

Abstract
Front loading is a strategy used to optimize the pharmacodynamic profile of an antibiotic through the administration of high doses early in therapy for a short duration. Our aims were to evaluate the impact of front loading of linezolid regimens on bacterial killing and suppression of resistance and on RNAIII, the effector molecule of the accessory gene regulator system (encoded by agr) in methicillin-resistant Staphylococcus aureus (MRSA). Time-killing experiments over 48 h were utilized for linezolid against four strains of MRSA: USA100, USA300, USA400, and ATCC 29213. A hollow-fiber infection model simulated traditional and front-loaded human therapeutic regimens of linezolid versus USA300 at 106 CFU/ml over 240 h. Over 48 h in time-kill experiments, linezolid displayed bacteriostatic activity, with reductions of >1 log10 CFU/ml for all strains. Front-loaded regimens that were administered over 5 days, 1,200 mg every 12 h (q12h) (total, 10 doses) and 2,400 mg q12h (total, 10 doses) followed by 300 mg q12h thereafter, resulted in sustained bactericidal activity, with reductions of the area under the CFU curve of −6.15 and −6.03, respectively, reaching undetectable limits at the 10-day study endpoint. All regimens displayed a reduction in RNAIII relative expression at 24 h and 240 h compared with that of the growth control. Monte Carlo simulations predicted a <1.27× increase in the fractional decreases in platelets for all front-loaded regimens versus the 600 mg q12h regimen, except for the highest-dose front-loaded regimen. Front-loading strategies for linezolid are promising and may be of utility in severe MRSA infections, where early aggressive therapy is necessary.
INTRODUCTION
Staphylococcus aureus is a primary human pathogen which can cause a plethora of infections ranging from asymptomatic nasal carriage to rapidly lethal, severe necrotizing pneumonia. It has been suggested that one strain in particular, USA300, which is the predominant clone in the United States responsible for the majority of infections by community-associated methicillin-resistant S. aureus (CA-MRSA), possesses exceptional virulence characteristics (8, 15, 18). In USA300, recent investigations suggest that virulence in CA-MRSA is largely mediated by genome-encoded virulence factors, including phenol-soluble modulins and alpha-toxins, which are largely mediated by the two-component regulator, accessory gene regulator (encoded by agr), which modulates extracellular and cell wall-mediated virulence factors in S. aureus (9, 31, 32).
To combat the exceptional virulence of S. aureus, efforts have been directed, largely unsuccessfully, toward conventional vaccine development (1). Although there has been a renewed interest in identifying putative S. aureus vaccine antigens using newer approaches, such as those that directly impact alpha-toxin, the clinical application of such a strategy is many years away (17). Therefore, some experts have recommended the use of traditional antibiotics, such as protein synthesis inhibitors, which have shown the potential benefit in earlier studies of reducing expression of alpha-hemolysin, Panton-Valentine leukocidin (PVL) toxin, and toxic shock syndrome 1 (26, 27). However, current clinical guidelines do not routinely recommend the use of protein synthesis inhibitors as adjunctive therapy for the management of invasive MRSA disease. In fact, a recent study suggests that subinhibitory concentrations of antibiotics may actually increase agr expression and phenol-soluble modulin (PSM) production (7). Collectively, this suggests that the role of protein synthesis inhibitors in suppressing the toxigenic profile of MRSA infections has not been fully elucidated. Furthermore, the effect of increasing the exposure of protein synthesis inhibitors on bacterial burden and impact on virulence has not been defined.
Front loading is a strategy to optimize the pharmacodynamic profile of an antibiotic through the administration of high doses early in therapy, for a short duration, to achieve maximal bactericidal activity and suppress growth of less-susceptible populations. Linezolid is an oxazolidinone antibiotic that has been utilized in clinical practice for more than a decade at a dose of 600 mg every 12 h (q12h). It exerts its action by preventing the formation of the 70S ribosomal complex in bacteria, inhibiting protein synthesis. Linezolid doses (up to 1,250 mg/day) were well tolerated in early open-label, uncontrolled, phase II dose-finding trials, with no evidence of a dose-response relationship for adverse events in adults receiving <1 g versus 1 to 1.25 g of linezolid for 7 to 14 days (23). We hypothesized that front loading of linezolid may have potential utility to rapidly reduce bacterial burden, suppress resistance, and alter agr expression in vitro. The effect of administering linezolid in this fashion has not been examined. The objective of this study was to utilize an in vitro hollow-fiber model to evaluate the impact of front-loaded linezolid regimens on bacterial killing and resistance and on RNAIII, the effector molecule of the agr quorum-sensing system, to gain insight into linezolid's role in invasive MRSA disease.(This work was presented in part at the 48th Annual Meeting of the Infectious Diseases Society of America, Vancouver, Canada, October 2010.)
MATERIALS AND METHODS
Bacterial isolates.
The following strains of MRSA were evaluated: pulsed-field gel electrophoresis (PFGE) types of USA100, USA300, and USA400, obtained from the Network on Antimicrobial Resistance in S. aureus (NARSA), and ATCC 29213 (methicillin-susceptible S. aureus [MSSA]). MICs to linezolid for all strains were 2.0 mg/liter as determined by broth microdilution in quadruplicate according to Clinical Laboratory and Standards Institute (CLSI) standards. All 4 strains were evaluated in time-killing experiments, while USA300 was selected as a representative strain, as the most common strain in the United States, to be studied using the hollow-fiber infection model and RNAIII expression.
Antibiotic, susceptibility testing and medium.
Linezolid analytical-grade powder was obtained from PGRD, Pfizer, Groton, CT. Stock solutions were freshly prepared at the beginning of each experiment. MIC values were determined by broth microdilution in Mueller-Hinton broth (Difco Laboratories, Detroit, MI) supplemented with calcium (25 mg/liter) and magnesium (12.5 mg/liter) (SMHB) according to CLSI guidelines. Brain heart infusion (BHI) broth (Difco, Detroit, MI) and tryptic soy agar (TSA) with 5% sheep blood were used for all time-kill experiments and hollow-fiber experiments.
Time-kill experiments.
To first evaluate the full concentration-effect relationship and select human linezolid regimens to be studied in the in vitro hollow-fiber model, time-kill experiments were performed as previously described against a starting inoculum of 106 CFU/ml over 48 h (30). In brief, for bacterial inoculum preparation, fresh bacterial colonies from overnight growth were added to BHI broth standard to provide a bacterial suspension, which was diluted with BHI broth to achieve a starting inoculum of approximately 106 CFU/ml. Quantitative cultures were determined on TSA plates with 5% sheep blood. The theoretical limit of detection was 102 CFU/ml. Time-kill experiments were conducted at concentrations of 0, 0.5, 1, 2, 4, 8, 16, 32, 64, 128, and 256 mg/liter to characterize linezolid pharmacodynamics against each strain. Bactericidal activity (99.9% kill) was defined as a ≥3-log10-CFU/ml reduction in colony count from the initial inoculum. Bacteriostatic activity was defined as a <3-log10-CFU/ml reduction in colony count from the initial inoculum.
HFIM.
A hollow-fiber infection model (HFIM), adapted from the method of Louie et al. (19) was used to evaluate the effect of selected linezolid dosing regimens on the change in bacterial burden and suppression of resistance of MRSA USA300 over 240 h. A cellulosic cartridge (C3008; FiberCell Systems Inc., Frederick, MD) was utilized for all experiments. The volume of the central reservoir was 100 ml, and the extracapillary space in the cartridge was 13 ml. The determination of bacterial counts for each experiment was performed by obtaining samples at 0, 24, 48, 72, 96, 144, 192, and 240 h. Samples (0.5 ml) were diluted in 0.9% sodium chloride, and total bacterial counts were determined by plating 20-μl aliquots of each diluted sample in quintuplicate on drug-free agar in order to enumerate the total population. Plates containing the bacteria were incubated at 37°C for 24 h, after which the bacterial colonies were counted and the CFU/ml were determined. Antimicrobial carryover from the model was taken into account by centrifuging the bacterial samples for at 5,000 × g for 5 min and reconstituting the centrifuged samples to their original volumes with sterile normal saline. Using samples obtained every 24 h for the quantification of the total population, 20-μl aliquots of the diluted sample were plated in quintuplicate on BHI plates containing linezolid at 4, 8, and 16 times the MIC in order to quantify the resistant subpopulation(s). The plates were incubated at 37°C for approximately 48 h, the bacterial colonies were counted, and the CFU/ml were determined.
Simulated linezolid regimens.
The following linezolid regimens were administered using an apparent half-life of 4.8 h and a protein binding level of 31%: (i) traditional regimen, 600 mg every 12 h (maximum concentration of free, unbound fraction of drug in serum [fCmax], 10.4 mg/liter; area under the concentration-time curve from 0 to 24 h for the free, unbound fraction of drug [fAUC0-24], 124); front-loaded regimens, (ii) 1,200 mg every 12 h (total, 2 doses) on days 0 to 1 (fCmax, 20.8; fAUC0-24, 248), followed by 600 mg every 12 h (total, 18 doses) on days 1 to 10 (fCmax, 10.4; fAUC0-24, 124); (iii) 2,400 mg every 12 h (total, 2 doses) on days 0 to 1 (fCmax, 41.6; fAUC0-24, 495), followed by 600 mg every 12 h (total, 18 doses) on days 1 to 10 (fCmax, 10.4; fAUC0-24, 124); (iv) 1,200 mg every 12 h (total, 10 doses) on days 0 to 5 (fCmax, 20.8; fAUC0-24, 248), followed by 300 mg every 12 h (total, 10 doses) on days 5 to 10 (fCmax, 5.2; fAUC0-24, 62); and (v) 2,400 mg (total, 10 doses) on days 0 to 5 (fCmax, 41.6; fAUC0-24, 495), followed by 300 mg every 12 h (total, 10 doses) on days 5 to 10 (fCmax, 5.2; fAUC0-24, 62).
RNA extraction and quantitative real-time PCR.
Cells were collected at 0, 24, and 240 h from the hollow-fiber infection model for MRSA USA300 before and after exposure of the traditional and front-loaded regimens. The samples were centrifuged at 14,000 rpm for 5 min at room temperature. The supernatant was aspirated, and the pellet was immediately frozen at −80°C until RNA isolation. Total RNA was isolated from the pellet (SV total RNA isolation system; Promega, Madison WI) following the manufacturer's protocol for Gram-positive bacteria. From the total RNA pool, mRNA was purified (MICROBExpress; Ambion, Austin TX). Reverse transcription (RT) of the mRNA (315 ng) was carried out using random hexamer primers and the AccuScript high-fidelity RT-PCR system (Stratagene, La Jolla CA).
RNAIII gene expression was assessed by quantitative real-time PCR using 1.5 mM MgCl2, 0.2 mM (each) deoxynucleoside triphosphates (dNTPs), 0.5 mM (each) primers (RNA III 1, 5′-GAATTTGTTCACTGTGTCGATAATCCATTT-3′; RNA III 2, 5′-GAAGGAGTGATTTCAATGGCACAAGATAT-3′); 0.025 U Taq, 0.5 μl (each) SYBR green (1/750 dilution) and ROX (1/500 dilution), and PCR buffer. The final volume of the reaction mix was 40 μl, containing 4 μl of the target sample, which was amplified on the Stratagene MX3000P real-time PCR instrument. The thermocycle contained an initial incubation at 95°C for 8 min, followed by 40 cycles of 94°C for 30 s, 58°C for 45 s, and 72°C for 30 s. This PCR cycle was followed by a dissociation curve analysis to confirm the presence of a single PCR product. gyrB gene expression was determined for normalization of the gene expression data. The PCR for gyrB was made to a final volume of 40 μl using 3 mM MgCl2, 0.2 mM (each) dNTPs, 0.5 mM (each) primers (forward, 5′-TTAGTGTGGGAAATTGTCGATAAT-3′; reverse, 5′-AGTCTTGTGACAATGCGTTTACA-3′); 0.025 U Taq, 0.5 μl(each) SYBR green (1/750 dilution) and ROX (1/500 dilution), and PCR buffer. The thermocycle started with an initial incubation at 95°C for 4 min, followed by 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min. The PCR cycle was once again followed by a dissociation curve as described above. A PCR product of the expected length for RNAIII (53 bp) was cloned into a plasmid vector (pCR2.1-TOPO) using the Topota cloning kit (Invitrogen Corp., Carlsbad CA). The cloned PCR product was additionally used to build a standard curve of known concentrations for absolute quantification of RNAIII gene expression levels.
Pharmacokinetic and pharmacokinetic/pharmacodynamic (PK/PD) analysis.
Samples from the central reservoir from the hollow-fiber infection model were stored at −80°C until they were assayed for concentrations of linezolid determined using a validated high-performance liquid chromatography (HPLC) assay (12). In brief, samples were measured using a system consisting of a ThermoFinnegan P4000 HPLC pump (San Jose, CA) with a model AS1000 fixed-volume autosampler, a model UV2000 UV detector, a Gateway series e computer (Poway, CA), and the Chromquest HPLC data management system. The plasma standard curve for linezolid ranged from 0.5 to 30 μg/ml. The within-sample precision (percent coefficient of variation [CV%]) of validation in a single standard concentration was 0.69%, and the overall validation precision across all standards was 1.04 to 4.39%. The measured drug concentrations were within 10% of the targeted values.
For PK/PD analyses, an integrated area measure (log ratio area) was applied to all CFU data to quantify the drug effect as shown in equation 1, as previously described (30).
| (1) |
where AUC CFUdrug or AUC CFUgrowth control is the area under the CFU curve for the drug regimen or the growth control, respectively.
Using nonlinear regression, a four-parameter concentration-effect Hill-type model was fit to the effect parameter using the software program Systat (version 11; Systat Software, Richmond, VA) using equation 2, as previously described (30):
| (2) |
where E is effect, E0 is the effect measured at a drug concentration of zero, Emax is the maximal effect, fAUC/MIC is the free drug AUC-to-MIC ratio, H is the Hill or sigmoidicity constant, is and EC50 is the fAUC/MIC ratio for which there is 50% maximal effect.
For population pharmacodynamic analysis, candidate models were fit to all CFU-versus-time data simultaneously using the MC-PEM algorithm in the parallelized S-ADAPT software program (version 1.57) and the SADAPT-TRAN interface (3, 4). A log-normal likelihood was assumed for all parameter values. The candidate structural PD models included a life cycle model to describe bacterial growth, as described earlier (5). Three subpopulations, differing in susceptibilities to drug, were considered. Drug effects were modeled as inhibition of successful replication and slowing of the growth of MRSA.
Monte Carlo simulations.
A series of 10,000 subject Monte Carlo simulations (MCS) were performed in order to provide clinical context to the simulated regimens that were studied in the HFIM using Systat (version 13; Systat Software, Richmond, VA). Simulations were based on the population model from a large, previously published population pharmacokinetic analyses of 318 adult patients, all of whom received 600 mg of linezolid every 12 h (20). A protein binding level of 31% was utilized to calculate the free-drug AUC from the total AUC. The resulting fAUCs were expressed as a log normal distribution. Since the original data were “fat tailed,” in order to conduct an MCS that closely related to the original patient population, the means and variances of the population model were utilized and the upper and lower tails at the minimum and maximum as seen in the original data were truncated (20).
To evaluate the toxicodynamics of linezolid regimens, the population pharmacokinetic/toxicodynamic model from Sasaki et al. (25) was utilized to simulate the full time course of linezolid concentrations and of platelet counts following various linezolid dosage regimens using the software program Berkeley Madonna (version 8.3.18). Simulations were performed for 70-kg patients with a creatinine clearance of 80 ml/min. Patients with low creatinine clearance will need dose adjustment to achieve the same AUC of linezolid. Since the baseline platelet counts are considered in clinical practice for linezolid therapy, we expressed the simulation results as a decline of platelet counts to a fraction of the platelet baseline.
RESULTS
Impact of linezolid front-loaded regimens on the total and resistant bacterial populations.
Linezolid time-kill experiments were first conducted to characterize the pharmacodynamic profile of linezolid against four strains of S. aureus to select potential regimens to be evaluated in the HFIM. Linezolid displayed bacteriostatic activity with >1 log10 CFU/ml reduction for all strains at 48 h compared to the baseline (Fig. 1). A concentration-dependent response at lower concentrations with a plateau of effect at higher concentrations of >8 mg/liter against all strains was evident. Interestingly, bacterial reduction was the greatest against MRSA USA100, the hospital-associated MRSA PFGE subtype, approaching 3 log10 CFU/ml at 24 h at linezolid concentrations of >8 mg/liter.
Fig 1.

Time-kill experiments for linezolid versus ATCC 29213 (A), MRSA USA100 (B), MRSA USA300 (C), or MRSA USA400 (D). MICs to linezolid for all strains were 2.0 mg/liter.
Humanized traditional and front-loaded regimens of linezolid were subsequently evaluated against MRSA USA300 using a hollow-fiber infection model (Fig. 2). The traditional regimen of linezolid at 600 mg q12h demonstrated a gradual reduction in bacterial counts over the study duration. The maximal reduction in area under the CFU curve compared with results for the growth control was −4.03. There were modest improvements in bacterial killing for short-duration front-loaded regimens of 1,200 mg and 2,400 mg q12h (total, 2 doses) followed by 600 mg q12h with area-under-the-CFU-curve reductions compared with results for the growth control of −5.43 and −5.42, respectively. Interestingly, front-loaded regimens that were administered over a 5-day extended duration, 1,200 mg and 2,400 mg q12h (total, 10 doses), followed by 300 mg q12h thereafter, resulted in the greatest reduction in bacterial counts. The reductions in area under the CFU curve compared with data for the growth control were −6.15 and −6.03, respectively. Specifically, at the end of the 10-day endpoint, these extended-duration front-loaded regimens resulted in bacterial counts approaching nondetectable limits, with the regimen of 2,400 mg q12h (total, 10 doses) resulting in complete eradication of MRSA. All regimens suppressed the amplification of resistant subpopulations.
Fig 2.

Hollow-fiber infection model experiments simulating linezolid traditional regimen (A) and front-loaded regimens (B to E) versus MRSA USA300. (A) Traditional regimen: 600 mg every 12 h. (B) Front-loaded regimen: 1,200 mg every 12 h (total, 2 doses) on days 0 to 1 followed by 600 mg every 12 h (total, 18 doses) on days 1 to 10. (C) Regimen of 2,400 mg every 12 h (total, 2 doses) on days 0 to 1 followed by 600 mg every 12 h (total, 18 doses) on days 1 to 10. (D) regimen of 1,200 mg every 12 h (total, 10 doses) on days 0 to 5 followed by 300 mg every 12 h (total, 10 doses) on days 5 to 1. (E) Front-loaded regimen: 2,400 mg (total, 10 doses) on days 0 to 5 followed by 300 mg every 12 h (total, 10 doses) on days 5 to 10.
Alterations in RNAIII secondary to linezolid exposure.
Differences between front-loaded and traditional regimens were compared for the relative expression of RNAIII, the primary transcript of agr (Fig. 3). There was a temporal relationship, with progressive decreased expression of RNAIII expression at 24 h and 240 h compared with that at 0 h in the presence of linezolid regimens. Although there were significant differences between the linezolid 600-mg, front-loaded 1,200-mg q12h and front-loaded 2,400-mg q12h regimens compared with results for the growth control, there were no significant differences between regimens. Increasing linezolid exposure from 600 mg to 1,200 mg or 2,400 mg q12h did not result in an additional benefit with regard to decreases in RNAIII expression at the early and later time points which were selected for analysis.
Fig 3.
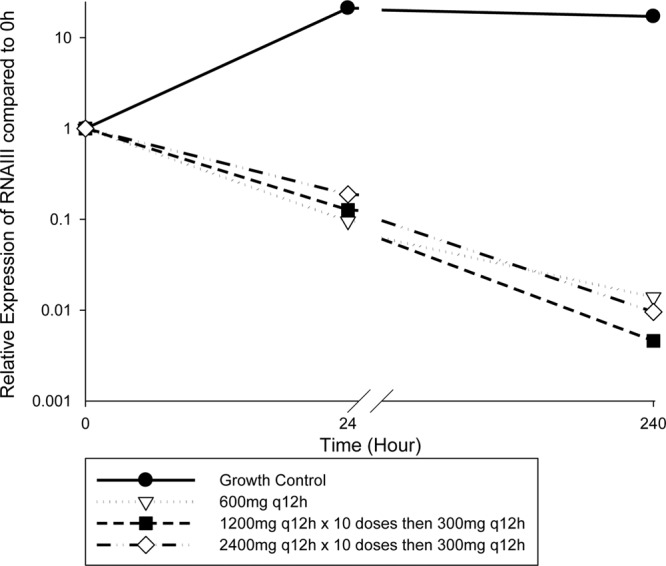
Relative expression of RNAIII, the primary transcript of the agr quorum-sensing system, in response to front-loaded and traditional dosing regimens of linezolid.
PK/PD analyses.
Model fits based on the killing curves were excellent, and the mechanism-based model was utilized to provide insight into the selection of regimens for the hollow-fiber infection model. A population-based mathematical model with two subpopulations that incorporated the drug effect by an inhibition of growth best described the CFU-versus-time profiles described in the time-kill experiment data, as shown in Fig. 4. The candidate structural PD models included a life cycle model to describe bacterial growth (13). Drug effects were modeled as inhibition of successful replication and slowing of the growth of MRSA. For front-loaded regimens from the hollow-fiber infection model, analysis of pharmacodynamics revealed acceptable model fits (r2 > 0.88) of the data to the Hill-type mathematical model, although the log ratio area dose-response relationship was not evenly distributed. Linezolid killing occurred in an exposure-dependent manner against MRSA USA300 (Fig. 5).
Fig 4.

(A) Mechanism-based based pharmacodynamic mathematical modeling. The final model contained two subpopulations with different susceptibilities. (B) Observed versus population-predicted plot for all time-kill experiments for linezolid versus ATCC 29213, MRSA USA100, USA300, and USA400.
Fig 5.

Dose-response relationship for all linezolid regimens. The y axis in panel A represents the log ratio area-under-the-curve approach as a measure of response, while panel B represents the point estimates comparing the bacterial reductions at 240 h to levels at 0 h. The x axis represents the derived mean 24-h fAUC, which was averaged over the 240 h. “R2” represents the coefficient of determination and values of model-fitted parameter estimates from equation 2.
Monte Carlo simulations.
Results from the Monte Carlo simulation are shown in Fig. 6. The 24-h fAUCs resultant from a 600-mg q12h dose from the original population pharmacokinetic study ranged from 39.2 to 601, with a median of 130. The 1,200-mg q12h (24-h fAUC of 248) and 2,400-mg q12h (24-h fAUC of 495) regimens simulated in the HFIM were toward the upper end of this range and represented the percentiles 86.7 and 99.4.
Fig 6.
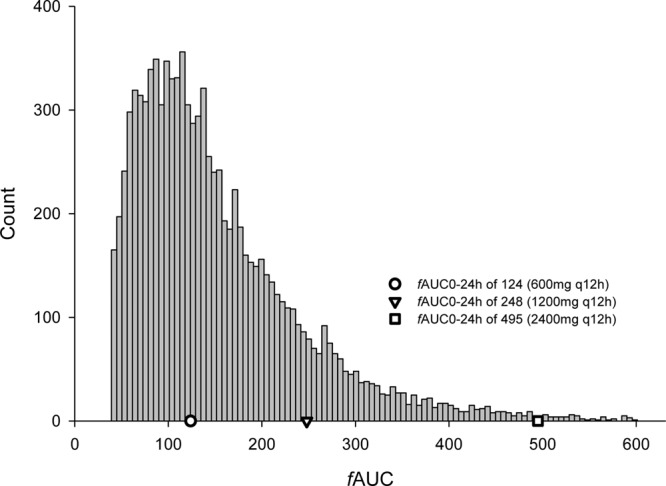
Monte Carlo simulations displaying the distribution of fAUCs based on previously published population pharmacokinetic analysis from the linezolid compassionate-use trials (20). All patients in the previously published study received 600 mg q12h. A protein binding level of 31% was utilized to calculate the 24-h free-drug AUC from the total AUC in the population PK study. The simulated doses studied in the in vitro hollow-fiber model are within this distribution and are overlaid as symbols.
For toxicodynamic analyses, the median (5th to 95th percentiles) of the fractional decline in platelet counts for each dose were as follows: 22% (2 to 57%) for 600 mg q12h for 10 days, 25% (2 to 58%) for 1,200 mg q12h for 1 day followed by 600 mg q12h for 9 days, 28% (2 to 69%) for 2,400 mg q12h for 1 day followed by 600 mg q12h for 9 days; 28% (4 to 72%) for 1,200 mg q12h for 5 days followed by 300 mg q12h for 5 days; and 49% (4 to 92%) for 2,400 mg q12h for 5 days followed by 300 mg q12h for 5 days.
DISCUSSION
MRSA continues to be a highly adaptable pathogen capable of exceptional virulence. The treatment of MRSA also continues to pose challenges for clinicians, with reports of failure and heterogeneous resistance patterns with first-line agents such as vancomycin (29). In particular, the rapid dissemination of USA300 in the community and hospital settings highlights the need to evaluate alternative strategies that combat highly virulent strains of MRSA (6, 28). Simulating human concentration-time profiles of linezolid of increasing dose intensities in the in vitro hollow-fiber system, we determined that the killing profiles of short-duration, 1-day, front-loaded linezolid regimens (1,200 mg or 2,400 mg q12h on the first day) followed by 600 mg q12h demonstrated killing profiles similar to those of the traditional regimens. There were no differences in resistance, since both short-duration, front-loaded regimens and a traditional regimen completely suppressed resistant subpopulations over the 10-day study. However, when front-loaded regimens were administered for an extended duration over 5 days (1,200 mg or 2,400 mg q12h for 5 days), followed by a regimen of decreased intensity (300 mg q12h), these regimens resulted in bactericidal activity by 96 h. Remarkably, the highest dose studied (2,400 mg q12h over 5 days) resulted in complete eradication of MRSA, although linezolid is largely considered to be a bacteriostatic agent. The 5-day front-loaded regimens resulted in greater killing compared to the 1-day front-loaded regimens over the first 24 h, although the exposure profiles were identical during that period, which may be due to the slightly lower inoculum (approximately 0.4 log10 CFU/ml lower) in the 5-day regimens and random variability. Overall, these findings suggest that there is a potential benefit of increasing doses of linezolid over an extended duration from a pharmacodynamic standpoint of killing, although no differences were evident from a resistance perspective.
Linezolid has shown significant pharmacokinetic variability as it relates to the exposure profiles that are resultant from a 600-mg q12h dose. From a population pharmacokinetic analysis with 318 adult patients treated in linezolid compassionate-use trials (20), who all received a dose of 600 mg of linezolid every 12 h, the derived 24-h AUC ranged between 57 and 871. In this study, it was clear that linezolid exhibited Michaelis-Menten or parallel first-order plus Michaelis-Menten pharmacokinetics. Although the simulated regimens utilized in the HFIM were indeed within the range of fAUC profiles obtained with a-600 mg dose in the population pharmacokinetic study (the 2,400-mg q12h dose was at the upper end at percentile 99.3), they did not account for linezolid's Michaelis-Menten pharmacokinetic profile, since linear increases in fAUC were utilized. In particular, in patients with reduced renal function, increased linezolid concentrations in plasma caused saturation of the Michaelis-Menten pathway and a further decrease in the nonrenal clearance (20). Therefore, if these doses are considered clinically, the resulting exposure profiles in humans after administration of high-dose linezolid (2,400 mg q12h) may result in fAUCs that are in fact higher than those that were simulated in the in vitro model. As long as such high AUCs are achieved only for a short duration, the expected platelet toxicity was predicted to be tolerable in patients with normal baseline platelet counts, since the model by Sasaki et al. (25) assumed that linezolid inhibits the production of platelets and does not directly kill platelets. Furthermore, the highest dose of linezolid that has been studied in humans was 750 mg q8h for 5 days in 2 healthy volunteers. Regimens including 625 mg every 8 h and 750 mg q8h described above were discontinued from future study since they resulted in aspartate transaminase (AST) and alanine aminotransferase (ALT) increases and in decreases in red blood cells (Pfizer data on file). Therefore, these results should be taken in context as it relates to the lack of safety data that exist at these high exposures.
Linezolid is indicated for infections for up to a 2-week course of therapy due to S. aureus, including nosocomial and community-acquired pneumonia and skin structure infections, both complicated and uncomplicated. It has been documented that the thrombocytopenia due to linezolid is dependent on a duration of therapy greater than 2 weeks: other hematologic toxicities have also been documented, including anemia and neutropenia, which appear to be duration dependent and reversible (10, 11, 23). Therefore, strategies that have been directed toward decreasing cumulative exposure while not compromising efficacy may hold promise, since AUC is the driver of toxicity. For example, reduction of linezolid from 600 mg q12h to 300 mg improved the occurrence of thrombocytopenia and neutropenia (2). In our in vitro studies, early high-dose therapy up to 5 days (up to 2,400 mg q12h), followed by reduced doses (300 mg q12h), was beneficial in reducing bacterial counts, achieving bactericidal activity and complete eradication at the 10-day endpoint.
The Monte Carlo simulations based on the model by Sasaki et al. (25) highlighted the time and concentration dependence of platelet-related toxicity of linezolid. All front-loaded regimens (except for the highest-dose regimen) resulted in slightly higher predicted toxicity than the traditional regimen of 600 mg q12h: fractional decreases in platelets for front-loaded regimens were up to a median of 28%, versus 22% for the traditional regimen. In particular, the highest studied doses, of 2,400 mg linezolid q12h for 10 doses followed by 300 mg linezolid q12h for 5 days, resulted in a 49% predicted fractional decline of platelets. The model by Sasaki et al. (25) assumes that linezolid inhibits platelet synthesis and that maturation of platelets takes several days. Therefore, regimens with only a short duration of high-dose linezolid therapy (i.e., 2,400 mg q12h administered for 1 day) were predicted to result in smaller increases in platelet suppression, since synthesis is inhibited for only a short time. Overall, this suggested that front-loaded dosage regimens given for a short duration may be feasible in patients with a normal baseline platelet count and normal renal function. Alternatively, high-dose front-loaded regimens administered for a longer duration (i.e., 2,400 mg q12h administered for 5 days) resulted in a predicted platelet toxicity dramatically higher (median decline of platelets, 49%) than that with traditional regimens (median decline, 22%). Therefore, although these higher-dose regimens are microbiologically promising, resulting in dramatic bactericidal activity, they should not be considered in clinical practice.
The use of protein synthesis inhibitors, such as linezolid, has been shown to suppress staphylococcal toxins (27). Although linezolid is not recommended by clinical guidelines as adjunctive therapy for invasive MRSA disease, some experts consider the administration of linezolid or clindamycin in severe disease, such as necrotizing pneumonia (16). Linezolid acts on the elongation complex of the 30S and 50S ribosomal subunits, which inhibits the growth of MRSA. The agr locus has been the central regulator of key virulence determinants in community-associated MRSA strain USA300 (9, 21, 24). Expression of phenol-soluble modulins (PSMα and PSMβ) and alpha-hemolysin are under direct control of the agr quorum-sensing system (22). Therefore, to evaluate the impact of linezolid on virulence, we measured transcript levels of RNAIII, the effector molecule of the agr system. Earlier reports by Stevens et al. (27) and Herbert et al. (14) determined that subinhibitory concentrations of protein synthesis inhibitors, such as clindamycin, inhibit alpha-toxin, PVL, and toxic shock syndrome toxin (TSST). These data were contradicted by the work of Joo et al. (15a), who determined that subinhibitory concentrations of clindamycin significantly increased the expression of agr and PSMs in USA300. To our knowledge, the results of the current study are the first to demonstrate that clinically achievable concentrations of linezolid have a significant impact on the suppression of agr activity. This represents an important finding, since they support the role of linezolid in invasive severe toxin-mediated disease due to MRSA, although no differences among increasing exposure profiles were determined.
The present study had potential limitations. First, although we utilized three PFGE subtypes of MRSA to capture the most common U.S. types, additional geographically diverse clinical isolates are needed in future studies. Although USA300 is the single most predominant clone in the United States, the benefits of linezolid front loading on agr expression and bacterial killing should not be generalized to other strains. Second, simulated doses in the hollow-fiber model were based on linear increases in fAUC and fCmax, which may underestimate the exposure profile in humans due to linezolid's Michaelis-Menten pharmacokinetics. However, this is unlikely to affect the bacterial killing profiles at the reported fAUC for linezolid. Third, studies using animal models, such as pneumonia, are needed to provide additional data to assess the impact of front loading on bacterial killing and agr in vivo, since numerous host defense mechanisms and the interaction with the host were not taken into account in our in vitro hollow-fiber system. Finally, there have been no human studies evaluating linezolid at such high exposure levels, since such caution is warranted. While the present study presents promising results for the bactericidal activity of front-loaded linezolid regimens, further in vivo studies are necessary to strengthen the translation of these in vitro findings to humans before these results are applied to clinical practice to guide selection of optimal antibiotic therapy against MRSA.
ACKNOWLEDGMENTS
We are grateful to Arnold Louie and George Drusano for providing insight into the hollow-fiber infection model system.
This work was supported by investigator-initiated research grants from Pfizer Global Research and Development and the ASPIRE Research Award, Pfizer.
Footnotes
Published ahead of print 23 April 2012
REFERENCES
- 1. Banerjee R, Gretes M, Basuino L, Strynadka N, Chambers HF. 2008. In vitro selection and characterization of ceftobiprole-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 52:2089–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bishop E, Melvani S, Howden BP, Charles PG, Grayson ML. 2006. Good clinical outcomes but high rates of adverse reactions during linezolid therapy for serious infections: a proposed protocol for monitoring therapy in complex patients. Antimicrob. Agents Chemother. 50:1599–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bulitta JB, Bingolbali A, Shin BS, Landersdorfer CB. 2011. Development of a new pre- and post-processing tool (SADAPT-TRAN) for nonlinear mixed-effects modeling in S-ADAPT. AAPS J. 13:201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bulitta JB, Landersdorfer CB. 2011. Performance and robustness of the Monte Carlo importance sampling algorithm using parallelized S-ADAPT for basic and complex mechanistic models. AAPS J. 13:212–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bulitta JB, et al. 2009. Development and qualification of a pharmacodynamic model for the pronounced inoculum effect of ceftazidime against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53:46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chambers HF. 2001. Methicillin-resistant Staphylococcus aureus. Mechanisms of resistance and implications for treatment. Postgrad. Med. 109:43–50 [DOI] [PubMed] [Google Scholar]
- 7. Deleo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diep BA, et al. 2010. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. U. S. A. 107:5587–5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diep BA, et al. 2008. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 197:1523–1530 [DOI] [PubMed] [Google Scholar]
- 10. French G. 2003. Safety and tolerability of linezolid. J. Antimicrob. Chemother. 51(Suppl 2):ii45–ii53 [DOI] [PubMed] [Google Scholar]
- 11. Green SL, Maddox JC, Huttenbach ED. 2001. Linezolid and reversible myelosuppression. JAMA 285:1291. [DOI] [PubMed] [Google Scholar]
- 12. Gunderson BW, Ibrahim KH, Peloquin CA, Hovde LB, Rotschafer JC. 2003. Comparison of linezolid activities under aerobic and anaerobic conditions against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecium. Antimicrob. Agents Chemother. 47:398–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harigaya Y, et al. 2009. Pharmacodynamics of vancomycin at simulated epithelial lining fluid concentrations against methicillin-resistant Staphylococcus aureus (MRSA): implications for dosing in MRSA pneumonia. Antimicrob. Agents Chemother. 53:3894–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herbert S, Barry P, Novick RP. 2001. Subinhibitory clindamycin differentially inhibits transcription of exoprotein genes in Staphylococcus aureus. Infect. Immun. 69:2996–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huggan PJ, et al. 2010. Population-based epidemiology of Staphylococcus aureus bloodstream infection in Canterbury, New Zealand. Intern. Med. J. 40:117–125 [DOI] [PubMed] [Google Scholar]
- 15a. Joo HS, Chan JL, Cheung GYC, Otto M. 2010. Subinhibitory concentrations of protein synthesis-inhibiting antibiotics promote increased expression of the agr virulence regulator and production of phenol-soluble modulin cytolysins in community-associated methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 54:4942–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Katayama Y, Zhang HZ, Chambers HF. 2003. Effect of disruption of Staphylococcus aureus PBP4 gene on resistance to beta-lactam antibiotics. Microb. Drug Resist. 9:329–336 [DOI] [PubMed] [Google Scholar]
- 17. Kobayashi SD, DeLeo FR. 2011. A MRSA-terious enemy among us: boosting MRSA vaccines. Nat. Med. 17:168–169 [DOI] [PubMed] [Google Scholar]
- 18. Liu C, et al. 2008. A population-based study of the incidence and molecular epidemiology of methicillin-resistant Staphylococcus aureus disease in San Francisco, 2004–2005. Clin. Infect. Dis. 46:1637–1646 [DOI] [PubMed] [Google Scholar]
- 19. Louie A, et al. 2010. The combination of meropenem and levofloxacin is synergistic with respect to both Pseudomonas aeruginosa kill rate and resistance suppression. Antimicrob. Agents Chemother. 54:2646–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meagher AK, Forrest A, Rayner CR, Birmingham MC, Schentag JJ. 2003. Population pharmacokinetics of linezolid in patients treated in a compassionate-use program. Antimicrob. Agents Chemother. 47:548–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48:1429–1449 [DOI] [PubMed] [Google Scholar]
- 22. Peddie BA, Lever M, Randall K, Chambers ST. 1999. Osmoprotective activity, urea protection, and accumulation of hydrophilic betaines in Escherichia coli and Staphylococcus aureus. Antonie Van Leeuwenhoek 75:183–189 [DOI] [PubMed] [Google Scholar]
- 23. Rubinstein E, et al. 2003. Worldwide assessment of linezolid's clinical safety and tolerability: comparator-controlled phase III studies. Antimicrob. Agents Chemother. 47:1824–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakoulas G, et al. 2002. Accessory gene regulator (agr) locus in geographically diverse Staphylococcus aureus isolates with reduced susceptibility to vancomycin. Antimicrob. Agents Chemother. 46:1492–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sasaki T, et al. 2011. Population pharmacokinetic and pharmacodynamic analysis of linezolid and a hematologic side effect, thrombocytopenia, in Japanese patients. Antimicrob. Agents Chemother. 55:1867–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schlievert PM, Kelly JA. 1984. Clindamycin-induced suppression of toxic-shock syndrome-associated exotoxin production. J. Infect. Dis. 149:471. [DOI] [PubMed] [Google Scholar]
- 27. Stevens DL, et al. 2007. Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 195:202–211 [DOI] [PubMed] [Google Scholar]
- 28. Tattevin P, Basuino L, Bauer D, Diep BA, Chambers HF. 2010. Ceftobiprole is superior to vancomycin, daptomycin, and linezolid for treatment of experimental endocarditis in rabbits caused by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 54:610–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsuji BT, Harigaya Y, Lesse AJ, Sakoulas G, Mylotte JM. 2009. Loss of vancomycin bactericidal activity against accessory gene regulator (agr) dysfunctional Staphylococcus aureus under conditions of high bacterial density. Diagn. Microbiol. Infect. Dis. 64:220–224 [DOI] [PubMed] [Google Scholar]
- 30. Tsuji BT, von Eiff C, Kelchlin PA, Forrest A, Smith PF. 2008. Attenuated vancomycin bactericidal activity against Staphylococcus aureus hemB mutants expressing the small-colony-variant phenotype. Antimicrob. Agents Chemother. 52:1533–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Villaruz AE, et al. 2009. A point mutation in the agr locus rather than expression of the Panton-Valentine leukocidin caused previously reported phenotypes in Staphylococcus aureus pneumonia and gene regulation. J. Infect. Dis. 200:724–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wright JS, III, Jin R, Novick RP. 2005. Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc. Natl. Acad. Sci. U. S. A. 102:1691–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]