

Abstract
Sontochin was the original chloroquine replacement drug, arising from research by Hans Andersag 2 years after chloroquine (known as “resochin” at the time) had been shelved due to the mistaken perception that it was too toxic for human use. We were surprised to find that sontochin, i.e., 3-methyl-chloroquine, retains significant activity against chloroquine-resistant strains of Plasmodium falciparum in vitro. We prepared derivatives of sontochin, “pharmachins,” with alkyl or aryl substituents at the 3 position and with alterations to the 4-position side chain to enhance activity against drug-resistant strains. Modified with an aryl substituent in the 3 position of the 7-chloro-quinoline ring, Pharmachin 203 (PH-203) exhibits low-nanomolar 50% inhibitory concentrations (IC50s) against drug-sensitive and multidrug-resistant strains and in vivo efficacy against patent infections of Plasmodium yoelii in mice that is superior to chloroquine. Our findings suggest that novel 3-position aryl pharmachin derivatives have the potential for use in treating drug resistant malaria.
INTRODUCTION
With an annual toll of 1 to 2 million people killed, malaria remains one of the most significant threats to human life in the tropics, and for every victim, there are several hundred more individuals who are severely stricken by the disease. For some of these survivors there can be significant long-term consequences, including developmental and neurological impairment (11). The impact of malaria is particularly devastating in sub-Saharan Africa, where its victims are primarily young children and pregnant women. This situation is worsened by the spread of Plasmodium parasites that are resistant to multiple drugs (22). The list of drugs that are losing potency against malaria includes the quinolines (chloroquine, quinine, and mefloquine), the antifolates (pyrimethamine and sulfadoxine), and the anti-respiratory combination of atovaquone and proguanil. In many parts of the world treatment of multidrug-resistant malaria relies on the endoperoxide artesunate, leaving a razor-thin wall of protection against a total collapse of malaria chemotherapy. One of the greatest challenges in global health today is the development of a safe and affordable replacement for chloroquine, the best and least expensive antimalarial ever developed.
Hans Andersag worked for Bayer IG Farbenindustrie in Elberfeld, Germany, where he discovered chloroquine (resochin) in 1934 (3). Other scientists associated with Bayer's drug development program evaluated the new compound's safety in animals and humans and considered it too toxic for clinical advancement (6). A few years later Andersag succeeded in developing a chloroquine derivative, sontochin, with an acceptable safety profile (2). As a result, this compound was advanced to clinical trials in a collaborative arrangement with the French firm Specia in North Africa in the early 1940s (Fig. 1). After Allied forces arrived in Tunis in May 1943, sontochin, along with reports of its efficacious use in humans, fell into the hands of Americans, who sent the material back to the United States for analysis (6). This analysis revealed sontochin's structural details and stimulated interest in the 4-aminoquinoline class of antimalarials, leading to the rediscovery of chloroquine. Clinician scientists soon recognized chloroquine's superior antimalarial properties, rapid parasite clearance, lack of skin discoloration (relative to quinacrine, an antimalarial in use at the time), and extended duration of protection (4, 5, 9). Sontochin, the original chloroquine replacement drug, was laid aside.
Fig 1.

Chemical structures of chloroquine, sontochin, and two short-chain analogs that are active against chloroquine-resistant strains of Plasmodium falciparum.
Chloroquine's antimalarial mode of action is through binding of free heme and inhibition of the formation of hemozoin, a nontoxic repository of heme dimers that accumulate as hemoglobin is progressively cleaved to salvageable peptides and amino acids (21). This process occurs in a specialized acidic digestive vacuole (18). Resistance to chloroquine is linked to the acquisition of point mutations in the gene that encodes the Plasmodium falciparum chloroquine resistance transporter, PfCRT, which are associated with diminished accumulation of chloroquine in the digestive vacuoles of resistant parasites (12, 15, 25, 28). Over the past 50 years, scientists have explored the chemical space around the 4-aminoquinoline nucleus for clues to overcome PfCRT-mediated resistance. Investigators have discovered that short-chain chloroquine analogs retain activity against chloroquine-resistant parasites (7, 24), and more recent studies suggest that greater metabolic stability is gained by replacing the terminal diethylamine group with a single tert-butylamine in these short-chain variants (19, 20, 27). Examples of 4-aminoquinolines with these structural motifs (AQ-13 and F2Bu) in comparison to chloroquine and sontochin are shown in Fig. 1.
We have discovered that the long-forgotten sontochin (also known as 3-methyl-chloroquine and SN-6911) retains significant activity against multidrug-resistant strains of Plasmodium falciparum. This is consistent with earlier reports (13, 14) that drew attention to the fact that certain 4-aminoquinolines, including sontochin, exhibited equipotency against chloroquine-resistant parasites. For this report we prepared structural analogs of sontochin (“pharmachins”) with shorter side chains at the 4-amino position, incorporating additional design optimization features to enhance intrinsic potency against drug-resistant strains and in vivo efficacy against murine malaria. One design imperative was to work toward optimization of the structural feature at the 3 position of the quinoline core, since it appears to be critical in circumventing chloroquine resistance.
MATERIALS AND METHODS
Chemical synthesis and analytical procedures.
Unless otherwise stated, all chemicals and reagents were from Sigma-Aldrich Chemical Company, St. Louis, MO, and were used as received. N-tert-butyl-propane-1,3-diamine was synthesized by the method of Tarbell et al. (29). 4,7-Dichloro-3-methylquinoline was synthesized according as described by Andersag (2). Melting points were obtained with an Optimelt automated melting point system from Stanford Research Systems, Sunnyvale, CA. Gas chromatography-mass spectrometry (GC-MS) profiles were obtained using a Hewlett-Packard HP5890 series II gas chromatograph (30-m DB5 column set at 150°C, 2 min, then increasing at 11°C/min to 280°C) with an HP5970 mass-selective ion detector operating at 70 eV. 1H nuclear magnetic resonance (NMR) spectra were obtained using a Bruker AMX-400 NMR spectrometer operating at 400.14 MHz in CDCl3 unless otherwise stated. The NMR data were processed using DataChord Spectrum Analyst software from One Moon Scientific, Inc., Westfield, NJ. Chemical shifts were recorded in parts million units (d) relative to tetramethylsilane (TMS) as the internal standard. J coupling constants values are in hertz. High-resolution mass spectra were acquired with a Thermo LTQ-Orbitrap Discovery hybrid mass spectrometer (San Jose, CA) equipped with an electrospray ionization source operating in the positive or negative mode. The Orbitrap was externally calibrated prior to data acquisition, allowing accurate mass measurements for (M + H)+ or (M − H)− ions to be obtained to within 4 ppm. In this paper we include the details for synthesis of pharmachin 128 (PH-128) as representative of the approaches used to prepare each of the pharmachins in this study.
Synthesis of PH-128.
The synthesis of pharmachin 128 (PH-128) [N-tert-butyl-N′-(7-chloro-3-methyl-quinolin-4-yl)-propane-1,3-diamine] was adapted from the procedure used by Andersag in 1948 (2) for synthesis of sontochin. The mixture of 4,7-dichloro-3-methylquinoline (2.12 g, 10.0 mmol), N-tert-butyl-propane-1,3-diamine (1.95 g, 15.0 mmol), and phenol (2.82 g, 30.0 mmol) was stirred in a 120°C oil bath for 24 h (Fig. 2). After cooling to room temperature, the resulting viscous brown oil was dissolved in methylene chloride (200 ml) and 10% sodium hydroxide (20 ml). The organic layer was separated from the aqueous layer and then washed with 10% sodium hydroxide (twice, 20 ml) and brine (three times, 20 ml), dried over Na2SO4, filtered, and rotary evaporated to afford 3.00 g of a brown solid. This material was crystallized from hexane-ethyl acetate to give 2.18 g of white crystals. A second crop from the mother liquor give an additional 210 mg (78% combined yield), mp 114 to 116°C. GC-MS: 305 M+, 25%; 205 (M− C6H14N), 100%. 1H NMR (400 MHz, CDCl3): δ 8.39 (s; 1H); 8.01 (d; J = 9.08 Hz; 1H); 7.91 (d; J = 2.22 Hz; 1H); 7.29 (d of d; J = 9.08, J = 2.22; 1H); 6.14 (broad s; 1H); 3.72 (q; J = 5.57 Hz; 2H); 2.82 (t; J = 5.77 Hz; 2H); 2.38 (s; 3H); 1.80 (quintuplet; J = 5.89 Hz; 2H); 1.15 (s; 9H). High-resolution mass spectrum calculated for (M + H)+ = 306.1732, observed for (M + H)+ = 306.1738, calculated for (M − H)− = 304.1575, observed for (M − H)− = 304.1571. Detailed methods for synthesis of other molecules profiled in this report, which are too lengthy for inclusion here, will be published elsewhere.
Fig 2.

PH-128 was obtained by the procedure applied by Hans Andersag for the synthesis of sontochin.
Analytical data for PH-203.
A detailed procedure for the synthesis of PH-203 [N1-(tert-butyl)-N3-(7-chloro-3-(4-(trifluoromethoxy)phenyl)quinolin-4-yl)propane-1,3-diamine] will be published separately. The following are the analytical data used to characterize and confirm the structure of PH-203 in the final product that was used for experimentation here. The initial product following flash chromatography presented as a white crystalline solid upon evaporation. This material was dissolved in ethyl acetate, and the solvent was allowed to slowly evaporate to give transparent white rectangular plates. GC-MS reveals a single peak with 451 M+, 28%; 351 (M-C6H14N), 100%. 1H NMR: 8.37 (1H, s), 8.07 (1H, d, J = 9.05 Hz), 7.96 (1H, d, J = 2.19 Hz), 7.45 to 7.44 (2H, m), 7.37 (1H, dd, J = 9.00, 2.22 Hz), 2.95 (2H, q, J = 5.35 Hz), 2.73 (2H, t, J = 5.35 Hz), 1.60 (2H, q, J = 3.22 Hz) 1.14 (9H, s).
Biochemicals and assay procedures.
SYBR green I dye and Albumax II were purchased from Invitrogen, Carlsbad, CA. RPMI 1640, gentamicin, and hypoxanthine were purchased from Sigma-Aldrich, St. Louis, MO. Blood, a source for red cells used to culture the parasite, was purchased from Lampire Biologicals, Pipersville, PA. White blood cells were removed by centrifugation, followed by removal of the buffy coat and uppermost red blood cells.
Parasites.
Plasmodium falciparum strains D6 (Sierra Leone), Dd2 (Indochina/Laos), and 7G8 (Brazil) were obtained from the MR4 Malaria Reagent Repository (ATCC, Manassas, VA). D6 is sensitive to chloroquine but moderately resistant to mefloquine (17). 7G8 is resistant to chloroquine, pyrimethamine, and cycloguanil. The Dd2 strain is resistant to the quinolines chloroquine and mefloquine and the folate antagonist pyrimethamine.
Parasite culture and drug sensitivity.
Three different laboratory strains of P. falciparum were cultured in human erythrocytes by standard methods under a low-oxygen atmosphere (5% O2, 5% CO2, 90% N2) in an environmental chamber (31). The culture medium was RPMI 1640 supplemented with 25 mM HEPES buffer, 25 mg/liter gentamicin sulfate, 45 mg/liter hypoxanthine, 10 mM glucose, 2 mM glutamine, and 0.5% Albumax II (complete medium). The parasites were maintained in fresh human erythrocytes suspended at a 2% hematocrit in complete medium at 37°C. Stock cultures were subpassaged every 3 to 4 days by transfer of infected red cells to a flask containing complete medium and uninfected erythrocytes.
The in vitro antimalarial activities of the pharmachin derivatives were assessed by the SYBR green I fluorescence-based method (the “MSF assay”) described previously by us (26) with minor modifications (32). Briefly, experiments were set up in triplicate in 96-well plates (Costar; Corning) with 2-fold dilutions of each drug across the plate in a total volume of 100 μl and at a final red blood cell concentration of 2% (vol/vol). The dilution series was initiated at a concentration of 1 μM, and the experiment was repeated beginning with a lower initial concentration for those compounds for which the 50% inhibitory concentration (IC50) was below 10 nM. Automated pipetting and dilution was carried out with the aid of a programmable Precision 2000 robotic station (BioTek, Winooski, VT). An initial parasitemia of 0.2% was attained by addition of normal uninfected red cells to a stock culture of asynchronous parasite-infected red cells (PRBC). The plates were incubated for 72 h at 37°C in an atmosphere of 5% CO2, 5% O2, and 90% N2. After this period, the SYBR green I dye-detergent mixture (100 μl) was added and the plates were incubated at room temperature for an hour in the dark and then placed in a 96-well fluorescence plate reader (Spectramax Gemini-EM; Molecular Diagnostics) for analysis, with excitation and emission wavelength bands centered at 497 and 520 nm, respectively. The fluorescence readings were plotted against the logarithm of the drug concentration, and curve fitting by nonlinear regression analysis (GraphPad Prism software) yielded the drug concentration that produced 50% of the observed decline relative to the maximum readings in drug-free control wells (IC50).
In vitro cytotoxicity (T-cell lymphocyte proliferation assay).
We assessed the cytotoxicities of selected quinoline derivatives in vitro against murine splenic lymphocytes (MSL) induced to proliferate and differentiate following concanavalin A stimulation. Briefly, 2 × 105 splenic cells were incubated at 37°C in 200 μl of RPMI 1640 supplemented with 5% heat-inactivated fetal calf serum, antibiotics (penicillin and streptomycin), and the test drug (in concentrations ranging from 0 to 25 μM). Concanavalin A was added at 10 μg/ml. Two-fold drug dilutions were made starting at 25 μM. After 72 h of incubation, 20 μl of PrestoBlue cell viability reagent (Invitrogen) was added, and plates were incubated for another 60 min. This reagent consists of a resazurin-based dye that is reduced by cellular dehydrogenases to a fluorescent product (excitation, 560 nm; emission, 590 nM). Cytotoxic IC50s were calculated from the dose-response curves using Prism 5 software and reflect the concentration of drug required to inhibit lymphocyte proliferation by 50% relative to drug-free controls. A primary goal of the drug testing studies was the determination of IC50s for drug activity (versus parasite-infected red blood cells) and drug toxicity (versus splenic lymphocytes). The ratio of the 2 IC50s, i.e., IC50 (versus lymphocytes)/IC50 (versus PRBC), was used to generate the in vitro selectivity index (SI) for selected pharmachins.
In vivo efficacy against murine malaria.
The in vivo activities of selected pharmachin derivatives were assessed against the blood stages using a modified 4-day test (1). Mice (female, CF1; Charles River Labs) were infected intravenously with 2.5 to 5.0 ×105 Plasmodium yoelii (Kenya strain, MR4 MRA-428)-parasitized erythrocytes from a donor animal. Drug administration commenced the day after the animals were inoculated (day 1). The test compounds were converted to corresponding hydrochloride salt form, dissolved in water, and administered by oral gavage once daily for 4 successive days; the hydrochloride salt of chloroquine was used as a positive control. On the fifth day, blood films were prepared and the extent of parasitemia was determined by microscopic examination of Giemsa-stained smears. Fifty percent effective dose (ED50) and ED90 values (mg/kg/day) were derived graphically from the dose required to reduce the parasite burden by 50% and 90%, respectively, relative to drug-free controls. Animals remaining parasite free for 30 days after the last drug dose were considered cured of the infection. As described, the malaria infection in this model system is rapidly fulminate, producing average parasitemias of ≈30% in untreated control animals by day 5. The procedures involved, together with all matters relating to the care, handling, and housing of the animals used in this study, were approved by the Portland VA Medical Center Institutional Animal Care and Use Committee.
Metabolic stabilities of pharmachin 128 and pharmachin 203.
The metabolic stabilities of chloroquine, sontochin, PH-128, and PH-203 were evaluated by Apredica Inc., Watertown, MA. Test compounds were incubated at 37°C with pooled human liver microsomes. The incubation mix consisted of test compounds (1 μM), 2.5 mM NADPH, and 3 mM MgCl2 in 100 mM phosphate buffer (pH 7.4) and pooled human microsomal protein (0.3 mg/ml). All incubations were performed in the presence and absence of NADPH in order to detect NADPH-independent degradation. Reactions were stopped at the end of incubation by the addition of an equal volume of ice-cold stop solution (0.3% acetic acid in acetonitrile containing an internal standard for quantitation purposes). The rate of loss of the parent compound was determined at 20-min intervals up to 3 h by liquid chromatography-tandem MS (LC-MS/MS) analysis. The amount of compound in the samples was expressed as a percentage of remaining compound compared to that at time zero (100%). The in vitro degradation half life was calculated from the degradation rate constant (k) obtained from the initial slope of the exponential decay of the loss of substrate using the relationship t1/2 = ln2/k at 1 μM. Warfarin and verapamil were included alongside the test samples as reference controls for high and low metabolic stabilities, respectively.
RESULTS AND DISCUSSION
Antiplasmodial activities.
Table 1 shows the IC50s of several standard antimalarial agents in comparison to sontochin and a number of 3-position-substituted 4-aminoquinoline derivatives, which we refer to as pharmachins. As shown, the D6 strain of P. falciparum is sensitive to the antiplasmodial action of chloroquine, but strains Dd2 and 7G8 are resistant to this drug. It is evident from the results that sontochin, which differs from chloroquine only in the 3-position methyl group, retains significant activity against the drug-resistant strains of P. falciparum, with IC50s ranging from 8 to 20 nM. AQ-13, a short-chain analog of chloroquine under clinical development, also shows a high level of antiplasmodial activity against all tested drug-resistant strains. However, reports suggest that AQ-13 is rapidly metabolized in vivo (16, 24). The pharmachin analogs containing both the 3-methyl substituent and the short chain extending from the 4-amino position show a range of inhibitory activities, with PH-128 appearing as a superior structural lead, with IC50s ranging from 5.1 to 11.3 nM against all 3 P. falciparum strains. Note that replacement of the 3-methyl group with an ethyl substituent is accompanied by a significant loss of activity (i.e., compare PH-137 and PH-128).
Table 1.
In vitro activities of pharmachin analogs in comparison to known antimalarials against drug-sensitive (D6) and chloroquine-resistant (Dd2 and 7G8) strains of Plasmodium falciparum
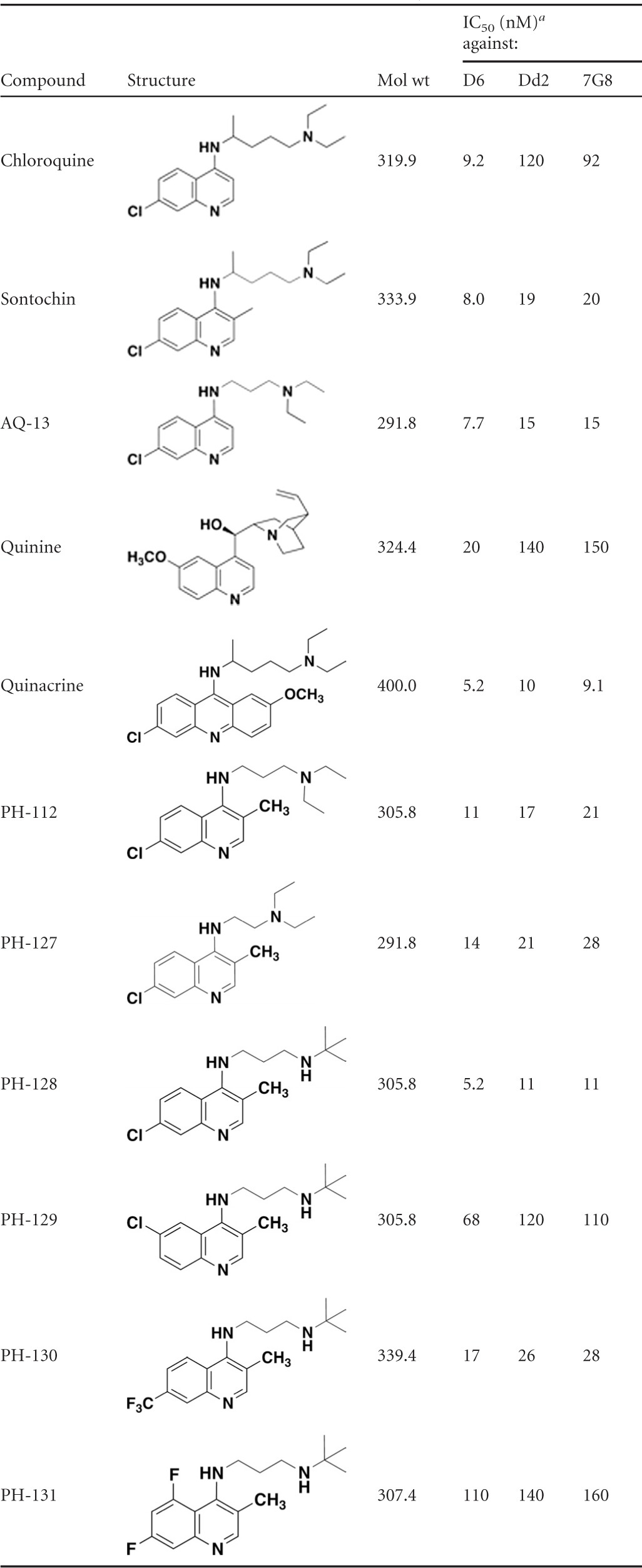
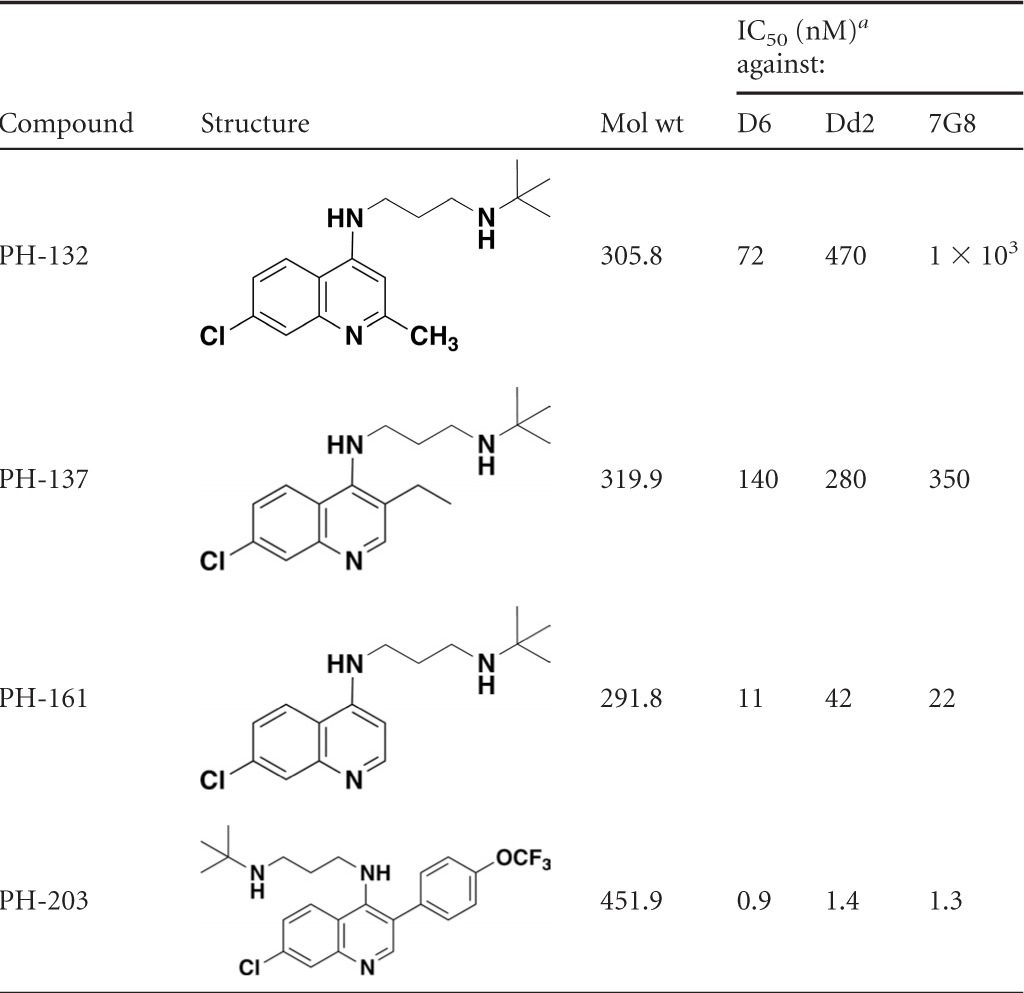
IC50s are averages of at least 2 determinations, each conducted in triplicate.
To demonstrate the importance of the methyl substituent on the quinoline core, we highlight 3 different 4-amino-quinoline derivatives varying only in the positioning of a CH3 group (Table 1). Notice that potency decreases and chloroquine cross-resistance increases in the molecule lacking a CH3 group at either the 2 or 3 position (PH-161). In PH-132 we made the PH-128 positional isomer in which the CH3 group is placed at the 2 position, and this molecule displays greatly diminished activity and significant cross-resistance in the two multidrug-resistant strains. Taken together, this information highlights the importance of placing a methyl group at the 3 position to circumvent chloroquine resistance.
Next we decided to introduce an aryl substituent at the 3 position to evaluate the effect on intrinsic antiplasmodial activity and cross-resistance. This concept originated from the idea that a large, structurally rigid group could impede interaction with the resistance-mediating PfCRT efflux protein. We also felt that such a group may help to guide the more hydrophobic drug into lipid droplets, where hemozoin is formed in the parasite's digestive vacuole (23). Notice that PH-203 exhibits enhanced antiplasmodial activity against chloroquine-sensitive and multidrug-resistant strains, with IC50s of ≈1 to 2 nM (Table 1).
Cytotoxicity and selectivity.
We determined the cytotoxicities of PH-128 and PH-203, two of the most active compounds in our pharmachin library. Murine splenic lymphocytes (MSL) were induced to proliferate following stimulation with concanavalin A in medium containing up to 25 μM drug incubated at 37°C for 3 days. In summary of our findings, PH-128 and PH-203 exhibited IC50s of >25 μM and 7.5 μM, respectively, against mitogen-stimulated lymphocyte proliferation. The IC50 for chloroquine in this assay is ≈25 μM. Considering that these two drugs exhibit antiplasmodial IC50s in the low nanomolar range, the cytotoxicity data establish a selectivity index (MSL IC50/P. falciparum IC50) of >4,000 for both drugs.
Safety.
Several antimalarial drugs, including halofantrine, are known to produce a QT interval prolongation via a blockade of the rapidly activating delayed rectifier K+ current (IKr), encoded by the human ether-a-go-go-related (hERG) gene (30). Thus, it is important to consider hERG channel inhibition in evaluation of our pharmachin lead molecules. The inhibitory effects of PH-128, PH-203, and chloroquine on the hERG potassium channel current expressed in mammalian cells were evaluated by ChanTest (Cleveland, OH) at room temperature using the QPatch HT (Sophion Bioscience A/S, Denmark), an automatic parallel patch clamp system. Each compound was evaluated in duplicate at 1, 4, and 12 μM. The duration of exposure was 5 min. Consistent with literature values, chloroquine inhibited the hERG channel with an IC50 of 2.5 μM. The calculated IC50s for PH-128 and PH-203 were 9.1 μM and 4 μM, respectively. These results indicate that the proarrhythmia risks of our two pharmachin leads are comparable to or slightly less than that for chloroquine, an antimalarial that is known to carry a moderate risk of cardiotoxicity at therapeutic dosages.
In vitro metabolic stability.
The potential for hepatic P450-dependent metabolism of PH-128 and PH-203 at 1 μM was assessed using pooled human liver microsomes in the presence of an NADPH-regenerating system. Chloroquine and sontochin were included for comparison purposes; the primary route of metabolism for chloroquine is via N dealkylation of the ethyl groups on the aminoalkyl side chain. The loss of substrate was monitored by LC-MS/MS. Verapamil and warfarin were monitored as well-characterized positive controls. Verapamil represents a drug with comparatively low metabolic stability, while warfarin represents a drug with a high degree of metabolic stability. Incubations were conducted in the absence of the NADPH-regenerating system to monitor for potential P450-independent metabolism. In the absence of NADPH, we did not observe significant metabolism of any of the test agents, including controls. Chloroquine had a longer half-life than the other antimalarial drugs in this study, and the degree of metabolism observed was negligible. Sontochin had a significantly shorter half-life (≈45 min) than chloroquine (>200 min), suggesting that the 3-position methyl group is attacked by P450 enzymes. This difference in metabolic stability may substantially account for the superior efficacy observed for chloroquine over sontochin in field trials with human volunteers conducted in the 1940s (4, 10). The t1/2 value recorded for PH-128 was 84.2 min in the presence of 0.3 mg/ml microsomal protein. Relative to the values obtained for the reference controls, verapamil and warfarin, this value predicts a moderate rate of metabolic degradation for PH-128 and suggests a low to intermediate rate of hepatic clearance in vivo. PH-203 displayed a slightly higher level of stability against metabolic degradation, with a half-life of 91.1 min. Thus, the 3-aryl pharmachin derivative PH-203 showed an extended half-life in comparison to sontochin as well as PH-128, although it is clear that additional structural modifications would be needed to increase the metabolic stability of this drug to the level observed for chloroquine.
In vivo antimalarial testing.
We evaluated PH-128 in vivo in mice infected with P. yoelii (strain K). Dosages ranged from 1 to 64 mg/kg/day and included a no-drug control Analysis of the results yielded an ED50 of 2.3 mg/kg/day and an ED90 of 3.9 mg/kg/day for PH-128; comparative values for chloroquine in the same study were 1.6 mg/kg/day (ED50) and 2.7 mg/kg/day (ED90). It is noteworthy that animals treated with 16 mg/kg PH-128 were aparasitemic on day 5 of the study; however, recrudescence occurred by day 13 in all of the animals in this group. Neither PH-128 or chloroquine demonstrated curative power in this assay over the tested dosage range.
We next evaluated the efficacy of PH-203 in vivo using the P. yoelii model. PH-203 proved to be highly efficacious, with an ED50 of 0.25 mg/kg/day, whereas chloroquine provided 50% suppression at 1.5 mg/kg/day. The parasitemia of animals treated with 4 mg/kg PH-203 was reduced by ≈95% relative to that of untreated controls. Perhaps the most impressive outcome of the study, however, relates to the superior curative power of PH-203. As before, chloroquine did not cure any of the animals at dosages of as high as 64 mg/kg/day, while PH-203 afforded 30-day cures in all animals at 16 and 64 mg/kg/day without any overt signs of toxicity or distress.
Taking the results together, with sontochin as our initial lead we established the importance of the 3-methyl feature in overcoming a significant degree of chloroquine resistance in P. falciparum. Replacement of the methyl group with an aryl feature as found in PH-203 yielded greater intrinsic potency against all tested strains, with IC50s in the low nanomolar range. While in vitro studies predict that this drug has an intermediate level of metabolic stability compared to chloroquine, it is superior in vivo against murine malaria. Possible explanations for the enhanced performance of PH-203 over chloroquine in the mouse model include (i) superior intrinsic antiplasmodial activity, (ii) enhanced metabolic stability and pharmacokinetics in mice, or (iii) conversion to active metabolites in vivo. We are currently evaluating these possibilities.
To gain insight into the orientation of the 3-position para-trifluoromethoxy-phenyl and the alkyl diamine group at the 4 position with respect to the planar quinoline core, a molecular mechanics calculation was performed on PH-203 using a Sybyl force field algorithm in Spartan software (Wavefunction, Irvine, CA). Full conformational analysis was calculated using a Monte Carlo approach to locate the lowest-energy conformer. A three-dimensional (3D) model of the lowest-energy conformer of PH-203 appears in Fig. 3, with the dihedral angle of 55.9 degrees around the bond between the quinoline ring and the para-trifluoromethoxy-phenyl group. For this model the dihedral angle at the 4 position between the quinoline ring and the nitrogen atom of the diaminoalkyl side chain was calculated to be 55.6 degrees. This lowest-energy conformer minimizes the steric hindrance between the three major structural elements. As shown, the para-trifluoromethoxy-phenyl group projects away from the 3 position and may sterically hinder binding to the chloroquine efflux protein PfCRT. It is also possible that this projection prevents the drug from adopting the preferred configuration that is recognized by PfCRT; i.e., the large aromatic ring would obstruct the free rotation of the 4-position side chain and restrict its lateral alignment off the quinoline nucleus occupied by the bulky protrusion.
Fig 3.
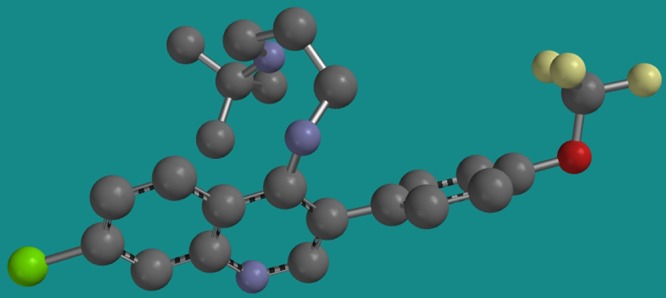
Structure representing the lowest-energy conformer obtained for PH-203. Notice that the 3-position trifluoromethoxy-phenyl group presents as a conformationally rigid projection that may sterically hinder association with the chloroquine-resistance associated protein PfCRT.
Here it seems appropriate to mention that we are not the first to place an aryl ring at position 3 of the 4-aminoquinoline nucleus. Compound SN-10,555 was prepared previously by Drake et al. (8). This molecule contained a simple phenyl ring at the 3 position [7-chloro-4-(4-diethylamino-1-methylbutylamino)-3-phenylquinoline, i.e., “3-phenyl-chloroquine”] and exhibited comparatively modest activity (relative to chloroquine) against avian malaria as reported in Wiselogle's 1946 A Survey of Antimalarial Drugs, 1941-1945 (33). Because of its rather unspectacular in vivo efficacy and since multidrug-resistant malaria had yet to emerge as a global health problem in 1946, the 3 position of 4-aminoquinoline antimalarials remains largely unexplored chemical space. It would appear from our findings that this portion of the molecule may hold the key to the development of a chloroquine replacement drug with potent activity against multidrug-resistant strains of P. falciparum. Here we show that the introduction of a lipophilic aromatic group at C-3 of the quinoline ring leads to a significant improvement in activity against multidrug-resistant P. falciparum strains and in vivo against lethal murine P. yoelii infections. These results provide encouragement for further investigation of this series as potential antimalarials for use in humans.
On the basis of its superior activity against multidrug resistant strains of P. falciparum in vitro and outstanding antimalarial performance in vivo in direct comparison to chloroquine, we consider PH-203 the current frontrunner for the pharmachin series. In PH-203 we have incorporated two structural features (i.e., the short side chain and 3-position aryl substituent) that independently circumvent chloroquine resistance in Plasmodium falciparum. The novel coexistence of these two structural features in a single molecule should greatly diminish the likelihood of emergence of resistant strains. Our efforts to further optimize the pharmachin chemotype for antimalarial activity, increased metabolic stability, and diminished propensity for emergence of resistance are ongoing and will continue in this laboratory.
ACKNOWLEDGMENTS
This project was supported in part by National Institutes of Health Challenge grant RC1 AI087011 (to M.K.R.) and with support from the Department of Veterans Affairs Merit Review Program (to M.K.R.). The National Science Foundation (grant 0741993) supported the purchase of the LTQ-Orbitrap Discovery MS system used for analysis of our samples.
Footnotes
Published ahead of print 16 April 2012
REFERENCES
- 1. Ager AJ. 1984. Rodent malaria models, vol 68/I. Springer-Verlag, Berlin, Germany [Google Scholar]
- 2. Andersag H. 1948. Antimalariamittel aus der Gruppe halogensubstituierter Chinolinverbindungen. Chem. Ber. 81:499–507 [Google Scholar]
- 3. Andersag H, Breitner S, Jung H. March 1941. Quinoline compound and process of making the same. US Patent 2,233,970
- 4. Berliner RW, et al. 1948. Studies on the chemotherapy of the human malarias. VI. The physiological disposition, antimalarial activity, and toxicity of several derivatives of 4-aminoquinoline. J. Clin. Invest. 27:98–107 [PubMed] [Google Scholar]
- 5. Blanchard K. 1947. Antimalarial drugs. Annu. Rev. Biochem. 16:587–604 [DOI] [PubMed] [Google Scholar]
- 6. Coatney GR. 1963. Pitfalls in a discovery: the chronicle of chloroquine. Am. J. Trop. Med. Hyg. 12:121–128 [DOI] [PubMed] [Google Scholar]
- 7. De D, Krogstad FM, Cogswell FB, Krogstad DJ. 1996. Aminoquinolines that circumvent resistance in Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 55:579–583 [DOI] [PubMed] [Google Scholar]
- 8. Drake N, et al. 1946. Synthetic antimalarials. The preparation of certain 4-aminoquinolines. J. Am. Chem. Soc. 68:1208–1213 [DOI] [PubMed] [Google Scholar]
- 9. Fairley N. 1949. Malaria. With special reference to certain experimental, clinical, and chemotherapeutic investigations. BMJ 1949:891–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fairley NH. 1946. Atebrin susceptibility of the Aitaipe-Wewak strains of P. falciparum and P. vivax; a field and experimental investigation by L. H. Q. Medical Research Unit, Cairns. Trans. R. Soc. Trop. Med. Hyg. 40:229–273 [DOI] [PubMed] [Google Scholar]
- 11. Fernando SD, Rodrigo C, Rajapakse S. 2010. The ‘hidden’ burden of malaria: cognitive impairment following infection. Malar. J. 9:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fidock AD, et al. 2000. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6:861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Geary TG, Divo AA, Jensen JB. 1987. Activity of quinoline-containing antimalarials against chloroquine-sensitive and -resistant strains of Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 81:499–503 [DOI] [PubMed] [Google Scholar]
- 14. Geary TG, Jensen JB. 1983. Lack of cross-resistance to 4-aminoquinolines in chloroquine-resistant Plasmodium falciparum in vitro. J. Parasitol. 69:97–105 [PubMed] [Google Scholar]
- 15. Martin RE, et al. 2009. Chloroquine transport via the malaria parasite's chloroquine resistance transporter. Science 325:1680–1682 [DOI] [PubMed] [Google Scholar]
- 16. Mzayek F, et al. 2007. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin. Trials 2:e6 doi:10.1371/journal.pctr.0020006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oduola AM, Milhous WK, Salako LA, Walker O, Desjardins RE. 1987. Reduced in-vitro susceptibility to mefloquine in West African isolates of Plasmodium falciparum. Lancet 330:1304–1305 [DOI] [PubMed] [Google Scholar]
- 18. Olliaro P, Goldberg D. 1995. The Pasmodium digestive vacuole: metabolic headquarters and choice drug target. Parasitol. Today 11:294–297 [DOI] [PubMed] [Google Scholar]
- 19. O'Neill PM, et al. 2003. Isoquine and related amodiaquine analogues: a new generation of improved 4-aminoquinoline antimalarials. J. Med. Chem. 46:4933–4945 [DOI] [PubMed] [Google Scholar]
- 20. O'Neill PM, et al. 2006. A medicinal chemistry perspective on 4-aminoquinoline antimalarial drugs. Curr. Top. Med. Chem. 6:479–507 [DOI] [PubMed] [Google Scholar]
- 21. Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK. 2000. The structure of malaria pigment beta-haematin. Nature 404:307–310 [DOI] [PubMed] [Google Scholar]
- 22. Petersen I, Eastman R, Lanzer M. 2011. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 585:1551–1562 [DOI] [PubMed] [Google Scholar]
- 23. Pisciotta JM, et al. 2007. The role of neutral lipid nanospheres in Plasmodium falciparum haem crystallization. Biochem. J. 402:197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ridley RG, et al. 1996. 4-Aminoquinoline analogs of chloroquine with shortened side chains retain activity against chloroquine-resistant Plasmodium falciparum. Antimicrob. Agents Chemother. 40:1846–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roepe PD. 2010. PfCRT-mediated drug transport in malarial parasites. Biochemistry 50:163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48:1803–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stocks PA, et al. 2002. Novel short chain chloroquine analogues retain activity against chloroquine resistant K1 Plasmodium falciparum. J. Med. Chem. 45:4975–4983 [DOI] [PubMed] [Google Scholar]
- 28. Summers RL, Martin RE. 2010. Functional characteristics of the malaria parasite's “chloroquine resistance transporter”: implications for chemotherapy. Virulence 1:304–308 [DOI] [PubMed] [Google Scholar]
- 29. Tarbell DS, et al. 1946. The synthesis of some 7-chloro-4-(3-alkylamino-propylamino) quinolines. J. Am. Chem. Soc. 68:1217–1219 [DOI] [PubMed] [Google Scholar]
- 30. Traebert M, et al. 2004. Inhibition of hERG K+ currents by antimalarial drugs in stably transfected HEK293 cells. Eur. J. Pharmacol. 484:41–48 [DOI] [PubMed] [Google Scholar]
- 31. Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675 [DOI] [PubMed] [Google Scholar]
- 32. Winter RW, et al. 2006. Evaluation and lead optimization of anti-malarial acridones. Exp. Parasitol. 114:47–56 [DOI] [PubMed] [Google Scholar]
- 33. Wiselogle FY. (ed). 1946. A survey of antimalarial drugs, 1941–1945. J. W. Edwards, Ann Arbor, MI [Google Scholar]