

Abstract
We previously reported that among a series of artemisinin-derived monomers and dimers, dimer diphenyl phosphate (838) was the most potent inhibitor of human cytomegalovirus (CMV) replication. Our continued investigation of a prototypic artemisinin monomer (artesunate [AS]) and dimer (838) now reveals that both compounds have specific activity against CMV but do not inhibit lytic replication of human herpesvirus 1 or 2 or Epstein-Barr virus. AS and 838 inhibited CMV replication during the first 24 h of the virus replication cycle, earlier than the time of ganciclovir (GCV) activities and prior to DNA synthesis. Neither compound inhibited virus entry. Quantification of DNA replication and virus yield revealed a similar level of inhibition by GCV, but AS and 838 had a 10-fold-higher inhibition of virus yield than of DNA replication, suggesting that artemisinins could inhibit CMV through multiple steps: a predominant early inhibition and possibly an additional step following DNA replication. During the strong early CMV inhibition, the transcription of immediate-early genes was not significantly downregulated, and viral protein expression was reduced only after 48 h. AS and GCV were reversible CMV inhibitors, but the inhibition of CMV replication by 838 was irreversible. Combinations of GCV and 838 as well as GCV and AS were highly synergistic. Finally, treatment with 838, but not AS, prior to CMV infection demonstrated strong anti-CMV activity. These findings illustrate the unique activities of dimer 838, including early and irreversible CMV inhibition, possibly by tight binding to its target.
INTRODUCTION
Human cytomegalovirus (CMV), a ubiquitous betaherpesvirus, is a major pathogen in transplant recipients and patients with AIDS (4, 9, 14, 20). It is also the most common congenitally acquired infection, resulting in neurological sequelae, deafness, and mental retardation (6).
All systemic anti-CMV drugs target the viral DNA polymerase and effectively inhibit CMV replication (26). However, their use is associated with toxicities to the bone marrow (ganciclovir [GCV]) and kidneys (foscarnet and cidofovir) (27, 29). GCV is the only agent approved for therapy of congenital CMV infection with central nervous system involvement, based on a phase III clinical trial in which hearing preservation and prevention of hearing loss were documented in the treated children (19). GCV-resistant mutants develop during prolonged courses of therapy in transplant recipients and in AIDS patients (14, 29). The widespread use of a limited number of drugs eventually leads to the emergence of resistant viral strains.
Anti-CMV agents that target viral proteins other than the DNA polymerase are in different stages of development. Among newly identified viral targets are the UL97 kinase inhibitor maribavir (21, 34) and the terminase inhibitor AIC246 (18, 24). Maribavir had promising results in a phase II clinical trial (35), but a multicenter phase III trial with bone marrow transplant recipients showed no significant difference in rates of CMV disease between subjects treated with maribavir and placebo. Therefore, maribavir is currently being evaluated to establish its role in CMV therapy. AIC246 is in phase II clinical trials (18, 24).
We recently reported on the highly potent anti-CMV activities of arteminisin-derived dimers (2). We also identified among a series of dimers the most potent anti-CMV compound, dimer diphenyl phosphate (molecular weight, 838). Its selectivity index was approximately 1,500, 130-fold higher than that of artemisinin-derived monomer artesunate (AS) and 15-fold higher than that of GCV (10). We therefore continued our investigation of the anti-CMV activity of dimer diphenyl phosphate (838) and compared it to AS and GCV.
We report here on the mechanism of CMV inhibition by dimer 838 compared to the activities of AS and GCV. Our results show that dimer 838 has unique mechanisms of action compared to the other compounds: it is an irreversible inhibitor of CMV replication, and exposure of cells to the compound prior to infection achieves significant CMV inhibition.
MATERIALS AND METHODS
Compounds.
GCV was purchased from Sigma Chemical Co. (St. Louis, MO). AS and dimer diphenyl phosphate were synthesized at Johns Hopkins University and solubilized in 100% dimethyl sulfoxide (DMSO) (1). Stocks of 10 mM were stored at −80°C. Synthetic compounds were at least 98% pure based on proton nuclear magnetic resonance (NMR) spectroscopy. Unless otherwise specified, the concentrations used for the experiments were as follows: AS, 30 μM; 838, 0.1 μM; and GCV, 10 μM. These concentrations were chosen based on full virus inhibition by plaque reduction assay and more than 90% inhibition of pp28-luciferase expression, while not causing cytotoxicity. The reported mean 50% effective concentrations (EC50) ± standard deviations (SD) (in μM) of AS, 838, and GCV are 6.6 ± 0.4, 0.04 ± 0.003, and 2.7 ± 0.1, respectively. The concentrations (in μM) of AS, 838, and GCV leading to 50% cell toxicity (CC50) are 71.7 ± 4, 55.8 ± 2.8, and 247 ± 33.4, respectively (10, 11).
Viruses and antiviral assays.
The pp28-luciferase Towne strain was constructed as previously described (11). This recombinant CMV expresses luciferase under the control of the UL99 (pp28) late promoter at 48 to 72 h postinfection (hpi). Human herpesvirus 1 and 2 strains were as follows: herpes simplex virus 1 (HSV-1) KOS, luciferase HSV-1-KOS/Dlux/oriS (32), and clinical isolates of HSV-1 and -2. The clinical isolates were obtained from the Johns Hopkins clinical virology laboratory with no identifiers that can be linked to a specific subject. The Johns Hopkins School of Medicine Office of Human Subject Research Institutional Review Board determined that the research does not involve human subjects under the DHHS or FDA regulations.
Human foreskin fibroblasts (HFF) and Vero cells were used for infections with CMV and HSV-1/2, respectively. Infection of HFF, plaque reduction, and luciferase assay have been previously described (10). Unless otherwise specified, all CMV infections were carried out at multiplicity of infection (MOI) of 1 PFU/cell.
Plaque assays were performed for HSV-1 and HSV-2. Vero cells were seeded at 3 × 105 cells per well in 12-well plates and were infected 24 h later with HSV-1 or HSV-2 strains at 200 PFU/well. Following 60 min of adsorption, the virus was aspirated, and Dulbecco modified Eagle medium (DMEM) containing 0.5% carboxymethyl cellulose, 4% fetal bovine serum (FBS), and compounds at the indicated concentrations were added to triplicate wells. After incubation at 37°C for 2 days, the overlay was removed, and plaques were counted after crystal violet staining. Plaque size was measured under a Nikon Eclipse E-200 microscope. Infection with HSV-1-luciferase followed the same infection protocols except that luciferase was quantified at 24 hpi.
Akata cells (4 × 106/well of a 24-well plate) latently infected with Epstein-Barr virus (EBV) were induced with 50 μg/ml of goat anti-human IgG (Sigma). AS, 838, and GCV were added to triplicate wells immediately after the addition of human IgG. Cells were collected at 48 h and 4 days after lytic induction for cell and supernatant DNA quantification, respectively. Cellular DNA was purified using a Wizard SV genomic kit (Promega, Madison, WI). Supernatants were treated with DNase (NEB) at 37°C for 1 h, and virion DNA was purified with a QIAamp MinElute virus spin kit (Qiagen, Valencia, CA) following the manufacturer's instructions. The effect of the compounds on lytic induction of EBV was analyzed by real-time PCR of the EBV DNA polymerase gene, BALF5 (23).
DNA synthesis.
HFF cells were infected with CMV Towne and treated with AS, 838, GCV, or DMSO as a control. Cells were collected at 48 hpi, and total DNA was extracted using the Wizard SV genomic kit (Promega). Viral DNA copies were quantified by real-time PCR of the highly conserved US17 gene as previously described (11, 33).
Immunofluorescence.
AS, 838, and a human-specific Toll-like receptor 9 (TLR9) ligand, CpG 2006 (13), were used to determine inhibition of virus entry. Compounds were diluted in serum-free medium and added to HFF cells seeded on chamber slides at 24 h prior to infection. After infection and treatment, cells were fixed, permeabilized, and air dried. Cells were then incubated with mouse monoclonal anti-pp65 antibody (Vector Laboratories, Burlingame, CA) at 37°C in humidified chambers for 1 h, washed three times with Tris-borate-EDTA plus Tween 20 (TBST) (0.1% Tween 20), incubated with fluorescein isothiocyanate (FITC)-conjugated donkey anti-mouse IgG (Sigma) at 37°C in humidified chambers for 1 h, and washed with TBST (0.1% Tween 20). A drop of mount oil containing DAPI (4′,6′-diamidino-2-phenylindole) (Santa Cruz Biotechnology, Santa Cruz, CA) was added to the slides before visualization with a Nikon Eclipse E-800 fluorescence microscope.
Time-of-addition and time-of-removal studies.
HFF cells were infected with Towne CMV as previously described. At 0, 6, 12, 24, 36, 48, and 72 hpi, the medium was replaced with a medium containing AS, 838, or GCV. For time-of-removal studies, the medium containing the compounds was removed at 0, 6, 12, 24, 36, 48, and 72 hpi, cells were washed three times with phosphate-buffered saline (PBS), and drug-free medium was added. Cells were incubated at 37°C, and the following assays were performed: DNA replication (by US17 real-time PCR in cell lysates at 48 and 72 hpi), late gene expression (luciferase assay at 72 hpi), and virus yield (by US17 real-time PCR in supernatants at 96 hpi).
MOI dependency.
HFF cells were infected with Towne CMV at MOIs of 0.05, 1, and 5. After infection, cells were treated with AS, 838, or GCV. Virus yield in supernatants of infected cells collected at day 6 postinfection was measured by real-time PCR.
CMV reversibility assay.
HFF cells were infected and treated with AS, 838 or, GCV. At 24, 48, and 72 hpi, media were removed and replaced with fresh media without compounds. Cells were incubated until 6 days postinfection, and supernatants were collected for viral DNA quantification by real-time PCR.
HFF reversibility assay.
HFF cells (5× 105) were seeded in each of four T25 tissue culture flasks and incubated for 3 days with fresh medium or medium containing AS, 838, or GCV. Cells were then trypsinized, and 2,500 cells were seeded in each well of 96-well tissue culture plates and grown in fresh medium without compounds. At 1, 2, 3, 4, 5, and 7 days after reseeding, cells were collected and cell viability was assayed by CellTiter-Glo luminescence (Promega) according to the manufacturer's instructions.
Western blot analysis.
HFF cells were infected with Towne CMV and treated with AS, 838, and GCV. At the indicated times, cells were harvested in sample buffer (radioimmunoprecipitation assay [RIPA] buffer, 50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP-40, and protease inhibitor cocktail) and boiled. Proteins were separated by electrophoresis and transferred to polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA). After blocking, blots were probed with the following primary antibodies: mouse anti-human CMV immediate-early (IE) protein (IE1 and IE2) MAb810 (Millipore, Billerica, MA), mouse anti-human CMV UL83 (pp65) (Vector Laboratories Inc., Burlingame, CA), and mouse anti-human CMV UL44 and UL84 (Santa Cruz). After being washed five times, blots were probed with horseradish peroxidase (HRP)-conjugated anti-mouse antibody (Sigma) at 1:10,000 for 1 h at room temperature. Blots were washed five times with TBST (0.1% Tween 20), and detection was by enhanced chemiluminescence (Pierce Chemical, Rockford, IL) according to the manufacturer's protocol.
RNA quantification.
Total RNA was purified from HFF cells using the RNeasy minikit (Qiagen) according to the manufacturer's instructions. One microgram of purified total RNA was reverse transcribed into cDNA using the RevertAid Premium first-strand cDNA synthesis kit (Thermo Scientific, Glen Burnie, MD). The mRNA levels of specific genes were quantified by real-time PCR using Maxima SYBR green/ROX quantitative PCR (qPCR) master mix (Thermo Scientific); GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as an internal control. Primer sequences were as follows: IE1/IE2 forward (F), 5′-ACAAGTGACCGAGGATTGCAACGA-3′; IE1 reverse (R), 5′-CAGCATGTGCTCCTTGATTCTATGC-3′; IE2 R, 5′-GTGGGTTGTCAGCGTGGGGC-3′; UL37 F, 5′-GCACCGGTTCCTGTGGCAA-3′; UL37 R, 5′-GTCAACAACAGCCACACAGACACC-3′; UL36 F, 5′-GCAAGATGGGCACCGTCTGTT-3′; UL36 R, 5′-CGATGAGCACGATGAGTTGGTAGC-3′; TRS1 F, 5′-CAGAACCACCGTTGACTCC-3′; TRS1 R, 5′-CCCAGAACCCAGAAAGTCTC-3′; US3 F, 5′-AGAGGAGCTGTTTCCCTCCT-3′; and US3 R, 5′-AGAGGTCCGTCTTCTTCGTC-3′.
Drug combination studies.
The combined inhibitory effects of AS and GCV as well as 838 and GCV on CMV replication were evaluated by adding compounds to infected HFF cells at the indicated dilutions. Analysis was performed using the Bliss independency model and the Bliss equation (3):
where fu is the fraction of unaffected cells, D is drug concentration, and m is the slope. The Bliss model assumes that two inhibitors act via independent mechanisms. Drug combination is represented as the product of two probabilistically independent events, FUA1 and FUA2, identical to the mutually nonexclusive case: FUA = FUA1 × FUA2. FUA is the fractional unaffected or the expected combined effect. If the ratio of observed fold inhibition divided by the expected fold inhibition is greater than 1, the compounds are synergistic. If the ratio is less than 1, the combination is considered antagonistic, and if it equals 1, the compounds are independent of each other. The Bliss model has been used to analyze antiretroviral combination therapy for HIV (16).
RESULTS
CMV-specific inhibition by AS and 838.
We evaluated whether AS and 838 could inhibit the replication of alphaherpesviruses (HSV-1 and -2) and gammaherpesviruses (EBV). AS and 838, at concentrations higher than those required for complete inhibition of CMV replication (10 μM and 50 μM for AS and 1 μM and 10 μM for 838), did not reduce luciferase expression of HSV-1-KOS/Dlux/oriS or the number of plaques of the KOS1 strain and clinical isolates of HSV-1 and -2. (Fig. 1A and B). Higher concentrations of AS (50 μM) and 838 (10 μM) inhibited Vero cell growth, and a reduction in plaque diameter (less than 50%) was observed for some of the clinical isolates (see Table S1 in the supplemental material). AS (50 μM) had mild activity against EBV (Fig. 1C). Thus, the strong inhibitory effect of artemisinin-derived monomer and dimer on CMV replication appears to be specific.
Fig 1.

Artemisinin derivatives specifically inhibit CMV replication, (A) Vero cells infected with HSV-1-luciferase (KOS/Dlux/oriS), KOS1, or a clinical isolate of HSV-1 and were treated with AS, 838, and GCV at the indicated concentrations. The anti-HSV-1 activities of compounds were determined by luciferase and plaque reduction assays performed at 24 and 48 hpi, respectively. (B) Vero cells were infected with two different HSV-2 clinical isolates (1 and 4) and treated with AS, 838, and GCV at the indicated concentrations. The anti-HSV-2 activities of compounds were determined by a plaque reduction assay performed at 48 hpi. (C) Akata (EBV-positive) cells were induced by goat anti-human IgG, and cells were treated with AS, 838, and GCV at the indicated concentrations. EBV DNA replication and virus yield were quantified by real-time PCR in cell lysates at 48 h and in supernatants at day 4. VND, virus with no drug. Data represent mean values (±SD) of triplicate determinations from three independent experiments.
CMV inhibition by AS and 838 occurs at early stages of virus replication.
To identify the time during which CMV is inhibited by AS and 838, we tested the ability of the compounds to inhibit virus entry. Cells were treated with AS, 838, GCV, or the TLR9 ligand CpG 2006 as a control (10 μM). Using immunofluorescence assay for the tegument protein pp65, we found that CMV entry was not affected by AS or 838, while CpG 2006 prevented virus entry (Fig. 2) (13).
Fig 2.

AS and 838 do not inhibit CMV entry into HFF. HFF were treated with AS, 838, GCV, and CpG 2006 (10 μM) at 24 h prior to infection. Cells were infected with CMV and treated with compounds for 90 min. Immunofluorescence staining was performed with mouse monoclonal anti-pp65 antibody. The fluorescence of FITC-conjugated donkey anti-mouse IgG and DAPI was visualized and merged using an Nikon Eclipse E-800 fluorescence microscope. VND, virus with no drug; NVND, no virus and no drug.
Next, AS and 838 were added to or removed from infected HFF cells at different times after infection. Virus DNA replication was quantified at 48 and 72 hpi (see Fig. S1C to F in the supplemental material), pp28 luciferase activity was quantified at 72 hpi (Fig. 3A), and infectious progenies in supernatants of infected cells were quantified by real-time PCR at 96 hpi (see Fig. S1A and B in the supplemental material). Using these three methods, the inhibition curves of the tested compounds correlated well. Starting from 24 hpi, the anti-CMV activity of 838 had gradually decreased, suggesting that it targets an event earlier than DNA synthesis. Removal of 838 at as early as 12 hpi showed that 838 had already achieved more than 50% of its anti-CMV activity, while the activity of AS was decreased to more than 50% only after removal at 36 h (Fig. 3B). When AS and 838 were added at 72 hpi, their anti-CMV activities measured as virus yield at 96 hpi were decreased to more than 50% (see Fig. S1A in the supplemental material). Similarly, when compounds were added at 48 hpi and DNA replication was measured at 72 hpi, the anti-CMV activity was decreased to more than 50% (see Fig. S1E in the supplemental material). The add-on data suggest that both AS and 838 are active at an early stage of CMV replication, while the removal data point to possible differences in their activity. CMV-infected cells require a short exposure to 838, but in the case of AS, a longer exposure may be required for effective CMV inhibition.
Fig 3.

Add-on, removal, and MOI dependency assays of AS, 838, and GCV. (A and B) AS, 838, and GCV were added (A) or removed (B) from CMV-infected HFF cells at the indicated time points, and luciferase activity was assayed at 72 hpi. (C) HFF cells were infected with Towne CMV at MOIs of 0.05, 1, and 5. Infected cells were treated with AS, 838, and GCV for 6 days. Virus yield in supernatants collected at 6 days postinfection was quantified by US17 real-time PCR. Data represent mean values (±SD) of triplicate determinations from three independent experiments.
To further support the early CMV inhibition by AS and 838, their activities were assayed at different MOIs (MOI dependency) (15, 22). Virus yields in supernatants collected at 6 days postinfection from HFF cells infected with CMV at MOIs of 0.05, 1, and 5 were quantified by real-time PCR. As expected, CMV inhibition with GCV was not MOI dependent, but there was a significant decrease in CMV inhibition with AS and 838 at higher MOIs (Fig. 3C). Taking these findings together, the antiviral activities of AS and 838 occur early during the virus replication cycle.
838 is an irreversible inhibitor of CMV replication.
To understand the early activities of 838, a reversibility assay was performed. CMV-infected HFF cells were treated with AS, 838, and GCV. At 24, 48, and 72 hpi, media containing the compounds were removed and replaced with fresh media. Cells were incubated for 6 days postinfection, media were collected, and US17 real-time PCR was performed. The anti-CMV activity of 838 was irreversible (Fig. 4A) even when the compound was removed at as early as 24 hpi. However, AS and GCV were both reversible inhibitors, even when removed at 3 days after infection.
Fig 4.

Reversibility of CMV infection and HFF cell growth after treatment with AS, 838, and GCV. (A) Compounds were present in CMV-infected cells at the indicated intervals. Virus DNA in supernatants was quantified by real-time PCR at 6 days postinfection. VND, virus with no drug. (B) HFF cells were treated with AS, 838, and GCV for 3 days, and then 2,500 treated or untreated cells were seeded on 96-well plates and cultured in DMEM with 10% FBS. Cell viability was measured by CellTiter-Glo at 1 to 7 days after seeding. Data represent mean values (±SD) of triplicate determinations from more than three independent experiments.
To determine whether the irreversibility of 838 inhibition of CMV replication was secondary to a cellular effect, a reversibility assay was performed in noninfected HFF cells treated with AS, 838, and GCV. HFF cells were treated with each compound for 3 days, after which equal numbers of treated cells were seeded in each well of 96-well plates and were grown in fresh media without compounds. At 1, 2, 3, 4, 5, and 7 days postseeding, cell viability was assayed (Fig. 4B). During the first day after release from AS- and 838-treated cells, there was somewhat slower cell growth than that of normal and GCV-treated cells, but starting at the second day and up until 7 days after seeding, the rate of cell growth was not different between the treated conditions. Since the effect of 838 on cell growth was reversible, the irreversible inhibition of CMV could not simply be explained by inhibition of cell growth.
Pretreatment of HFF with 838 is sufficient to inhibit CMV replication.
To further assess the early CMV inhibition with 838, HFF cells were treated with AS or 838 prior to infection with CMV. After 24 h, the compounds were removed and cells were infected with CMV. DNA replication, pp28 late gene expression, and virus yield were measured at the indicated times. Pretreatment with 838, but not AS or GCV, resulted in CMV inhibition as measured by all three assays (Fig. 5) but was not as potent as treatment following virus inoculation (60- versus 160-fold inhibition of virus yield). Since CMV inhibition with pretreatment could not have resulted from a block of entry (Fig. 2), this suggests that 838 could bind to a cellular target which may be necessary for CMV replication.
Fig 5.
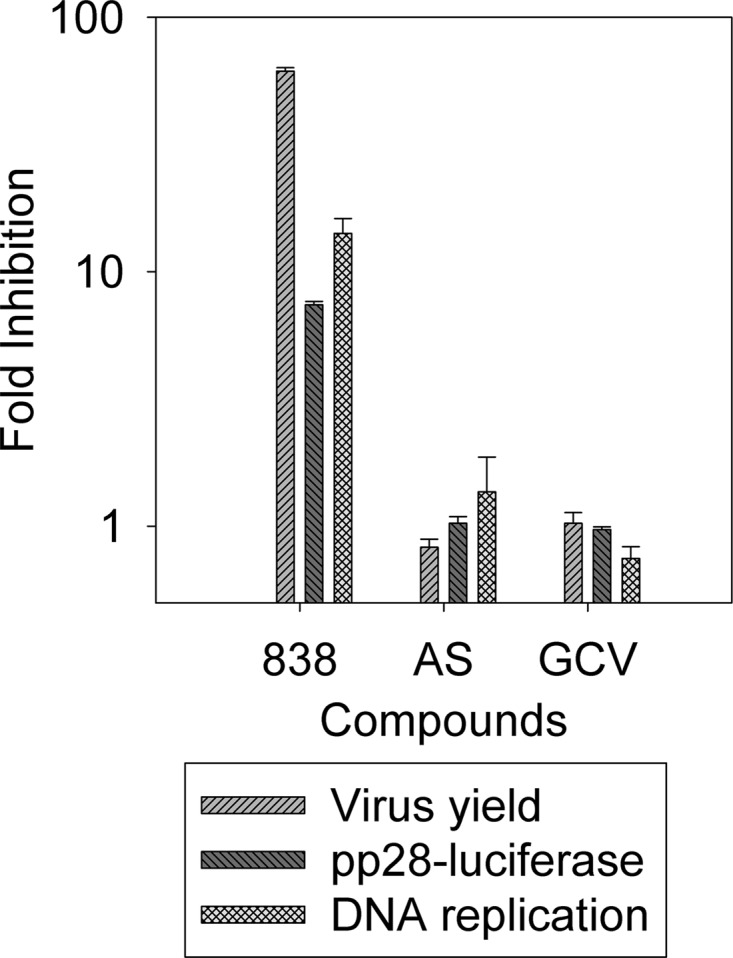
Effects of pretreatment with AS, 838, and GCV on CMV replication. HFF cells were treated with AS, 838, and GCV 24 h prior to infection. Compounds were removed just before infection. DNA replication in cells collected at 48 hpi was measured by real-time PCR. pp28-luciferase activity was measured in cell lysates collected 72 hpi. Virus yield in supernatants collected at 96 hpi was measured by real-time PCR. Data represent mean values (±SD) of triplicate determinations from three independent experiments.
Inhibition of DNA replication and virus yield by AS and 838.
We previously quantified the anti-CMV activities of AS and 838 using the pp28-luciferase system, which correlates well with the plaque reduction assay (11). Here we conducted a detailed evaluation of CMV inhibition by the tested compounds using additional assays. Quantification of infectious progeny (virus yield) based on plaque reduction and real-time PCR from supernatants correlated well (Fig. 6). Therefore, virus yield based on real-time PCR was used in subsequent experiments. GCV, AS, and 838 at concentrations sufficient to inhibit more than 90% of pp28 late gene expression inhibited virus yield to more than 100-fold (Fig. 6). However, inhibition of DNA replication varied between the compounds. While the DNA polymerase inhibitor CV, exhibited similar inhibition of virus yield and DNA replication, the inhibition of virus yield by AS and 838 was approximately 10-fold higher than the inhibition of DNA replication at both 48 and 72 hpi, suggesting that in addition to the early effect, a late process may also contribute to their CMV inhibition.
Fig 6.

Inhibition of DNA replication, pp28 expression, and virus yield by AS, 838, and GCV. DNA replication in cells collected at 48 and 72 hpi was quantified by real-time PCR. pp28-luciferase activity was measured in cell lysates collected at 72 hpi. Virus yield in supernatants of infected cells collected at 96 hpi was measured by plaque reduction and real-time PCR. Data represent mean values (±SD) of triplicate determinations from three independent experiments.
AS and 838 inhibit CMV protein expression at a late time during virus replication.
The effects of AS and 838 on the overall CMV protein expression were examined. AS and 838 strongly inhibited the expression of the late gene pp65, while the early polymerase accessory gene (UL44) was only moderately inhibited. The expression of IE1 protein was not significantly decreased until after 72 h, while IE2 expression was already reduced at 48 h.
To evaluate whether AS and 838 had a direct late effect on the expression of late viral proteins, as opposed to a late effect resulting from an earlier event, the compounds were added at 0, 24, and 48 hpi and Western blotting was performed at 72 hpi (Fig. 7B). The expression of late CMV proteins was not affected when compounds were added at later time points.
Fig 7.

Effects of AS, 838, and GCV on CMV gene expression. (A) CMV protein expression in AS-, 838-, and GCV-treated cells. Compounds were added after virus adsorption, and cell lysates were collected for Western blotting at 24, 36, 48, and 72 hpi. (B) Compounds were added at different time points following infection, and cell lysates were collected for Western blotting at 72 hpi. (C) HFF cells were infected with CMV and treated with the compounds. Cell lysates were collected at 6, 12, and 24 hpi. Depicted are mRNA levels of the IE genes IE1, IE2, and TRS1 as quantified by real-time RT-PCR. Data represent mean values (±SD) of triplicate determinations from two independent experiments.
Because of a predominant early inhibition of CMV, the transcription of immediate-early (IE) genes was quantified in the presence of the compounds. RNA extracts were prepared from infected and treated cells at 6, 12, and 24 hpi, and reverse transcription-PCR (RT-PCR) amplification of the CMV IE genes IE1, IE2, TRS1, US3, UL36, and UL37 was performed. Human β-actin was used as an internal control. AS and 838 did not show a significant effect on immediate-early gene transcripts at 6 and 12 hpi. 838 showed moderate inhibition of IE1 and TRS1 mRNAs at 24 hpi, while IE2 transcripts were not decreased (Fig. 7C). Similarly, US3, UL36, and UL37 transcripts were unchanged during treatment with the compounds (data not shown).
The combination of 838 and GCV is synergistic.
Since AS and 838 clearly demonstrated a mechanism of action distinct from that of GCV, we investigated whether the combination of the compounds could result in additive/synergistic activity against CMV replication. The Bliss independency model was used, based on the assumption that inhibitors could bind mutually nonexclusively through distinct mechanisms (8). The combined effect of two inhibitors is computed as the product of the individual effects of the two inhibitors. Initially, all compounds were tested at their EC50. The combination of GCV and 838 as well as of GCV and AS was highly synergistic (Tables 1 and 2).
Table 1.
Combinations of GCV and 838
| GCV |
838 |
Combination |
|||
|---|---|---|---|---|---|
| Concn (μM) | Fold inhibitiona | Concn (μM) | Fold inhibitiona | Fold inhibitiona |
|
| Theoretical | Actual | ||||
| 1 | 1.50 ± 0.06 | 0.03 | 1.62 ± 0.40 | 2.42 ± 0.24 | 3.90 ± 0.14 |
| 1 | 1.50 ± 0.06 | 0.04 | 3.01 ± 0.05 | 4.51 ± 0.10 | 10.0 ± 1.49 |
| 1 | 1.50 ± 0.06 | 0.06 | 5.06 ± 0.74 | 7.58 ± 0.59 | 13.6 ± 0.27 |
| 1.7 | 2.10 ± 0.21 | 0.03 | 1.62 ± 0.40 | 3.39 ± 0.0.51 | 7.50 ± 0.53 |
| 1.7 | 2.10 ± 0.21 | 0.04 | 3.01 ± 0.05 | 6.31 ± 0.30 | 12.5 ± 1.40 |
| 1.7 | 2.10 ± 0.21 | 0.06 | 5.06 ± 0.74 | 10.6 ± 1.23 | 24.1 ± 0.73 |
Mean values (±SD) of triplicate determinations from three independent experiments are shown.
Table 2.
Combinations of GCV and AS
| GCV |
AS |
Combination |
|||
|---|---|---|---|---|---|
| Concn (μM) | Fold inhibitiona | Concn (μM) | Fold inhibitiona | Fold inhibitiona |
|
| Theoretical | Actual | ||||
| 1 | 1.69 ± 0.04 | 2.5 | 1.23 ± 0.06 | 2.10 ± 0.06 | 6.99 ± 0.62 |
| 1 | 1.69 ± 0.04 | 5 | 1.36 ± 0.05 | 2.30 ± 0.05 | 10.0 ± 0.30 |
| 1 | 1.69 ± 0.04 | 10 | 2.27 ± 0.10 | 3.84 ± 0.13 | 16.9 ± 0.63 |
| 1.7 | 2.62 ± 0.17 | 2.5 | 1.23 ± 0.06 | 3.24 ± 0.18 | 14.4 ± 1.75 |
| 1.7 | 2.62 ± 0.17 | 5 | 1.36 ± 0.05 | 3.57 ± 0.17 | 21.2 ± 3.94 |
| 1.7 | 2.62 ± 0.17 | 10 | 2.27 ± 0.10 | 5.94 ± 0.32 | 28.8 ± 1.88 |
Mean values (±SD) of triplicate determinations from three independent experiments are shown.
DISCUSSION
We report here that CMV inhibition by artemisinin-derived monomer, AS, and dimer diphenyl phosphate (838) is specific to CMV among other representative herpesviruses, that it occurs predominantly at a stage prior to DNA replication, and that dimer diphenyl phosphate is an irreversible inhibitor of CMV replication.
Artemisinin derivatives were originally reported to inhibit CMV replication (7, 17), and we have shown that artemisinin-derived dimers are highly potent inhibitors of CMV replication compared to their monomeric counterparts (2). We now report that AS and 838 have no activities against selected alpha- and gammaherpesviruses. Although we cannot exclude their activities against other herpesviruses, it is possible that these compounds are specific inhibitors of CMV or betaherpesviruses. Thus, artemisinins bind either to a specific CMV target or to cellular ligands that are highly specific for CMV. Other anti-CMV agents, such as maribavir, have also been reported to have activities against specific members of the herpesvirus family (34). Although in our assays neither AS nor 838 inhibited HSV-1 and -2 replication or EBV lytic reactivation, others have reported moderate inhibition of HSV and EBV with AS (7, 25). The discrepancy may result from differences in methodology and materials, such as the viral strains and the assays used for evaluation of inhibition.
We demonstrate that artemisinins inhibit CMV replication at an early stage of virus replication and prior to DNA synthesis. This was clearly demonstrated by the add-on and removal and the MOI dependency assays. The early inhibition could result from direct interaction with immediate-early (IE) or early CMV proteins or from binding to cellular regulatory proteins that subsequently reduce the expression of CMV-encoded IE and early proteins.
A detailed comparison of DNA synthesis, late gene expression, and virus yield in the presence of the compounds revealed that while the inhibition of DNA replication and virus yield by GCV correlated, the inhibition of virus yield by AS and 838 was 10-fold higher than that of DNA replication. This result also supports a difference between GCV and artemisinins in their anti-CMV activities. The discrepancy between the moderate (20- to 30-fold) inhibition of DNA replication and the strong (200- to 300-fold) inhibition of virus yield suggests that inhibition of a step following DNA replication may also contribute to the potent reduction of viral progeny by AS and 838 (Fig. 6). However, the add-on assay and the Western blot (Fig. 3 and 7B) showed that when added at 48 hpi, AS and 838 did not inhibit late gene expression or virus yield. Therefore, the potential late effect of AS and 838 could reflect an early recruitment of a protein that has ancillary effects later in the virus cycle (Fig. 3; see Fig. S1 in the supplemental material).
Although AS and 838 showed similar patterns of CMV inhibition, 838 appeared to be distinct from AS not only by virtue of its highly potent EC50 and slope (10) but also by its irreversible CMV inhibition. In addition, treatment of noninfected HFF cells with 838 followed by drug removal and CMV infection revealed strong inhibition of viral DNA replication and virus yield. These data may suggest that at least one ligand of 838 could be a cellular protein (Fig. 5). Proof that AS and 838 target a specific viral protein will require demonstration that AS and 838 can select for resistant viral mutants (12). These studies are in progress. Dimer 838 may represent a highly potent “anticellular antiviral” compound possessing complex mechanisms of action involving several steps in the virus replication cycle. Despite the activity differences between AS and 838, the similarity in the timing of CMV inhibition suggests that these compounds may share the same ligand. The high slope of 838 is indicative of strong target binding affinity or a multitarget mechanism (28). Interestingly, the removal of 838 from noninfected HFF delayed cell growth for less than 1 day; thus, the cells could clearly tolerate the effects of 838 binding to its ligand, while CMV replication could not recover.
The expression pattern of immediate-early, early, and late genes revealed a strong effect on late protein expression (Fig. 7); the expression of IE1 was slightly reduced at 48 hpi, a time at which a significant reduction in IE2 was observed. Despite the very early effect on CMV replication, the transcription of IE genes was not significantly reduced at the early time points (Fig. 7C). Only moderate inhibition of IE1 and TRS1 transcripts was observed for 838 at 24 hpi. These data indicate that the effects of 838 on gene expression are time dependent and may be executed following IE gene transcription or are secondary to cellular effects.
While modestly decreasing IE1 expression, 838 strongly downregulated IE2 protein expression. IE2 is the major regulatory gene for late gene expression, as well as for DNA replication (30, 31). The strong inhibition of IE2 expression may explain the downregulation of early-late and late genes at 48 and 72 hpi.
Lastly, the combinations of GCV and 838 and of GCV and AS were highly synergistic. Similarly, the combination of AS and maribavir was reported to be synergistic (5).
We conclude that artemisinins inhibit CMV during an early stage of virus replication, that their effects are specific for CMV, and that dimer 838 has unique activities resulting in irreversible virus inhibition. These activities could be due to the dimer backbone or the diphenyl phosphate group. Future studies will dissect the cellular and viral activities of 838 compared to other artemisinin dimers with known anti-CMV activity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants 1R01AI093701 and KO8 AI074907 and March of Dimes grant 6-FY11-268 (R.A.-B.) and by NIH grant AI 34885 (G.H.P.).
We are grateful to our colleagues for providing the following viruses: Prashant Desai (Johns Hopkins) provided HSV-1 KOS, David A. Leib (Dartmouth Medical School) provided KOS/Dlux/oriS, and Diane Hayward and Renfeng Li provided EBV Akata.
Footnotes
Published ahead of print 30 April 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Alagbala AA, et al. 2006. Biological mechanisms of action of novel C-10 non-acetal trioxane dimers in prostate cancer cell lines. J. Med. Chem. 49:7836–7842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arav-Boger R, et al. 2010. Artemisinin-derived dimers have greatly improved anti-cytomegalovirus activity compared to artemisinin monomers. PLoS One 5:e10370 doi:10.1371/journal.pone.0010370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bliss CI. 1939. The toxicity of poisons applied jointly. Ann. Appl. Biol. 26:585–615 [Google Scholar]
- 4. Boeckh M, et al. 2003. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol. Blood Marrow Transplant. 9:543–558 [DOI] [PubMed] [Google Scholar]
- 5. Chou S, et al. 2011. The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antiviral Res. 92:364–368 [DOI] [PubMed] [Google Scholar]
- 6. Demmler GJ. 1991. Infectious Diseases Society of America and Centers for Disease Control. Summary of a workshop on surveillance for congenital cytomegalovirus disease. Rev. Infect. Dis. 13:315–329 [DOI] [PubMed] [Google Scholar]
- 7. Efferth T, et al. 2002. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 80:233–242 [DOI] [PubMed] [Google Scholar]
- 8. Greco WR, Bravo G, Parsons JC. 1995. The search for synergy: a critical review from a response surface perspective. Pharmacol. Rev. 47:331–385 [PubMed] [Google Scholar]
- 9. Griffiths PD, Clark DA, Emery VC. 2000. Betaherpesviruses in transplant recipients. J. Antimicrob. Chemother. 45 (Suppl. T3):29–34 [DOI] [PubMed] [Google Scholar]
- 10. He R, et al. 2011. An artemisinin-derived dimer has highly potent anti-cytomegalovirus (CMV) and anti-cancer activities. PLoS One 6:e24334 doi:10.1371/journal.pone.0024334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He R, et al. 2011. Recombinant luciferase-expressing human cytomegalovirus (CMV) for evaluation of CMV inhibitors. Virol. J. 8:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herrmann EC, Jr, Herrmann JA. 1977. A working hypothesis—virus resistance development as an indicator of specific antiviral activity. Ann. N. Y. Acad. Sci. 284:632–637 [DOI] [PubMed] [Google Scholar]
- 13. Iversen AC, et al. 2009. A proviral role for CpG in cytomegalovirus infection. J. Immunol. 182:5672–5681 [DOI] [PubMed] [Google Scholar]
- 14. Jabs DA, Martin BK, Forman MS. 2010. Mortality associated with resistant cytomegalovirus among patients with cytomegalovirus retinitis and AIDS. Ophthalmology 117:128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jacobson JG, et al. 1999. Nonnucleoside pyrrolopyrimidines with a unique mechanism of action against human cytomegalovirus. Antimicrob. Agents Chemother. 43:1888–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jilek BL, et al. 2012. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat. Med. 18:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaptein SJ, et al. 2006. The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antiviral Res. 69:60–69 [DOI] [PubMed] [Google Scholar]
- 18. Kaul DR, et al. 2011. First report of successful treatment of multidrug-resistant cytomegalovirus disease with the novel anti-CMV compound AIC246. Am. J. Transplant. 11:1079–1084 [DOI] [PubMed] [Google Scholar]
- 19. Kimberlin DW, et al. 2003. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: a randomized, controlled trial. J. Pediatr. 143:16–25 [DOI] [PubMed] [Google Scholar]
- 20. Kovacs A, et al. 1999. Cytomegalovirus infection and HIV-1 disease progression in infants born to HIV-1-infected women. Pediatric Pulmonary and Cardiovascular Complications of Vertically Transmitted HIV Infection Study Group. N. Engl. J. Med. 341:77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krosky PM, Baek MC, Coen DM. 2003. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 77:905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lewis AF, et al. 1994. Inhibition of human cytomegalovirus in culture by alkenyl guanine analogs of the thiazolo[4,5-d]pyrimidine ring system. Antimicrob. Agents Chemother. 38:2889–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li R, et al. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host. Microbe 10:390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lischka P, et al. 2010. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 54:1290–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milbradt J, Auerochs S, Korn K, Marschall M. 2009. Sensitivity of human herpesvirus 6 and other human herpesviruses to the broad-spectrum antiinfective drug artesunate. J. Clin. Virol. 46:24–28 [DOI] [PubMed] [Google Scholar]
- 26. Prichard MN, Kern ER. 2011. The search for new therapies for human cytomegalovirus infections. Virus Res. 157:212–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schreiber A, et al. 2009. Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin. Pharmacother. 10:191–209 [DOI] [PubMed] [Google Scholar]
- 28. Shen L, et al. 2008. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14:762–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steininger C. 2007. Novel therapies for cytomegalovirus disease. Recent Pat. Antiinfect. Drug Discov. 2:53–72 [DOI] [PubMed] [Google Scholar]
- 30. Stenberg RM, Kerry JA. 1995. Cytomegalovirus genes: their structure and function. Scand. J. Infect. Dis. Suppl. 99:3–6 [PubMed] [Google Scholar]
- 31. Stinski MF, Thomsen DR, Stenberg RM, Goldstein LC. 1983. Organization and expression of the immediate early genes of human cytomegalovirus. J. Virol. 46:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Summers BC, Margolis TP, Leib DA. 2001. Herpes simplex virus type 1 corneal infection results in periocular disease by zosteriform spread. J. Virol. 75:5069–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanaka Y, et al. 2002. Monitoring cytomegalovirus infection by antigenemia assay and two distinct plasma real-time PCR methods after hematopoietic stem cell transplantation. Bone Marrow Transplant. 30:315–319 [DOI] [PubMed] [Google Scholar]
- 34. Williams SL, et al. 2003. In vitro activities of benzimidazole d- and l-ribonucleosides against herpesviruses. Antimicrob. Agents Chemother. 47:2186–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Winston DJ, et al. 2008. Maribavir prophylaxis for prevention of cytomegalovirus infection in allogeneic stem cell transplant recipients: a multicenter, randomized, double-blind, placebo-controlled, dose-ranging study. Blood 111:5403–5410 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.