence of the antifungal was completely inhibited by concentrations of doxycycline that only mildly affected wild-type cells (Fig. 7), indicating that doxycycline further compounds the hypersusceptibility of cna1Δ/cna1Δ mutants to fluconazole. Identical results were obtained using cnb1Δ/cnb1Δ mutants (13) lacking subunit B of calcineurin (our unpublished data).
Fig 7.

Synergism between doxycycline and fluconazole is not mediated by calcineurin. Cells of the indicated C. albicans strains were spotted onto SD plates containing combinations of fluconazole and doxycycline. Fluconazole was maintained at the standard 10-μg/ml concentration, whereas the concentration of doxycycline was decreased as indicated (μg/ml) to provide a sensitized background for the assay. Plates were incubated for 3 days before being scanned.
Calcineurin is inactive under normal conditions, being activated only in the presence of certain external cues (52). A strain of C. albicans in which calcineurin was permanently activated by removal of the self-inhibitory C-terminal domain of Cna1 was less susceptible to fluconazole (55). While we confirmed a moderately increased tolerance to fluconazole in this strain (our unpublished observations), we also found this strain to be as susceptible to the fluconazole-doxycycline combination as an isogenic control strain carrying a reintegrated copy of wild-type CNA1 (Fig. 7), demonstrating that constitutively active calcineurin is incapable of rescuing the cells' susceptibility to fluconazole plus doxycycline. Taken together, these observations suggest that the synergism of doxycycline with fluconazole is not mediated by inhibition of calcineurin activity.
DISCUSSION
We found that doxycycline, and to a lesser extent tetracycline, two licensed antimicrobials that prevent bacterial protein synthesis, potentiate the antifungal activity of fluconazole against Candida albicans in a dosage-dependent manner. Doxycycline converts the action of fluconazole from fungistatic to fungicidal and prevents the onset of drug resistance. Addition of doxycycline appears to have a major impact on prevention of fluconazole tolerance, defined as incomplete growth inhibition at supra-MICs of fluconazole. This finding is consistent with the role of doxycycline in converting fluconazole to a fungicidal drug and may also have implications in the prevention of drug resistance.
Other authors have reported on alteration of C. albicans susceptibility to fluconazole in the presence of tetracyclines (42, 43). Odds and collaborators (42) observed a strong reduction of the MIC for fluconazole and elimination of trailing growth by doxycycline when they used diagnostic susceptibility test agar medium, but not when tissue culture-based medium was used. On the other hand, Oliver and collaborators (43) reported on a positive effect of tetracycline on a fluconazole MIC of strain SC5314 in broth microdilution tests and a negative one when MIC was measured using Etest strips. Given the similarity between the bacterial and mitochondrial ribosomes, these authors proposed that increased susceptibility to fluconazole might result from the action of tetracycline against mitochondrial translation. Consistently, they reported inhibition of respiratory growth of C. albicans in the presence of 200 μg/ml tetracycline (43). We report here that the doxycycline MIC is higher than 200 μg/ml. Furthermore, the 50-μg/ml concentration of doxycycline used throughout this study does not affect growth of C. albicans on any medium tested, including medium containing ethanol and glycerol, carbon sources that can only be metabolized via mitochondrial respiration. It therefore appears unlikely that the synergistic effect of fluconazole plus doxycycline can be attributed to a perturbation of respiration by doxycycline. Furthermore, we tested whether inhibition of mitochondrial respiration could also be synergistic with fluconazole. Although a substantial growth defect could be observed in the presence of fluconazole when respiration was blocked by cyanide, this growth impairment appeared to be the sum of individual contributions of either chemical to the cells' fitness, rather than a synergistic effect, and did not resemble the strong effect of fluconazole plus doxycycline (our unpublished data).
Addition of iron to the growth medium restored growth of C. albicans, as well as that of S. cerevisiae, in the presence of fluconazole plus doxycycline, suggesting that hypersusceptibility to the drugs in combination could be due to antibiotic-mediated titration of iron. Consistent with this hypothesis, we confirmed that doxycycline and, to a minor extent, tetracycline act as iron chelators. Interestingly, while iron was found to be a chemical suppressor of fluconazole-doxycycline hypersusceptibility in baker's yeast as well as in C. albicans, an increased dosage of ERG11 rescued only the growth defect of S. cerevisiae. Iron depletion by doxycycline may result in lower incorporation of heme in the Erg11 protein in yeast, and an increase in the copy number of Erg11 may help the protein compete for heme. Alternatively, since iron deprivation has been demonstrated to result in downregulation of ERG11 in C. albicans (25, 50), it is possible that a similar situation occurs in S. cerevisiae and that ERG11 overexpression may simply restore gene expression to wild-type levels. Whatever the mechanism in baker's yeast, the situation appears to be more complex in C. albicans. In this organism, iron depletion as the major cause of growth inhibition has been demonstrated by reversal upon addition of ferric iron to the medium. However, the failure of an increased dosage of CaERG11 to suppress growth inhibition in C. albicans, despite full competence of the corresponding protein to act as a suppressor in yeast, suggests that doxycycline-mediated iron depletion may have more severe effects in this fungus. This interpretation is also corroborated by the higher susceptibility to fluconazole plus doxycycline observed in C. albicans with respect to S. cerevisiae (see the text above and the panels with the fluconazole-doxycycline combination in Fig. 5A and B). Alternatively, lower levels of gene overexpression obtained in C. albicans compared to those achieved in the yeast system could provide a simpler explanation for our observations. In fact, we found that overexpression of CaERG11 in our C. albicans strains was 3-fold higher than that under normal conditions (Fig. 5C), whereas overexpression in yeast transformants carrying ERG11 on a multicopy plasmid was ∼10- to 11-fold higher than that in the untransformed control (our unpublished data). However, several observations argue against this simple threshold hypothesis for failed suppression by CaERG11 in C. albicans. First, the C. albicans overexpressers show no signs of suppression even when mild, low-stringency conditions of fluconazole plus doxycycline are applied (Fig. 5B) (our unpublished observations). Second, if high levels of CaERG11 were necessary and sufficient per se to confer a resistant phenotype, one would expect the onset of stable, spontaneous mutants of C. albicans resistant to fluconazole plus doxycycline at high frequency, as predicted from the frequency of mutations in single genetic loci. Mutations in CaFKS1 conferring resistance to the antifungal caspofungin, for instance, have been reported to occur at a frequency of <10−8 mutations per cell per generation (4). The fact that we never recovered spontaneous mutants resistant to fluconazole plus doxycycline (our unpublished observations) favors the hypothesis that multiple genetic alterations may be necessary for such resistance to occur. On the other hand, while strong suppression of susceptibility to fluconazole upon overexpression of ERG11 in baker's yeast is reported in this article and has also been described by others (24, 32), a similar clear-cut correlation has not been established in C. albicans. The only published paper, to our knowledge, describing resistance to fluconazole upon overexpression of CaERG11 in C. albicans reports a moderate resistance in cells with ∼11-fold increased levels of CaERG11 (14). Although certain mutations in CaERG11 (54), as well as increased CaERG11 mRNA levels (46, 61), have been associated with resistance to fluconazole in C. albicans, it has become increasingly clear in recent years that decreased susceptibility to fluconazole in this fungus derives from complex genetic alterations that comprise and are possibly not limited to genomic rearrangements and deregulated expression of several genes (11, 30, 57).
Iron depletion has been proposed to decrease ergosterol content in C. albicans, leading to higher fluidity in cell membranes, with consequent increased passive diffusion of fluconazole (50). Some of the results reported in this paper appear similar to those obtained using BPS as the iron chelator: addition of 200 μM BPS lowered the MIC of C. albicans for fluconazole (50), and so did that for 200 μg/ml doxycycline. Importantly, the iron chelation properties of the two chemicals at the indicated concentrations are similar (our unpublished observations). Minor differences could be due to different experimental conditions.
We report here that fluconazole and doxycycline are also effective against a clinical strain of C. albicans with previously acquired resistance to fluconazole, although a higher dosage of the chemicals is required in this case. While elevated dosage of doxycycline alone has a negative impact on growth of the reference strain SC5314 (without reaching the MIC), presumably by iron depletion, clinical strain FH5 is insensitive to these conditions, suggesting that the ability of this strain to thrive in a low-iron environment and resistance to fluconazole may be linked. Interestingly, transcription of the iron transporters FRE9 and FTR1—the latter required for growth under low-iron conditions (51)—is upregulated in response to exposure to fluconazole (27).
Recently, a role for C. albicans Als3 in iron acquisition from host ferritin has been proposed (2). We therefore tested whether als3Δ/als3Δ mutants were differentially susceptible to fluconazole plus doxycycline and found them to be as susceptible as their wild-type counterpart (our unpublished observations), a result that argues against a role for Als3 in the establishment of doxycycline-mediated potentiation of fluconazole activity. Similarly, our experiments argue against a role for calcineurin in this process.
From a practical point of view, since experiments with doxycycline-regulated promoters are an established reality in the field of C. albicans molecular genetics, we propose that the interesting results obtained using this system should be further confirmed by experiments performed using media with additional iron, in order to discriminate between phenotypes that can be attributed to altered dosage of the gene of interest and phenotypes that may have arisen from doxycycline-mediated perturbation of iron homeostasis.
ACKNOWLEDGMENTS
We thank Ilse Palmans for excellent technical assistance and Nico Vangoethem for help with the preparation of figures. We also thank Judith Berman, Joe Heitman, and Dominique Sanglard for strains.
This work was supported by SBO grant 060839 from the Flemish Institute for Science and Technology (IWT) and the research fund of the K.U. Leuven (GOA/2007/08).
Footnotes
Published ahead of print 7 May 2012
REFERENCES
- 1. Akins RA. 2005. An update on antifungal targets and mechanisms of resistance in Candida albicans. Med. Mycol. 43:285–318 [DOI] [PubMed] [Google Scholar]
- 2. Almeida RS, et al. 2008. The hyphal-associated adhesin and invasin Als3 of Candida albicans mediates iron acquisition from host ferritin. PLoS Pathog. 4:e1000217 doi:10.1371/journal.ppat.1000217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Backen AC, et al. 2000. Evaluation of the CaMAL2 promoter for regulated expression of genes in Candida albicans. Yeast 16:1121–1129 [DOI] [PubMed] [Google Scholar]
- 4. Balashov SV, Park S, Perlin DS. 2006. Assessing resistance to the echinocandin antifungal drug caspofungin in Candida albicans by profiling mutations in FKS1. Antimicrob. Agents Chemother. 50:2058–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barelle CJ, et al. 2004. GFP as a quantitative reporter of gene regulation in Candida albicans. Yeast 21:333–340 [DOI] [PubMed] [Google Scholar]
- 6. Berman J, Sudbery PE. 2002. Candida albicans: a molecular revolution built on lessons from budding yeast. Nat. Rev. Genet. 3:918–930 [DOI] [PubMed] [Google Scholar]
- 7. Braun BR, et al. 2005. A human-curated annotation of the Candida albicans genome. PLoS Genet. 1:36–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Canton E, et al. 2003. Minimum fungicidal concentrations of amphotericin B for bloodstream Candida species. Diagn. Microbiol. Infect. Dis. 45:203–206 [DOI] [PubMed] [Google Scholar]
- 9. Chopra I, Roberts M. 2001. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 65:232–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clinical and Laboratory Standards Institute 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A3, 3rd ed Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 11. Coste A, et al. 2007. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot. Cell 6:1889–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Craven RJ, Mallory JC, Hand RA. 2007. Regulation of iron homeostasis mediated by the heme-binding protein Dap1 (damage resistance protein 1) via the P450 protein Erg11/Cyp51. J. Biol. Chem. 282:36543–36551 [DOI] [PubMed] [Google Scholar]
- 13. Cruz MC, et al. 2002. Calcineurin is essential for survival during membrane stress in Candida albicans. EMBO J. 21:546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Du W, Coaker M, Sobel JD, Akins RA. 2004. Shuttle vectors for Candida albicans: control of plasmid copy number and elevated expression of cloned genes. Curr. Genet. 45:390–398 [DOI] [PubMed] [Google Scholar]
- 15. Enjalbert B, et al. 2009. A multifunctional, synthetic Gaussia princeps luciferase reporter for live imaging of Candida albicans infections. Infect. Immun. 77:4847–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ernst EJ. 2005. Susceptibility testing methods of antifungal agents. Methods Mol. Med. 118:3–12 [DOI] [PubMed] [Google Scholar]
- 16a. Fonzi WA, Irwin MY. 1983. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527–534 [DOI] [PubMed] [Google Scholar]
- 18. Gillum AM, Tsay EY, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet. 198:179–182 [DOI] [PubMed] [Google Scholar]
- 19. Grenier D, Huot MP, Mayrand D. 2000. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob. Agents Chemother. 44:763–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griffin MO, Ceballos G, Villarreal FJ. 2011. Tetracycline compounds with non-antimicrobial organ protective properties: possible mechanisms of action. Pharmacol. Res. 63:102–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heckman DS, et al. 2001. Molecular evidence for the early colonization of land by fungi and plants. Science 293:1129–1133 [DOI] [PubMed] [Google Scholar]
- 22. Johnson MD, MacDougall C, Ostrosky-Zeichner L, Perfect JR, Rex JH. 2004. Combination antifungal therapy. Antimicrob. Agents Chemother. 48:693–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelly SL, Arnoldi A, Kelly DE. 1993. Molecular genetic analysis of azole antifungal mode of action. Biochem. Soc. Trans. 21:1034–1038 [DOI] [PubMed] [Google Scholar]
- 24. Kontoyiannis DP, Sagar N, Hirschi KD. 1999. Overexpression of Erg11p by the regulatable GAL1 promoter confers fluconazole resistance in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 43:2798–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lan CY, et al. 2004. Regulatory networks affected by iron availability in Candida albicans. Mol. Microbiol. 53:1451–1469 [DOI] [PubMed] [Google Scholar]
- 26. Lass-Florl C. 2009. The changing face of epidemiology of invasive fungal disease in Europe. Mycoses 52:197–205 [DOI] [PubMed] [Google Scholar]
- 27. Lepak A, et al. 2006. Time course of microbiologic outcome and gene expression in Candida albicans during and following in vitro and in vivo exposure to fluconazole. Antimicrob. Agents Chemother. 50:1311–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leuker CE, Sonneborn A, Delbruck S, Ernst JF. 1997. Sequence and promoter regulation of the PCK1 gene encoding phosphoenolpyruvate carboxykinase of the fungal pathogen Candida albicans. Gene 192:235–240 [DOI] [PubMed] [Google Scholar]
- 29. Lewis RE. 2009. Overview of the changing epidemiology of candidemia. Curr. Med. Res. Opin. 25:1732–1740 [DOI] [PubMed] [Google Scholar]
- 30. MacCallum DM, et al. 2010. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrob. Agents Chemother. 54:1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maesaki S, et al. 1998. Synergic effects of tacrolimus and azole antifungal agents against azole-resistant Candida albicans strains. J. Antimicrob. Chemother. 42:747–753 [DOI] [PubMed] [Google Scholar]
- 32. Mallory JC, et al. 2005. Dap1p, a heme-binding protein that regulates the cytochrome P450 protein Erg11p/Cyp51p in Saccharomyces cerevisiae. Mol. Cell. Biol. 25:1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marchetti O, et al. 2000. Potent synergism of the combination of fluconazole and cyclosporine in Candida albicans. Antimicrob. Agents Chemother. 44:2373–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marr KA, White TC, van Burik JA, Bowden RA. 1997. Development of fluconazole resistance in Candida albicans causing disseminated infection in a patient undergoing marrow transplantation. Clin. Infect. Dis. 25:908–910 [DOI] [PubMed] [Google Scholar]
- 35. Miller CA, Martinat MA, Hyman LE. 1998. Assessment of aryl hydrocarbon receptor complex interactions using pBEVY plasmids: expression vectors with bi-directional promoters for use in Saccharomyces cerevisiae. Nucleic Acids Res. 26:3577–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morschhauser J. 2002. The genetic basis of fluconazole resistance development in Candida albicans. Biochim. Biophys. Acta 1587:240–248 [DOI] [PubMed] [Google Scholar]
- 37. Murad AM, et al. 2000. CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast 16:325–327 [DOI] [PubMed] [Google Scholar]
- 38. Nakayama H, et al. 2000. Tetracycline-regulatable system to tightly control gene expression in the pathogenic fungus Candida albicans. Infect. Immun. 68:6712–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nelson ML. 1998. Chemical and biological dynamics of tetracyclines. Adv. Dent. Res. 12:5–11 [DOI] [PubMed] [Google Scholar]
- 40. Noble SM, Johnson AD. 2007. Genetics of Candida albicans, a diploid human fungal pathogen. Annu. Rev. Genet. 41:193–211 [DOI] [PubMed] [Google Scholar]
- 41. Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52:1. [DOI] [PubMed] [Google Scholar]
- 42. Odds FC, Abbott AB, Pye G, Troke PF. 1986. Improved method for estimation of azole antifungal inhibitory concentrations against Candida species, based on azole/antibiotic interactions. J. Med. Vet. Mycol. 24:305–311 [DOI] [PubMed] [Google Scholar]
- 43. Oliver BG, et al. 2008. Tetracycline alters drug susceptibility in Candida albicans and other pathogenic fungi. Microbiology 154:960–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ostrosky-Zeichner L, et al. 2010. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 9:719–727 [DOI] [PubMed] [Google Scholar]
- 45. Park YN, Morschhauser J. 2005. Tetracycline-inducible gene expression and gene deletion in Candida albicans. Eukaryot. Cell 4:1328–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perea S, et al. 2001. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 45:2676–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20:133–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pfaller MA, Messer SA, Karlsson A, Bolmstrom A. 1998. Evaluation of the Etest method for determining fluconazole susceptibilities of 402 clinical yeast isolates by using three different agar media. J. Clin. Microbiol. 36:2586–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pfaller MA, Sheehan DJ, Rex JH. 2004. Determination of fungicidal activities against yeasts and molds: lessons learned from bactericidal testing and the need for standardization. Clin. Microbiol. Rev. 17:268–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prasad T, Chandra A, Mukhopadhyay CK, Prasad R. 2006. Unexpected link between iron and drug resistance of Candida spp.: iron depletion enhances membrane fluidity and drug diffusion, leading to drug-susceptible cells. Antimicrob. Agents Chemother. 50:3597–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramanan N, Wang Y. 2000. A high-affinity iron permease essential for Candida albicans virulence. Science 288:1062–1064 [DOI] [PubMed] [Google Scholar]
- 52. Rusnak F, Mertz P. 2000. Calcineurin: form and function. Physiol. Rev. 80:1483–1521 [DOI] [PubMed] [Google Scholar]
- 53. Ryan ME, et al. 2001. Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr. Med. Chem. 8:305–316 [DOI] [PubMed] [Google Scholar]
- 54. Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14α-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 42:241–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sanglard D, et al. 2003. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Mol. Microbiol. 48:959–976 [DOI] [PubMed] [Google Scholar]
- 56. Sanglard D, et al. 1995. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 39:2378–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Selmecki A, et al. 2008. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 68:624–641 [DOI] [PubMed] [Google Scholar]
- 58. Shapiro RS, Robbins N, Cowen LE. 2011. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev. 75:213–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sherman F. 2002. Getting started with yeast. Methods Enzymol. 350:3–41 [DOI] [PubMed] [Google Scholar]
- 60. Stettler S, et al. 1993. A general suppressor of RNA polymerase I, II and III mutations in Saccharomyces cerevisiae. Mol. Gen. Genet. 239:169–176 [DOI] [PubMed] [Google Scholar]
- 61. White TC. 1997. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 41:1482–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]