

Abstract
Arylimidamides (AIAs) have shown outstanding in vitro potency against intracellular kinetoplastid parasites, and the AIA 2,5-bis[2-(2-propoxy)-4-(2-pyridylimino)aminophenyl]furan dihydrochloride (DB766) displayed good in vivo efficacy in rodent models of visceral leishmaniasis (VL) and Chagas' disease. In an attempt to further increase the solubility and in vivo antikinetoplastid potential of DB766, the mesylate salt of this compound and that of the closely related AIA 2,5-bis[2-(2-cyclopentyloxy)-4-(2-pyridylimino)aminophenyl]furan hydrochloride (DB1852) were prepared. These two mesylate salts, designated DB1960 and DB1955, respectively, exhibited dose-dependent activity in the murine model of VL, with DB1960 inhibiting liver parasitemia by 51% at an oral dose of 100 mg/kg/day × 5 and DB1955 reducing liver parasitemia by 57% when given by the same dosing regimen. In a murine Trypanosoma cruzi infection model, DB1960 decreased the peak parasitemia levels that occurred at 8 days postinfection by 46% when given orally at 100 mg/kg/day × 5, while DB1955 had no effect on peak parasitemia levels when administered by the same dosing regimen. Distribution studies revealed that these compounds accumulated to micromolar levels in the liver, spleen, and kidneys but to a lesser extent in the heart, brain, and plasma. A 5-day repeat-dose toxicology study with DB1960 and DB1955 was also conducted with female BALB/c mice, with the compounds administered orally at 100, 200, and 500 mg/kg/day. In the high-dose groups, DB1960 caused changes in serum chemistry, with statistically significant increases in serum blood urea nitrogen, lactate dehydrogenase, aspartate aminotransferase, and alanine aminotransferase levels, and a 21% decrease in body weight was observed in this group. These changes were consistent with microscopic findings in the livers and kidneys of the treated animals. The incidences of observed clinical signs (hunched posture, tachypnea, tremors, and ruffled fur) were more frequent in DB1960-treated groups than in those treated with DB1955. However, histopathological examination of tissue samples indicated that both compounds had adverse effects at all dose levels.
INTRODUCTION
Leishmaniasis and Chagas' disease (CD) are two widespread neglected diseases that affect impoverished areas of developing countries. Visceral leishmaniasis (VL) accounts for approximately 25% of the estimated 2 million new cases of leishmaniasis that occur annually. VL is caused by the protozoan parasites Leishmania donovani in India and Africa, L. infantum in the Mediterranean region, and L. chagasi in the countries of the New World where the disease is endemic. Symptomatic VL results in hepatosplenomegaly, anemia, wasting, and susceptibility to other infections, which are almost always fatal if left untreated. Despite advances in treating VL on the Indian subcontinent, which include the registration of both the oral drug miltefosine (26) and the injectable antibiotic paromomycin (25), as well as the demonstration of the high efficacy of single-dose liposomal amphotericin B (24), these drugs are often too expensive or less effective when used in Africa or Brazil (5, 15–17). In addition, miltefosine is teratogenic (4, 20). Considering these weaknesses of existing agents used against VL, a new orally available, inexpensive, small-molecule antileishmanial drug is needed that would display high efficacy when given in a short course, either alone or in a combination. Likewise, new therapies are urgently needed for CD. CD is caused by the protozoan parasite Trypanosoma cruzi and affects more than 10 million people in the poorest areas of Latin America, as well as in other geographic regions. The reduviid vector transmits the parasite in Latin America; cases occurring in other regions are due mainly to the migration of infected individuals to areas such as the United States, Canada, and many European and some Western Pacific countries (9). CD therapy is based on two nitroaromatic drugs, nifurtimox and benznidazole (Bz), that are recommended for all acute-stage, early-chronic-stage, and reactivated cases (2, 6). However, both drugs are far from ideal and give variable results, depending on the area where the disease is endemic; present considerable toxicity; are administered over 30 or more days; and are not very effective against the later chronic phase of CD or against naturally resistant strains (7, 21). Another challenge for CD therapy is related to blood prophylaxis in areas where the disease is endemic since the only drug used, gentian violet, is toxic, gives blood a purple color, and may stain the skin and mucosa of recipients (6, 7). Thus, a new drug candidate against CD should display activity against a large panel of parasite isolates, including bloodstream forms, intracellular forms, and resistant organisms; should present oral efficacy equal to or better than that of Bz; and should exhibit low toxicity.
Our previous efforts have identified arylimidamides (AIAs) as compounds with outstanding in vitro antileishmanial (8, 22) and anti-T. cruzi (12, 18, 19) activities. In addition to excellent in vitro antileishmanial potency, the AIA hydrochloride salt 2,5-bis[2-(2-propoxy)-4-(2-pyridylimino)aminophenyl]furan (DB766) displayed good efficacy in both murine and hamster models of VL (27), was as active as Bz in experimental models of T. cruzi infection (3), was not mutagenic in the Ames test, and did not have an effect on the serum chemistry of mice when given orally at a dose of 100 mg/kg/day × 5, although higher dose levels were not tested due to the limited aqueous solubility of DB766 (27). In an effort to advance members of this class of compounds toward clinical development for the treatment of VL and CD, more-soluble AIA salts were synthesized (Fig. 1 shows their structures), including DB1960 (mesylate salt of DB766) and DB1955 {mesylate salt of DB1852, 2,5-bis[2-(2-cyclopentyloxy)-4-(2-pyridylimino)aminophenyl]furan hydrochloride}. We report here the in vitro and in vivo antileishmanial and anti-T. cruzi activities, pharmacokinetic (PK) profiles, and in vivo toxicities of these AIA mesylate salts.
Fig 1.

Structures of DB766, DB1852, DB1955, and DB1960.
MATERIALS AND METHODS
AIAs.
The preparation and characterization of DB766 (27) and DB1852 (1) have been reported earlier. For the procedures used for the synthesis of DB1955 and DB1960, along with the characterizations of these compounds, see the supplemental material.
Solubility measurements.
The water solubility of the AIA salts of interest was measured by the method of Zhou et al. (28), with some modifications. Briefly, serial dilutions of compounds were prepared in dimethyl sulfoxide and read at 370 nm using a SpectraMax Plus 384 spectrophotometer (Molecular Devices, Sunnyvale, CA) to construct a standard curve. A saturated solution of each compound containing some undissolved solid was then prepared and centrifuged. The supernatant from the saturated solution was collected, diluted 1:40 with water, and read at 370 nm as described above. The water solubility of each compound was determined by interpolation from the appropriate standard curve.
In vitro susceptibility assays.
The potency of AIAs and control compounds for VL (amphotericin B) and for CD (Bz) against intracellular L. amazonensis and L. donovani and bloodstream trypomastigotes and amastigotes of T. cruzi strain Y was determined as follows. Intracellular Leishmania assays were conducted as described previously for L. donovani (29) and L. amazonensis (13). For the analysis of the effects of the compounds upon bloodstream trypomastigote forms, 5 × 106/ml parasites were incubated for 24 h at 37°C in RPMI 1640 medium supplemented with 10% fetal calf serum in the presence or absence of serial dilutions of each compound. Parasite death was determined by light microscopy using a Neubauer chamber, which allows the direct visualization and quantification of the number of motile and live parasites, and then 50% inhibitory concentrations (IC50s) were calculated (3). For the analysis of the effects of compounds on intracellular forms of T. cruzi after 24 h of parasite-host cell interaction (10:1 ratio), infected cardiac cultures were washed to remove extracellular parasites and then maintained at 37°C in a 5% CO2–95% air atmosphere in the presence of nontoxic (to cardiac cells) concentrations of each compound with replacement of the medium (containing the respective AIA) every 24 h. Infected cultures lacking a test compound were used as controls. After 48 h, all cultures were fixed and stained with Giemsa solution. The endocytic index was used to compare compound activities and was calculated by multiplying the percentage of infected cells by the mean number of parasites per infected cell. IC50s were calculated as previously reported (3).
Primary cultures of embryonic cardiomyocytes were obtained from Swiss mice and purified as previously described (3). The toxicities of the test compounds for cardiac cell cultures (3) and murine peritoneal macrophages (27) were measured as described previously.
DNA binding experiments.
ΔTm studies were performed as previously described (22).
In vivo evaluation of antileishmanial efficacy.
In vivo murine VL model experiments were carried out as described previously (13), with some modifications for this study. Briefly, BALB/c mice were inoculated intravenously with 5 × 107 LV82 promastigotes. After infection on day 0, mice were marked for individual identification and randomly sorted into groups of 4. The treatment was started 1 week after infection (day 7) and continued for 5 days (days 7 to 11). In these experiments, compounds were dissolved in water and given either intraperitoneally (i.p.) or orally (p.o.) by gavage. One group received water as an untreated control, one group received miltefosine at 10-mg/kg/dose as a positive control, and other groups received either DB1852 at 10 or 30 mg/kg/dose i.p., DB1955 at 100 or 200 mg/kg/dose p.o., or DB1960 at 50, 100, or 200 mg/kg/dose p.o. On day 14 (i.e., 3 days after the last dose), animals were euthanized and Giemsa-stained liver smear slides were prepared. The number of amastigotes per 200 cell nuclei was determined by microscopy, and parasite burdens, expressed in Leishman-Donovan units (LDU), were calculated by using the equation LDU = number of amastigotes per 1,000 cell nuclei × total liver weight (grams). Compound efficacy was determined by comparing the LDU counts of mice in the treated and untreated groups.
In vivo evaluation of anti-T. cruzi activity.
Male Swiss mice were obtained from the Fundação Oswaldo Cruz (FIOCRUZ) animal facilities (Rio de Janeiro, Brazil). Mice were housed at a maximum of 8 per cage and kept in a conventional room at 20 to 24°C under a 12/12-h light-dark cycle. The animals were provided with sterilized water and chow ad libitum. Infection was performed by i.p. injection of 104 bloodstream trypomastigotes of strain Y. Animals (18 to 21 g) were divided into the following groups: uninfected (noninfected and nontreated), untreated (infected with T. cruzi but nontreated), and treated (infected and treated i.p. or p.o. at 12.5 to 100 mg/kg/day with DB1955 and DB1960 or at 100 mg/kg/day with Bz). For DB1955 and DB1960, mice received a 0.1-ml i.p. injection or a 0.2-ml oral dose, with five daily consecutive doses, starting at the onset of parasitemia. For Bz treatment, infected mice received a 0.2-ml oral dose (gavage) following the therapeutic schemes described above. Parasitemia was individually checked by direct microscopic counting of parasites in 5 μl of blood as described previously (3). At different days postinfection, body weight was measured and the number of deaths was recorded daily until 30 days posttreatment and expressed as a percentage of the cumulative mortality rate. All compounds were directly diluted with water.
Single-dose PKs and tissue distribution studies with uninfected mice.
Male Swiss Webster mice (20 to 30 g; n = 3 per time point) were given DB1955 or DB1960 via oral gavage at a dose volume of 5 ml/kg. Compounds were dissolved in sterile water to form a clear solution at 20 mM. Terminal blood and tissue (liver, spleen, kidney, heart, and brain) samples were collected at selected time points from 1 h to 72 h postdose and further processed for quantification of AIAs as described below using previously described methods (27), with modifications. Briefly, liver, spleen, kidney, heart, and brain samples from mice were weighed, diluted with 2 volumes of water (assuming that 1 g of wet tissue equals a volume of 1 ml), and then homogenized. Tissue homogenates or plasma samples were extracted and prepared for high-performance liquid chromatography (HPLC)/UV or HPLC-tandem mass spectrometry (MS/MS) analysis as previously described (27). HPLC/UV analyses of DB1960 and DB1955 from tissue homogenates were performed using a reversed-phase analytical column (Zorbax Bonus-RP, 2.1 by 50 mm, 3.5 μm; Agilent, Palo Alto, CA) and by monitoring analytes at a wavelength of 368 nm. The calibration curves ranged from 0.1 to 100 μM. HPLC-MS/MS analyses of DB1960 and DB1955 in plasma samples were performed with an Applied Biosystems (Foster City, CA) API 3000 triple quadrupole mass spectrometer equipped with a Turbo IonSpray interface (MDS Sciex, San Francisco, CA). The characteristic SRM transitions for DB1960 and DB1955 were m/z 575.3 → 454.1 and 627.5 → 610.4, respectively, in positive-ion mode. The calibration curve for DB1960 and DB1955 ranged from 10 to 4,000 nM using a quadratic equation with 1/x weighting.
Toxicity and toxicokinetic evaluations in female BALB/c mice.
Single-dose escalation and 5-day repeat-dose toxicity experiments were performed using 7- to 8-week-old BALB/c female mice. The no-observable-adverse-effect levels (NOAELs) and maximum tolerated doses (MTDs) of DB1955 and DB1960 were determined in female BALB/c mice following a single p.o. administration and five daily p.o. administrations. Dose formulations were prepared fresh daily under yellow light by dissolving appropriate amounts of compound in sterile water to achieve the target concentrations. The dosing volume was 10 ml/kg. For the single-dose escalation experiment, mice (n = 2 for each dose level per compound) were administered 0, 50, 100, 200, or 500 mg/kg and observed for 3 days. Since no adverse effects were observed in any of the groups, 100-, 200-, and 500-mg/kg dose levels were selected to conduct a 5-day repeat-dose experiment with the two compounds. In a repeat-dose range-finding study, DB1955 and DB1960 were administered to female BALB/c mice for 5 consecutive days at the selected doses. Mice were euthanized on day 6 for the main necropsy and day 12 for the recovery necropsy. For the experimental design of this range-finding study, see Table S1 in the supplemental material. A satellite group of mice were also included for evaluation of plasma drug levels and toxicokinetic parameters. Plasma drug levels were evaluated at 0.5, 1, and 6 h postdose on day 1 and predose and at 0.5, 1, 4, 6, and 24 h postdose on day 5. Body weights were recorded prior to day 1 and prior to each necropsy. Clinical hematology and serum chemistry were measured for blood samples collected 1 day after the last dose (day 6) prior to necropsy. Histopathologic examination of tissues retained in 10% neutral buffered formalin was performed by a board-certified veterinary pathologist. Toxicokinetic analysis was conducted using WinNonlin version 5.2 Professional by noncompartmental modeling. The following parameters were determined: the maximal plasma drug concentration (Cmax), time to maximum plasma drug concentration (Tmax), the area under the plasma drug concentration-time curve (AUC) from 0 to 6 h on days 1 and 5 (AUC0-6), and the AUC0-24 on day 5. The terminal elimination half-life (t1/2) was also determined in groups that had at least three sample collection times after the Tmax.
Statistical analysis.
Body weights, organ weights, and clinical pathology data were evaluated by one-way analysis of variance (ANOVA), followed by Dunnett's test (if the ANOVA result was significant). All other numeric parameters were evaluated by Student's t test unless specified otherwise. For clinical pathology data, values that were below the detection threshold were not included in the statistical evaluation.
RESULTS
Solubility of AIA salts.
Our initial observations suggested that mesylate salts DB1955 and DB1960 were more soluble than their corresponding hydrochloride salts DB1852 and DB766. Measurement of the solubilities of these AIA salts in water indicated that DB1955 was 3.1-fold more soluble than DB1852 (solubility limits of 54.2 mM and 16.7 mM, respectively), while DB1960 was 5.3-fold more soluble than DB766 (solubility limits of 55.5 mM and 10.4 mM, respectively).
In vitro antileishmanial and anti-T. cruzi efficacy studies with DB766, DB1852, DB1955, and DB1960.
The in vitro antileishmanial activities of DB766, DB1852, DB1955, and DB1960 are shown in Table 1. Dose-response curves were steeper for DB1960 than for DB1955 (IC90 = 0.038 ± 0.007 μM and 1.7 ± 0.2 μM [mean ± standard error], respectively). As expected, the biological activity of the hydrochloride salts was nearly identical to the activity of the corresponding mesylate salts in the intracellular Leishmania assay and the peritoneal macrophage toxicity assay. DB1852 did not display DNA binding, as judged by its negligible ΔTm value obtained using poly(dA·dT)2 DNA, consistent with the ΔTm data obtained for this compound using T. cruzi kinetoplast DNA (10). The mesylate salt of DB1852, DB1955, also gave no ΔTm increase. In a similar manner, DB766 and DB1960 have the same ΔTm values within the range of experimental error, indicating that the anion does not play a significant role in DNA interactions of these compounds, as expected. The low ΔTm values of all four compounds suggest that DNA binding does not play a significant role in their biological effects in the test systems. Evaluation of these compounds against T. cruzi revealed that although all of the AIAs studied (DB766, DB1955, and DB1852) displayed similar effects upon bloodstream trypomastigotes, DB766 was the most active against intracellular forms, exhibiting about 100 times more potency than the reference drug Bz (Table 2).
Table 1.
In vitro antileishmanial activities and ΔTm values of DB766, DB1852, DB1955, and DB1960
| Compound | IC50 (μM)a vs: |
ΔTm(AT)b (°C) | ||
|---|---|---|---|---|
| L. donovani LV82 intracellular amastigotes | L. amazonensis intracellular amastigotes | Murine peritoneal macrophages | ||
| DB766c | 0.036 ± 0.005 | 0.087 ± 0.015 | 2.8 ± 0.8 | 6.0 |
| DB1852 | 0.16 ± 0.03 | 0.17 ± 0.06 | 6.9 ± 0.9 | 1.0 |
| DB1955 | 0.25 ± 0.08 | 0.16 ± 0.03 | 8.3 ± 1.2 | 1.1 |
| DB1960 | 0.020 ± 0.002 | 0.056 ± 0.019 | 4.7 ± 0.8 | 5.3 |
| Amphotericin B | 0.05 ± 0.01 | 0.15 ± 0.03 | 3.9 ± 2.1 | NDd |
The values are means ± standard errors of at least three independent experiments.
ΔTm(AT), increase in thermal melting of poly(dA·dT)2 on addition of the compound to a saturation ratio; the reproducibility is ±0.5°C.
The values for DB766 are from reference 27.
ND, not determined.
Table 2.
In vitro activities of DB766, DB1852, and DB1955 against T. cruzi strain Y
In vivo antileishmanial efficacies of DB1852, DB1955, and DB1960.
In vivo evaluation of DB1852 given at 5 × 30 mg/kg/day by the i.p. route showed 61% ± 5% inhibition of liver parasitemia (Fig. 2A), which was comparable to the previously reported in vivo efficacy of DB766 (63% ± 11%) given by the same dosing regimen and route (27). Considering that DB766 showed in vivo activity in both the murine and hamster VL models when given orally (27), the efficacy of the more soluble mesylate salts of both of these compounds was examined in the murine model of VL by the oral route. DB1955 was evaluated at 5 × 100 mg/kg/day and 5 × 200 mg/kg/day, giving 57% ± 5% and 78% ± 4% inhibition of liver parasitemia, respectively (Fig. 2B). DB1960 was tested at oral doses of 5 × 50 mg/kg/day, 5 × 100 mg/kg/day, and 5 × 200 mg/kg/day, resulting in 29% ± 4%, 51% ± 6%, and 93% ± 6% inhibition of liver parasitemia, respectively (Fig. 2C). These compounds were also evaluated in hamsters infected with L. donovani and displayed good activity in this model as well (A. Srivastava and D. Kyle, unpublished data).
Fig 2.

Liver parasite burdens (LDU counts) of BALB/c mice infected with LV82 and treated with AIAs or miltefosine as determined by microscopy (n = 4). Panels: A, LDU counts of mice treated with DB1852 i.p.; B, LDU counts of mice treated with DB1955 p.o.; C, LDU counts of mice treated with DB1960 p.o. Miltefosine was given p.o. in each case. *, P < 0.05 (compared with untreated control); **, P < 0.01 (compared with untreated control).
In vivo trypanocidal efficacy of DB1955 and DB1960.
When given under different treatment schemes (five consecutive daily i.p. and p.o. doses), DB1955 did not show any trypanocidal efficacy as assessed by the peak parasitemia levels that occurred at 8 days postinfection (Fig. 3A). Instead, some schemes resulted in a slight increase in parasitemia compared to that in untreated animals (Fig. 3A). Regarding the mortality rate, only the 25-mg/kg i.p. dose resulted in 100% survival while the other doses (12.5 and 50 mg/kg/day) reduced mortality rates only slightly or increased mortality rates (100 mg/kg/day p.o.; data not shown). DB1960 was tested at oral doses of 5 × 12.5 to 100 mg/kg/day. Although the 12.5- and 25-mg/kg doses neither reduced parasitemia nor protected against death, treatments with DB1960 at 50 and 100 mg/kg/day resulted in 28% ± 7% and 46% ± 10% reductions of peak parasitemia, respectively (Fig. 3B). While DB1960 at 50 mg/kg/day did not reduce mortality rates, administration of this compound at 100 mg/kg/day resulted in an about 35% reduction in the mortality rates (data not shown), showing less activity than DB766 in experimental models of acute T. cruzi infection (3). The administration of Bz not only suppressed parasitemia but also resulted in 100% animal survival in both sets of assays (Fig. 3). Regarding body weight, neither DB1955 nor DB1960 was able to restore the weight loss induced in mice by acute T. cruzi infection, in contrast to Bz treatment (data not shown).
Fig 3.

Parasitemia levels of T. cruzi-infected mice treated with five consecutive daily doses of DB1955 and DB1960 via the i.p. and p.o. routes. The results of a representative study examining the efficacy of 12.5- to 100-mg/kg DB1955 (A; n = 5 per group) and DB1960 (B; n = 8 per group) are shown, with Bz given p.o. (100 mg/kg/day) used as the reference drug. Symbols and error bars denote the means and standard errors.
PKs and tissue distribution of DB1955 and DB1960 in mice.
The average plasma and tissue drug concentrations in uninfected mice given a single p.o. dose (100 μmol/kg or approximately 76 mg of salt/kg for DB1955 and 78 mg of salt/kg for DB1960) of DB1955 or DB1960 were determined to confirm their potential as orally active drug candidates. Both molecules quickly appeared in plasma with a Tmax of around 1 to 2 h, followed by a rapid decline during the distribution phase and a slower terminal elimination phase with t1/2s ranging from 14 to 18 h (Fig. 4 and Table 3). The maximal plasma DB1955 concentration was approximately 3-fold greater than that of DB1960 (3.13 μM versus 1.07 μM); however, similar plasma exposures (AUC) were observed due to the longer t1/2 of DB1960 (18 h versus 14 h).
Fig 4.
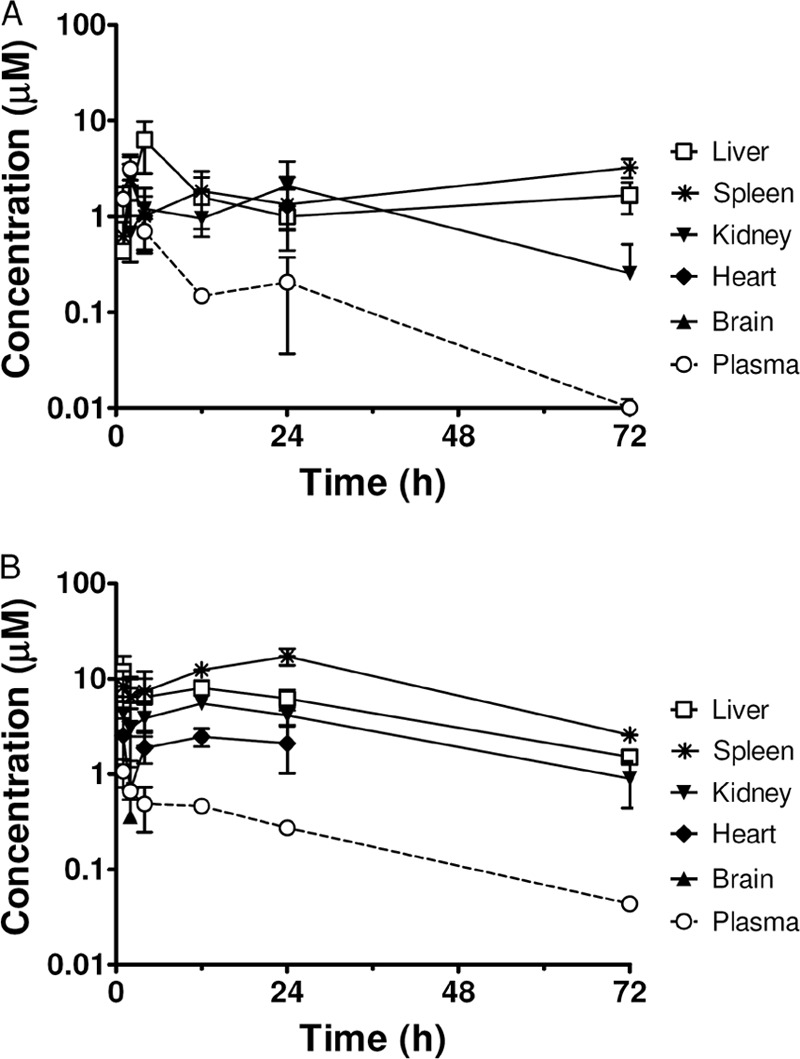
Plasma and tissue DB1955 (A) and DB1960 (B) concentrations of mice after a single oral administration of 100 μmol/kg (or 76 mg of salt/kg for DB1955 and 78 mg of salt/kg for DB1960). Tissue drug concentrations were calculated assuming that 1 g of wet tissue equals a volume of 1 ml. Symbols and error bars denote the means and standard errors, respectively, of triplicate determinations. Spleen DB1955 concentrations at 1 and 24 h were from duplicate determinations.
Table 3.
PK measurements of DB1955 and DB1960 after a single oral dose in mice
| Drug | Dose level (μmol/kg)/(mg/kg)a | Cmax (μM)b | Tmax (h) | AUClast (h·μmol/liter)b | AUC∞ (h·μmol/liter) | MRTlast (h) | t1/2 (h) |
|---|---|---|---|---|---|---|---|
| DB1955 | 100/76 | 3.1 ± 1.0 | 2 | 17.6 ± 5.5 | 17.8 | 12 | 14 |
| DB1960 | 100/78 | 1.07 ± 0.35 | 1 | 18.4 ± 1.7 | 19.5 | 19 | 18 |
Milligrams of salt per kilogram of body weight.
The values are means ± standard errors.
Average tissue DB1955 (Fig. 4A) or DB1960 (Fig. 4B) concentrations after a single oral administration in uninfected mice were determined using liver, spleen, kidney, heart, and brain tissue homogenates. As expected based on our previous observations with the AIAs DB745 and DB766 (27), the highest drug concentrations were detected in the spleen and liver, which are target organs for antileishmanial/anti-T. cruzi chemotherapy. The maximum DB1955 concentrations were 6.0, 2.3, and 1.9 μg/g (or 9.5, 3.6, and 3.1 μM) in the liver, spleen, and kidney, respectively. The maximum DB1960 concentrations were 6.8, 9.9, and 3.2 μg/g (or 11.9, 17.3, and 5.5 μM) in the liver, spleen, and kidney, respectively. DB1960 was also detected in the heart at a maximum concentration of 1.4 μg/g (or 2.5 μM) but was below the lower limit of quantitation (LLOQ) (0.06 μg/g or 0.1 μM) in the brain, except for one animal. In contrast, DB1955 was not detected in either the brains or the hearts of mice receiving this molecule (except that the compound was detected in the heart of one animal). The lower levels of DB1955 in the heart may be related to its poor efficacy in our T. cruzi-infected mouse model since this organ is one of the main targets of this parasite.
Single-dose range-finding toxicology study summary.
A single-dose range-finding study was conducted with DB1955 and DB1960 and 7- to 8-week-old female BALB/c mice at oral doses of 50, 100, 200, and 500 mg/kg. No adverse effects were observed in any of the groups. Based on the results of this study, the MTD after a single oral dose administration was estimated to be at least 500 mg/kg.
Five-day repeat-dose range-finding toxicology and toxicokinetic study.
Clinical findings and body weight changes. All animals in the DB1955 groups survived for the duration of the repeat-dose range-finding study. No overt responses were observed in the low-dose DB1955 group, and most of the animals in the medium- and high-dose groups also appeared normal. However, single animals in the medium-dose group showed lacrimation, dyspnea, and hypoactivity on day 5 or 6, while hunched posture (one animal), tachypnea (two animals), and ruffled fur (three animals) were observed in the high-dose group. Compared with the control group, there were no changes in body weight on day 6 in animals treated with DB1955 but on day 12, mice in the 500-mg/kg group had a 14% body weight reduction that was statistically significant. The average body weights of all groups are given in Table S2 in the supplemental material.
The incidences of observed clinical signs were generally higher in DB1960-treated groups than in those treated with DB1955, and the animals in the high-dose DB1960 recovery group were euthanized in moribund condition (on day 6) due to hypoactivity, hunched posture, and tremors. All other animals treated with DB1960 survived to their scheduled necropsy. Mice treated with DB1960 displayed tremors, ruffled fur, and tachypnea in a dose-dependent manner, with the effects occurring in an increasing number of animals per group as the dose was escalated. On day 6, mice treated with DB1960 at 500 mg/kg had a 21% reduction in body weight that was statistically significant. Except for those mentioned above, there were no other changes in body weight that could be attributed to treatment with DB1955 or DB1960.
Hematology and clinical chemistry.
DB1955 and DB1960 treatments had little effect on the hematocrit (HCT), hemoglobin (HGB) level, red blood cell (RBC) count, or white blood cell (WBC) count, although there was a statistically significant decrease in the HCT and the HGB level in mice in the medium-dose DB1960 group compared with those in the control group. The platelet count (PLC) was also decreased in mice treated with DB1960, and the changes in animals in the medium- and high-dose groups were statistically significant. The percentage of neutrophils (PNS) was increased on day 6 by 3-fold in mice treated with high-dose DB1960, possibly due to tissue damage and associated inflammation. A summary of these parameters is given in Table 4. By day 12, no changes in the above parameters were observed in animals treated with either test drug compared to those in untreated control animals (data not shown). Hematology parameters are not available for mice in the high-dose DB1960 recovery group since these animals were euthanized in moribund condition on day 6.
Table 4.
Hematology summary for test animals measured on day 6
| Group and dose (mg/kg/day) | HCT (%) | HGB level (g/dl) | RBC count (106/μl) | RDW (%) | WBC count (103/μl) | PLC (103/μl) | PNS (%) |
|---|---|---|---|---|---|---|---|
| Vehicle,a 0 | 40.8 ± 1.09 | 13.5 ± 0.30 | 9.18 ± 0.265 | 14.8 ± 0.53 | 3.14 ± 0.434 | 857 ± 183.6 | 19.8 ± 4.32 |
| DB1955 | |||||||
| 100a | 40.0 ± 1.68 | 13.3 ± 0.44 | 8.94 ± 0.247 | 14.8 ± 1.21 | 3.62 ± 0.658 | 958 ± 76.2 | 16.9 ± 5.59 |
| 200a | 39.6 ± 3.42 | 13.3 ± 1.19 | 8.91 ± 0.778 | 14.4 ± 1.29 | 3.21 ± 1.417 | 832 ± 191.9 | 16.6 ± 3.45 |
| 500a | 37.6 ± 1.84 | 12.6 ± 0.31 | 8.46 ± 0.297 | 14.7 ± 0.75 | 3.92 ± 1.068 | 832 ± 69.2 | 15.5 ± 1.65 |
| DB1960 | |||||||
| 100a | 39.5 ± 1.76 | 13.1 ± 0.58 | 8.91 ± 0.317 | 15.2 ± 1.29 | 3.35 ± 0.551 | 727 ± 119.4 | 16.6 ± 5.13 |
| 200a | 36.7 ± 1.46c | 12.2 ± 0.41c | 8.29 ± 0.363 | 15.3 ± 1.80 | 2.06 ± 0.666 | 577 ± 61.8d | 26.4 ± 7.02 |
| 500b | 38.4 ± 2.23 | 12.7 ± 0.76 | 8.80 ± 0.551 | 15.1 ± 0.82 | 4.04 ± 1.079 | 604 ± 136.7d | 63.6 ± 7.60d |
The values are means ± standard deviations of experimental groups of five animals.
The values are means ± standard deviations of experimental groups of eight animals.
Significant difference from control (P < 0.05).
Significant difference from control (P < 0.01).
Clinical chemistry measurements are provided in Table 5. Changes in lactate dehydrogenase (LDH), aspartate aminotransferase (AST), alanine aminotransferase (ALT), and cholesterol (CHO) levels were statistically significant in the high-dose DB1960 group compared with those of the controls. DB1960 also produced a 5-fold increase in the blood urea nitrogen (BUN) level in the high-dose DB1960 group compared with that in the control group. Triglycerides (TRI) were decreased on day 6 in a dose-dependent manner in mice treated with either DB1955 or DB1960, and this decrease was statistically significant for mice in all DB1960 groups and those in the high-dose DB1955 groups. Statistically significant decreases in albumin (ALB) and the albumin/globulin ratio (AGR) were observed in the medium- and high-dose DB1960 groups, suggesting malnutrition possibly due to enteropathy. On day 12, the AST and ALT levels remained elevated in mice treated with high-dose DB1955, while other parameters for DB1960 and DB1955 appeared to return to control levels by day 12 (data not shown). As with the hematology parameters, clinical chemistry values were not available for mice in the high-dose DB1960 recovery group since these animals were euthanized earlier in the study.
Table 5.
Clinical chemistry summary for test animals measured on day 6
| Group and dose (mg/kg/day) | LDH (U/liter) | AST (U/liter) | ALT (U/liter) | TRI (mg/dl) | ALB (g/dl) | AGR | CHO (mg/dl) | BUN (mg/dl) |
|---|---|---|---|---|---|---|---|---|
| Vehiclea, 0 | 165 ± 45 | 70 ± 7.5 | 40 ± 12 | 181 ± 22.8 | 3.9 ± 0.17 | 4 ± 0.9 | 90 ± 4.6 | 21 ± 2.6 |
| DB1955 | ||||||||
| 100a | 155 ± 49.3 | 64 ± 9.4 | 35 ± 4.7 | 188 ± 52.8 | 3.8 ± 0.35 | 3 ± 1.1 | 99 ± 6.3 | 22 ± 1.9 |
| 200a | 441 ± 487.3 | 143 ± 166.9 | 86 ± 57.6 | 131 ± 47.3 | 3.6 ± 0.35 | 3 ± 0.4 | 101 ± 6.1 | 25 ± 4.4 |
| 500a | 202 ± 73.5 | 75 ± 30.6 | 41 ± 27.8 | 119 ± 36.6d | 3.6 ± 0.23 | 3 ± 0.4 | 114 ± 8.6 | 36 ± 7.7 |
| DB1960 | ||||||||
| 100a | 228 ± 90 | 91 ± 49.7 | 42 ± 11.1 | 92 ± 21.9e | 3.5 ± 0.15 | 3 ± 0.5 | 87 ± 6.4 | 22 ± 4.2 |
| 200a | 257 ± 34.6 | 163 ± 42.8 | 109 ± 38.1e | 64 ± 14.3e | 3.2 ± 0.25d | 2 ± 0.0e | 109 ± 8.6 | 29 ± 6.8 |
| 500b | 820 ± 422.6e | 364 ± 134.7e | 91 ± 24.7d | 55 ± 14.2c,e | 3.3 ± 0.41e | 2 ± 0.5e | 129 ± 25.7e | 108 ± 57.5e |
The values are means ± standard deviations of experimental groups of five animals.
The values are means ± standard deviations of experimental groups of eight animals.
The values are means ± standard deviations of experimental groups of seven animals.
Significant difference from control (P < 0.05).
Significant difference from control (P < 0.01).
Gross necropsy findings and organ weights.
No gross necropsy findings could be attributed to DB1955 treatment. At the main necropsy (day 6), tissue changes that were attributed to treatment with DB1960 included discolored dark or pale kidneys; a discolored dark, pale, yellow, or mottled liver; discolored dark or yellow lymph nodes; a discolored dark spleen; discolored orange or red stomach contents; a discolored dark and pale thymus; and a small thymus. These changes were observed mainly in mice treated with DB1960 at 500 mg/kg. At the recovery necropsy (day 12), tissues in all groups appeared normal. There was no recovery necropsy for mice in the high-dose DB1960 group since they were euthanized in moribund condition on day 6. Most organ weights appeared to be within control ranges (data not shown).
Histopathology.
At the main necropsy, treatment-related changes associated with DB1955 were present in the stomach, mesenteric lymph nodes, and bone marrow smears (cytology) at all three dose levels. Forestomach epithelial hyperplasia was present at all dose levels, and inflammation, ulceration, and hyperkeratosis were present in the high-dose group only. The bone marrow smears had an increased myeloid/erythroid cell ratio in the medium- and high-dose groups. Treatment-related changes associated with all dose levels of DB1960 at the main necropsy were present in the spleen, liver, kidneys, duodenum, jejunum, ileum, mesenteric lymph nodes, bone marrow smear, bone marrow histology (sternum section), thymus, and pancreas. The kidneys showed renal nephrosis, which was present in the medium- and high-dose DB1960-treated groups. Protein casts were present only in the high-dose group. The splenic changes at all dose levels were lymphoid depletion, hemosiderosis, and proliferation of extramedullary hematopoiesis (EMH). Proliferation of reticuloendothelial (RE) cells and phagocytized (cellular) debris in RE cells were present in the medium- and high-dose groups. Splenic lymphocyte necrosis and hemosiderosis were present in the high-dose group, and proliferation of RE cells was present in the medium- and high-dose groups. Liver fatty change accompanied by decreased hepatocytic cytoplasmic vacuolation (glycogen vacuolation) compared to controls was present in all dose groups. Proliferation of RE cells, phagocytosis of debris by RE cells, apoptosis, and hepatocytic necrosis were present in high-dose-group livers. Hepatic basophilic foci were present in medium- and high-dose groups.
At the recovery necropsy, treatment-related changes associated with DB1955 were observed in the spleen, stomach, bone marrow, and ileum. The forestomach in all dose groups had epithelial hyperplasia. Treatment-related changes associated with DB1960 at the recovery necropsy involved the spleen, liver, duodenum, bone marrow cytology, and bone marrow histology. The spleen had proliferation of EMH and hemosiderosis in the low- and medium-dose groups and phagocytized debris in RE cells in the medium-dose group. Proliferation of EMH was observed in the livers of the mice in the medium-dose group. The above-listed changes associated with the test compounds were not present in all of the mice in the various groups in which they were observed. Also, the severity of the changes varied between individual mice. No test substance-related changes were present in other organs at either sacrifice time.
Plasma compound levels.
Figure 5 shows the mean plasma DB1955 and DB1960 concentrations on days 1 and 5. Both compounds were rapidly absorbed, reaching their maximum concentrations 0.5 to 1 h after dose administration in most of the treatment groups. In two of the DB1955 groups, however, the highest mean concentration was reached later on day 5, at 4 h (100 mg/kg) and 6 h (200 mg/kg). DB1955 was cleared from plasma more rapidly than DB1960, and at 24 h after dose administration, only the 500-mg/kg dose group displayed compound levels greater than the LLOQ. Toxicokinetic parameter data on DB1955 and DB1960 are presented in Table S3 in the supplemental material. The t1/2 of DB1955 was 6.1 h and could be determined only in the 500-mg/kg dose group on day 5. The t1/2s of DB1960 were substantially longer at 18.4 h (100 mg/kg), 31.3 h (200 mg/kg), and 29.5 h (500 mg/kg). Exposure to DB1955 and DB1960, based on Cmax and AUC values, increased with dose but not in a linear fashion. AUC0-6 values were slightly higher for DB1960 than for DB1955. The AUC0-24 is about 3-fold higher for DB1960 than for DB1955 at a dose of 500 mg/kg.
Fig 5.

Plasma DB1955 and DB1960 levels measured after compound administration on days 1 (A and B) and 5 (C and D).
DISCUSSION
AIAs (formerly known as reversed amidines) were inspired by antimicrobial diamidines such as pentamidine, with the distinction that AIAs contain an imino group attached to an anilino nitrogen rather than directly attached to an aromatic ring as in diamidines (23). AIAs display outstanding in vitro activity against both intracellular Leishmania (8, 22, 27) and intracellular T. cruzi (12, 18, 19). The high potency of AIAs against these intracellular kinetoplastid parasites could be explained in part by their lower pKa values and higher lipophilicity than those of diamidines (27), which may permit enhanced penetration into intracellular pathogens. DB766, one of the most potent AIAs in vitro, also showed promising efficacy in a murine VL model when given orally at a dose of 100 mg/kg/day × 5 without displaying any obvious adverse effects in mice or hamsters (27). Higher doses could not be given due to the solubility of DB766. Similarly, DB766 exhibited strong trypanocidal activity and excellent selectivity for bloodstream trypomastigotes and intracellular amastigotes of T. cruzi strain Y, with IC50s of 60 and 25 nM, respectively, and displayed an efficacy similar to that of Bz in mouse models of T. cruzi infection employing Y and Colombian strains (3). To determine whether a higher dose and/or improved solubility could enhance the antileishmanial and anti-T. cruzi efficacy of AIAs and to permit the use of higher doses in toxicology studies aimed at determining the therapeutic windows of candidate compounds, the mesylate salt of DB766 (DB1960) was prepared and tested. When evaluated in the murine VL model at an oral dose of 200 mg/kg/day × 5, DB1960 nearly eradicated liver parasitemia (Fig. 2C). For T. cruzi infection, DB1960 was also active at a 100-mg/kg dose (Fig. 3B), resulting in a decrease in parasitemia levels (about 50%) and protection against death (about 35%). Although the mesylate salts were expected to possess greater efficacy than their corresponding hydrochloride salts, a comparison of the in vivo antikinetoplastid efficacy and PK properties of the hydrochloride salt DB766 and its corresponding mesylate, DB1960, indicates that DB766 possesses antiparasitic properties superior to those of DB1960. When given at an oral dose of 100 mg/kg/day for 5 days, DB766 inhibits liver parasitemia in L. donovani-infected mice by 71% (27) while DB1960 displays 51% inhibition in this model at the same dose (Fig. 2). Also, the maximum concentration of DB766 in the liver after a single 100-μmol/kg dose in mice (at which dose DB766 formed a viscous solution) was 45.8 μM (28), much higher than the level of DB1960 found in the liver after the same dose of this mesylate salt (Fig. 4B). We speculate that the more soluble mesylate salt DB1960 may have a shorter gastrointestinal (GI) tract transit time than the more viscous solution of hydrochloride salt DB766, permitting greater absorption of the latter compound.
As shown in Fig. 4, DB1955 and DB1960 accumulate to high concentrations in the liver and spleen, organs that harbor infection during VL. Considering that these agents achieve concentrations in infected organs that are 2 orders of magnitude higher than their IC50s in the intracellular Leishmania assays, it is surprising that liver parasitemia is not cleared in our murine VL model. This observation is particularly puzzling for DB1960, which displays an in vitro IC90 similar to its IC50. In VL, Leishmania parasites infect Kupffer cells, the resident macrophages of the liver. Kupffer cells are estimated to account for only about 2% of the liver tissue volume, with hepatocytes comprising almost 80% (14). Since the distribution of the AIAs within infected liver tissue is not known, it is conceivable that the compounds do not reach the necessary location in sufficient levels to cure parasite infections. Further work is required to test this hypothesis.
Although the in vivo efficacies of DB1955 and DB1960 are similar in the murine VL model (Fig. 2B and C), the same was not observed for T. cruzi infection, given that DB1955 was inactive in vivo despite possessing good activity in vitro (Fig. 3 and Table 2). This could be related to the lower levels of DB1955 in the heart, which is an important target of parasite infection and inflammation (7, 21). However, DB1960 exhibited greater toxicity than DB1955 in mice, as illustrated by the following data. Statistically significant changes in ALT levels were observed in the medium-dose DB1960 group, and significant changes in BUN, ALT, AST, and LDH levels were measured in the medium- and high-dose DB1960 groups (Table 5). Such changes, which are indicative of adverse effects on the kidneys and liver, were not found in any of the DB1955 groups. At the main necropsy, discolorations in the livers, kidneys, lymph nodes, spleens, and stomachs of the high-dose DB1960 group, but not the high-dose DB1955 group, were observed. Body weights of the medium- and high-dose DB1960 groups were decreased compared to those of controls at the main necropsy, but only the high-dose DB1955 group exhibited a decrease in body weight. DB1960 groups, in general, displayed a higher level of histopathological changes than the corresponding DB1955 groups. Most importantly, the animals in the high-dose DB1960 recovery group had to be euthanized in moribund condition due to numerous adverse effects. In general, the adverse effects of DB1960 on hematology, clinical chemistry, and the liver and kidneys appeared to be higher than those observed for DB1955. This could be due to the following factors. (i) DB1955 was cleared more rapidly from the plasma, as the half-life of this compound (6.1 h) was 5-fold shorter than that of DB1960 (29.5 h) in the 500-mg/kg group (Fig. 5; see Table S3 in the supplemental material). (ii) Exposure levels of DB1960 were higher than those of DB1955. The AUC0-6 value of DB1960 was slightly higher (27.09 h·μg/ml) than that of DB1955 (17.37 h·μg/ml), and the AUC0-24 value of DB1960 was about 3-fold higher (99.15 h·μg/ml) than that of DB1955 (36.81 h·μg/ml) at the 500-mg/kg dose (see Table S3 in the supplemental material). (iii) Metabolites were formed in animals treated with DB1960. Three potential metabolites were detected in day 5 plasma samples at all time points in the 500-mg/kg DB1960 dose group, while no metabolite peaks were detected in plasma collected from animals that received DB1955 (data not shown). It is possible that one or more of these DB1960 metabolites are partially responsible for the toxicity of this compound. However, since individual metabolites were not identified and evaluated for toxicity as part of this study, this cannot be conclusively determined.
Based on the data presented here and given that irreversible microscopic findings were also present in the GI tracts of the mice in the low-dose treatment groups, the MTD of both DB1955 and DB1960 is estimated to be below 100 mg/kg/day. In this study, the NOAEL could not be determined in the 5-day repeat dose experiment because adverse effects were seen at the lowest dose level (100 mg/kg). Further development of these compounds has therefore been discontinued due to the lack of a therapeutic window for existing AIAs. Nonetheless, the AIAs remain among the most active classes of compounds against intracellular Leishmania and T. cruzi. Our efforts to improve the efficacy and reduce the toxicity of AIAs have resulted in the synthesis and testing of additional AIAs containing shorter linkers (unpublished data) and AIAs containing one AIA group (mono-AIAs), as opposed to the two AIA groups found in DB1955, DB1960, and other AIAs reported thus far (8, 22). Within the class of AIAs containing shorter linkers, the active compounds are toxic, with administration to mice resulting in acute, immediate toxicity (unpublished data), unlike the compounds examined here. However, none of the mono-AIA compounds possessing antikinetoplastid activity that have been tested in vivo to this point have exhibited overt toxicity to mice (X. Zhu and T. Pandharkar, unpublished data). A better understanding of the mechanisms of the antileishmanial and trypanocidal actions of the AIAs along with the determination of their mammalian toxicity targets could provide valuable clues for the design of new, selective antileishmanial and anti-T. cruzi drugs. Further studies regarding the antikinetoplastid activities and mechanisms of action of AIAs will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Bill and Melinda Gates Foundation, contract N01-AI-60011 with SRI International from the National Institute of Allergy and Infectious Diseases, FIOCRUZ, and by Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ) PPSUS, APQ1, and Pensa-Rio (16/2009-E-26/110-313/2010), Conselho Nacional Desenvolvimento científico e Tecnológico (CNPq), PDTIS/FIOCRUZ, and PROEP.
We thank the other members of the Consortium for Parasitic Drug Development for helpful discussions.
Footnotes
Published ahead of print 16 April 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Akay S. 2009. Diagnosis and inhibition tools in medicinal chemistry. Ph.D. dissertation Georgia State University, Atlanta, GA [Google Scholar]
- 2. Apt W. 2010. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des. Devel. Ther. 4:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Batista D, et al. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas' disease treatment. Antimicrob. Agents Chemother. 54:2940–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berman J. 2005. Miltefosine to treat leishmaniasis. Expert Opin. Pharmacother. 6:1381–1388 [DOI] [PubMed] [Google Scholar]
- 5. Berman J, et al. 1998. Efficacy and safety of liposomal amphotericin B (AmBisome) for visceral leishmaniasis in endemic developing countries. Bull. W.H.O. 76:25–32 [PMC free article] [PubMed] [Google Scholar]
- 6. Buckner F, Navabi N. 2010. Advances in Chagas disease drug development: 2009-2010. Curr. Opin. Infect. Dis. 23:609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clayton J. 2010. Chagas disease: pushing through the pipeline. Nature 465:S12–S15 [DOI] [PubMed] [Google Scholar]
- 8. Collar C, Zhu X, Werbovetz K, Boykin D, Wilson W. 2011. Governing inhibition of arylimidamides against Leishmania: conservative computational modeling to improve chemotherapies. Bioorg. Med. Chem. 19:4552–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coura J, Viñas P. 2010. Chagas disease: a new worldwide challenge. Nature 465:S6–S7 [DOI] [PubMed] [Google Scholar]
- 10. Daliry A, et al. 2011. The trypanocidal activity of amidine compounds does not correlate with their binding affinity to Trypanosoma cruzi kinetoplast DNA. Antimicrob. Agents Chemother. 55:4765–4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Da Silva C, et al. 2011. The efficacy of novel arylimidamides against Trypanosoma cruzi in vitro. Parasitology 138:1863–1869 [DOI] [PubMed] [Google Scholar]
- 12. Da Silva C, et al. 2011. In vitro trypanocidal activity of DB745B and other novel arylimidamides against Trypanosoma cruzi. J. Antimicrob. Chemother. 66:1295–1297 [DOI] [PubMed] [Google Scholar]
- 13. Delfín DA, Morgan RE, Zhu X, Werbovetz KA. 2009. Redox-active dinitrophenylthioethers against Leishmania: synthesis, structure-activity relationships and mechanism of action studies. Bioorg. Med. Chem. 17:820–829 [DOI] [PubMed] [Google Scholar]
- 14. Gumucio J, Berkovitz C, Webster S, Thornton A. 1996. Structural and functional organization of the liver, p 3–19 In Kaplowitz N. (ed), Liver and biliary diseases, 2nd edition Williams & Wilkins, Baltimore, MD [Google Scholar]
- 15. Hailu A, et al. 2010. Geographical variation in the response of visceral leishmaniasis to paromomycin in East Africa: a multicentre, open-label, randomized trial. PLoS Negl. Trop. Dis. 4:e709 doi:10.1371/journal.pntd.0000709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mueller M, et al. 2007. Unresponsiveness to AmBisome in some Sudanese patients with kala-azar. Trans. R. Soc. Trop. Med. Hyg. 101:19–24 [DOI] [PubMed] [Google Scholar]
- 17. Seaman J, et al. 1995. Liposomal amphotericin B (AmBisome) in the treatment of complicated kala-azar under field conditions. Clin. Infect. Dis. 21:188–193 [DOI] [PubMed] [Google Scholar]
- 18. Silva C, et al. 2007. Activity of “reversed” diamidines against Trypanosoma cruzi in vitro. Biochem. Pharmacol. 73:1939–1946 [DOI] [PubMed] [Google Scholar]
- 19. Silva C, et al. 2007. Cellular effects of reversed amidines on Trypanosoma cruzi. Antimicrob. Agents Chemother. 51:3803–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sindermann H, Engle J. 2006. Development of miltefosine as an oral treatment for leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 100(Suppl 1):S17–S20 [DOI] [PubMed] [Google Scholar]
- 21. Soeiro M, De Castro S. 2011. Screening of potential anti-Trypanosoma cruzi candidates: in vitro and in vivo studies. Open Med. Chem. J. 5:21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stephens C, et al. 2003. The activity of diguanidino and “reversed” diamidino 2,5-diarylfurans versus Trypanosoma cruzi and Leishmania donovani. Bioorg. Med. Chem. Lett. 13:2065–2069 [DOI] [PubMed] [Google Scholar]
- 23. Stephens C, et al. 2001. Diguanidino and “reversed” diamidino 2,5-diarylfurans as antimicrobial agents. J. Med. Chem. 44:1741–1748 [DOI] [PubMed] [Google Scholar]
- 24. Sundar S, Chakravarty J, Agarwal D, Rai M, Murray H. 2010. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. N. Engl. J. Med. 362:504–512 [DOI] [PubMed] [Google Scholar]
- 25. Sundar S, Jha T, Thakur C, Sinha P, Bhattacharya S. 2007. Injectable paromomycin for visceral leishmaniasis in India. N. Engl. J. Med. 356:2571–2581 [DOI] [PubMed] [Google Scholar]
- 26. Sundar S, Murray H. 2005. Availability of miltefosine for the treatment of kala-azar in India. Bull. W.H.O. 83:394–395 [PMC free article] [PubMed] [Google Scholar]
- 27. Wang M, et al. 2010. Novel arylimidamides for the treatment of visceral leishmaniasis. Antimicrob. Agents Chemother. 54:2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou T, et al. 2010. Anti-AIDS agents 79. Design, synthesis, molecular modeling and structure-activity relationships of novel dicamphanoyl-2′,2′-dimethyldihydropyranochromone (DCP) analogs as potent anti-HIV agents. Bioorg. Med. Chem. 18:6678–6689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu X, Pandharkar T, Werbovetz K. 2012. Identification of new antileishmanial leads from hits obtained by high-throughput screening. Antimicrob. Agents Chemother. 56:1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.