

Abstract
The nonimmunosuppressive cyclophilin (Cyp) inhibitor SCY-635 blocks hepatitis C virus (HCV) replication both in vitro and in vivo and represents a novel potent anti-HCV agent. However, its mechanism of action remains to be fully elucidated. A growing body of evidence suggests that cyclophilin A (CypA) is absolutely necessary for HCV replication and that the HCV nonstructural 5A (NS5A) protein serves as a main viral ligand for CypA. In this study, we examined the effect of SCY-635 on HCV replication. Specifically, we asked whether SCY-635 blocks HCV replication by targeting CypA-NS5A interactions. We also investigated the possibility that HCV can escape SCY-635 selection pressure and whether this resistance influences either CypA-NS5A interactions or the dependence of HCV on CypA. We found not only that SCY-635 efficiently inhibits HCV replication, but it is sufficient alone to clear HCV replicon-containing cells. We found that SCY-635 prevents CypA-NS5A interactions in a dose-dependent manner. SCY-635 prevents the contact between CypA and NS5A derived from genotypes 1 to 3. Together, these data suggest that NS5A-CypA interactions control HCV replication and that SCY-635 blocks viral replication by preventing the formation of these complexes. We also found that NS5A mutant proteins found in SCY-635-resistant HCV replicons behave similarly to wild-type NS5A in terms of both CypA binding and SCY-635-mediated dissociation and inhibition of CypA binding. However, the NS5A mutations found in SCY-635-resistant HCV replicons rescued viral replication in CypA-knockdown cells, suggesting that the NS5A mutations, which arose in vitro under SCY-635 selection, do not alter the binding affinity of CypA for NS5A. These specific mutations in NS5A eliminate the dependence of HCV RNA replication on the expression of host CypA
INTRODUCTION
Hepatitis C virus (HCV) is the major causative agent of acute and chronic liver diseases (1). The primary infection in humans is associated with no or mild symptoms, whereas individuals who are chronically infected are at high risk for chronic liver diseases—e.g., hepatocellular carcinoma and cirrhosis (8). Nearly 200 million people worldwide (3% of the population), including 4 to 5 million in the United States, are chronically infected with HCV, and 4 million new infections occur globally every year (1, 43). In the developed world, HCV accounts for two-thirds of all cases of liver cancer and transplants (41), and in the United States, an estimated 12,000 people die each year from end-stage liver disease associated with chronic HCV infection (2).
The 2011 regulatory approvals of the protease inhibitors Victrelis (boceprevir) and Incivek (telaprevir) represent a major milestone in the treatment of HCV. The addition of boceprevir or telaprevir to PEGylated alpha interferon (pIFN-α) and ribavarin (pIFN-α/RBV) will certainly improve cure rates and shorten the treatment duration of the earlier pIFN-α/RBV standard of care (SOC). However, it is likely that this triple therapy may still not be sufficient for patients who are intolerant to pIFN-α or RBV or for those who do not respond to pIFN-α/RBV. Because boceprevir is inactive against genotype 3 (GT3) (3, 13; J. Pawlotsky et al., presented at the 62nd Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, 2011) combined with the fact that all protease inhibitors and most of the nonnucleoside polymerase inhibitors in development are primarily active against GT1 (3, 37), pIFN-α/RBV will remain the SOC for non-GT1 until new classes of inhibitors enter clinical practice. GT1 patients who do not respond to the new triple therapy risk developing resistance to protease inhibitors that may limit future retreatment options. Thus, there remains an important need for the identification of additional anti-HCV agents.
A novel class of HCV inhibitors that has great potential for the treatment of HCV has recently emerged: the cyclophilin (Cyp) inhibitors (11, 16, 30, 36, 38, 45, 46). To date, three Cyp inhibitors—alisporivir, NIM-811, and SCY-635—have demonstrated safety and efficacy in patients with HCV in phase I and II studies (12, 23, 29; R. Flisiak et al., presented at the 43rd Annual Meeting of the European Association for the Study of the Liver, Milan, Italy, 2008; R. Flisiak et al., presented at the 46th Annual Meeting of the European Association for the Study of the Liver, Berlin, Germany, 2011; J. Pawlotsky et al., presented at the 62nd Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, 2011). SCY-635 is a nonimmunosuppressive Cyp inhibitor derived from cyclosporine (CsA) (24). SCY-635 has potent anti-HCV activity in vitro and binds Cyp with high affinity (24). SCY-635 was evaluated in a 15-day, phase I, randomized, double-blind, placebo-controlled, multidose study in GT1 patients (23). Participants were enrolled into one of 3 ascending-dose cohorts, receiving daily oral doses of 300, 600, or 900 mg of SCY-635 for 15 consecutive days. In the 900-mg cohort, all treated patients experienced a reduction in HCV viral load, with a group mean maximum decrease of 2.2 log10 on day 15 (23), demonstrating that SCY-635 has potent in vivo anti-HCV activity.
Although the in vitro and in vivo potencies of SCY-635 against HCV are accepted, its mechanisms of action remain to be fully understood. The main intracellular targets for CsA and derivatives such as SCY-635 are the Cyps (24). In 2003, Watashi et al. provided the first link between HCV and Cyps by showing that CsA and NIM811 suppress HCV replication in vitro (48). This important finding that CsA and derivatives block HCV replication was subsequently confirmed by several laboratories (10, 18, 25, 26, 32, 33, 35, 40). Supporting the notion that Cyps play a role in HCV replication, three independent studies clearly demonstrated using a stable knockdown approach that CypA is the major Cyp member vital for HCV replication (5, 28, 50). More recent studies showed that the peptidyl-prolyl isomerase (PPIase) activity of CypA is critical for HCV replication (5, 28, 31). Importantly, NS5A was found to be the major viral ligand for CypA. Specifically, several studies showed that CypA binds NS5A directly (6, 7, 9, 14, 21, 44, 47, 49). CypA binds to proline-rich regions in domains II and III of NS5A (7, 21, 44, 49). Importantly, CsA inhibits CypA-NS5A interactions in a dose-dependent manner (6, 9, 14, 21, 44, 47, 49). Altogether, these data suggest that CypA-NS5A interactions are important for HCV replication and that drug-mediated dissociation of the CypA-NS5A complex may account for the antiviral activity associated with the Cyp inhibitor class of compounds.
In this study, we examined the effect of the Cyp inhibitor SCY-635 on HCV replication and HCV clearance. Specifically, we asked whether SCY-635 blocks HCV replication by targeting CypA-NS5A interactions. We also investigated the possibility that HCV can escape SCY-635 selection pressure and whether this resistance influences either CypA-NS5A interactions or the dependence of HCV on CypA.
MATERIALS AND METHODS
HCV RNA replication and infection.
Ten micrograms of in vitro-transcribed genomic JFH-1 or Con1 RNA was electroporated into parental or CypA-knockdown (CypA-KD) Huh7.5.1 cells. The establishment of the CypA-KD cells was described previously (5). At the indicated time points, intracellular HCV RNA was analyzed via reverse transcription-quantitative PCR (RT-qPCR); the results are presented as genome equivalents (GE) per microgram of total RNA as described previously (27). After 7 days, cells electroporated with JFH-1 were fixed and stained with anti-NS5A human IgG (E1) (1:1,000) for 2 h at room temperature. After 3 washes, cells were incubated for 1 h at room temperature with secondary Alexa Fluor 555-conjugated goat anti-human IgG (1:1,000). Cells were washed 3 times; cells and plates were stored in the dark at 4°C until pictures were taken. The number of HCV-positive foci was counted by fluorescence microscopy. SCY-635 diluted in dimethyl sulfoxide (DMSO) was added to cells at the time of the cell plating, immediately after the electroporation.
HCV replicon clearance and rebound.
Huh7 cells expressing the subgenomic Con1 (GT1b) replicon were seeded in a 10-cm dish at a density of 3 × 105 cells per culture flask in complete Dulbecco's modified Eagle's medium (DMEM) (without G418) with or without SCY-635. Cells were grown until they reached 90% confluence, trypsinized, and reseeded at the same cell concentration in a new 10-cm dish with or without the same concentration of SCY-635. Cells (1.5 × 105) from each dish were lysed in RLT buffer (RNeasy minikit; Qiagen) and stored at −80°C until further use. Cells were passaged 8 times in the presence or the absence of SCY-635. After all samples had been collected, the RNA was extracted according to the manufacturer's instructions and the samples were analyzed by RT-PCR for replicon content. Telaprevir was obtained from Selleck Chemicals.
Development of SCY-635-resistant HCV replicons.
Huh7 cells expressing the subgenomic Con1 (GT1b) replicon were incubated with 0.5 μM SCY-635 (5-fold excess of SCY-635; 50% effective concentration [EC50], 100 nM) and 100 μg/ml of G418 in 10-cm CellBIND plates (Corning). Cells were split every 3 days at 1:6, and the G418 concentration was increased sequentially to 200 and 300 μg/ml. Most of the cells were dying 3 weeks after drug incubation. At this point, medium was changed every 3 to 4 days until the emergence of SCY-635-resistant clones. Clones were individually transferred to 24-well CellBIND plates using cloning disks. Each clone was split into 2 identical plates and was separately maintained under 0.5 or 1.0 μM SCY-635 and 100 μg/ml of G418. Once the colonies were established, the G418 concentration was increased to 300 μg/ml. Clones resistant to 1 μM SCY-635 were further screened for resistance to 2 μM SCY-635. Total RNA was prepared using the RNeasy Plus minikit as per the manufacturer's recommendation. Two hundred nanograms of total RNA was used for cDNA production using primers specific to NS4B, NS5B, or oligo(dT)18 using the AccuScript high-fidelity 1st strand cDNA synthesis kit (Stratagene). NS5A/B-specific amplicons were generated using primers defining the sequence at its 5′ and 3′ ends. The amplicons were gel purified, quantitated, and sequenced using primers located internally in NS5A or NS5B reading to the 5′ and 3′ ends. Sequences were assembled and analyzed using the ClustalW algorithm within the MacVector program.
Production of recombinant Cyp and NS5A proteins.
Recombinant glutathione S-transferase (GST)-CypA, GST-CypB, and full-length NS5A from various genotypes (pET-Ub-NS5A Con1-His) were produced and purified as described previously (6). NS5A mutants were created by PCR mutagenesis. The NS5A genes from genotype 1a (H77), 1b (Con1), 2a (JFH-1), and 2b (MD2b-1) were cloned and expressed as described previously (6).
Cyp-NS5A pull-down studies.
Glutathione beads were incubated for 2 h in dialysis buffer (50 mM Tris [pH 7.4], 100 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.5% NP-40, 1 mM dithiothreitol [DTT]) with 5 mg/ml bovine serum albumin (BSA) and washed twice at 4°C in binding buffer (20 mM Tris [pH 7.9], 0.5 M NaCl, 10% glycerol, 10 mM DTT, and 1% NP-40). Meanwhile, 1 μg of GST-Cyp or GST was mixed with 200 ng of NS5A-His in a total volume of 200 μl of binding buffer for 3 h at 4°C with mixing on a wheel rotator. Glutathione beads (25 μl) were added to the GST-Cyp–NS5A mixture for 30 min at 4°C and washed 3 times with 400 μl of binding buffer. Beads were pelleted for 30 s at 2,000 × g in a microcentrifuge, and bound material was eluted with 25 μl of 2× SDS sample buffer, heated for 5 min, and frozen at −20°C. Bound material was then analyzed by Western blotting using anti-GST, anti-Cyp, and anti-His antibodies as described previously (6).
Cyp-NS5A ELISA.
For the Cyp-NS5A enzyme-linked immunosorbent assay (ELISA), Nunc MaxiSorb 8-well strip plates were coated with GST or GST-Cyp for 16 h at 4°C and blocked as we described previously (6). Recombinant NS5A-His (1 ng/ml) was added to wells in 50 μl of binding buffer (20 mM Tris [pH 7.9], 0.5 M NaCl, 10% glycerol, 10 mM DTT, and 1% NP-40) for 16 h at 4°C. Captured NS5A-His was subsequently detected using mouse anti-His antibodies (1 μg/ml) (anti-6×His; Clontech) and rabbit anti-mouse horseradish peroxidase phosphatase (HRP)-conjugated antibodies (1:1,000 dilution) as we described previously (6).
RESULTS
SCY-635 blocks HCV RNA replication and infection.
We analyzed the effect of SCY-635 on HCV RNA replication and infection. Specifically, in vitro-transcribed subgenomic Con1 (genotype 1b) or genomic JFH-1 (genotype 2a) RNA was electroporated into parental Huh-7 cells in the presence or absence of increasing concentrations of SCY-635 (0.3125, 0.625, 1.25, and 2.5 μM). At the indicated time points, intracellular viral RNA replication was analyzed via reverse transcription-quantitative PCR; the results are presented as genome equivalents (GE) per microgram of total RNA. HCV RNA replication was profoundly reduced in the presence of SCY-635 (Fig. 1A). We found that 0.625 μM SCY-635 completely inhibited HCV RNA replication in Huh7 cells 6 to 8 days after initiation of drug treatment (Fig. 1A). After demonstrating that SCY-635 inhibits replication of both Con1 and JFH-1 RNAs, we examined its effect on JFH-1 infection. Specifically, 7 days postelectroporation, JFH-1 infectivity was quantified by indirect immunofluorescence using anti-NS5A IgG. Results are expressed as focus-forming units (FFU) per milliliter of supernatant, determined by the average number of NS5A-positive foci detected at the highest dilutions. We found that SCY-635 inhibits in a dose-dependent manner JFH-1 infection of Huh7 cells (Fig. 1B). Figure 1C shows typical positive focus immunostainings of Huh7 cells infected with JFH-1 in the presence or absence of SCY-635. These data further demonstrated that the Cyp inhibitor SCY-635 inhibits efficiently HCV RNA replication as well as HCV infection.
Fig 1.
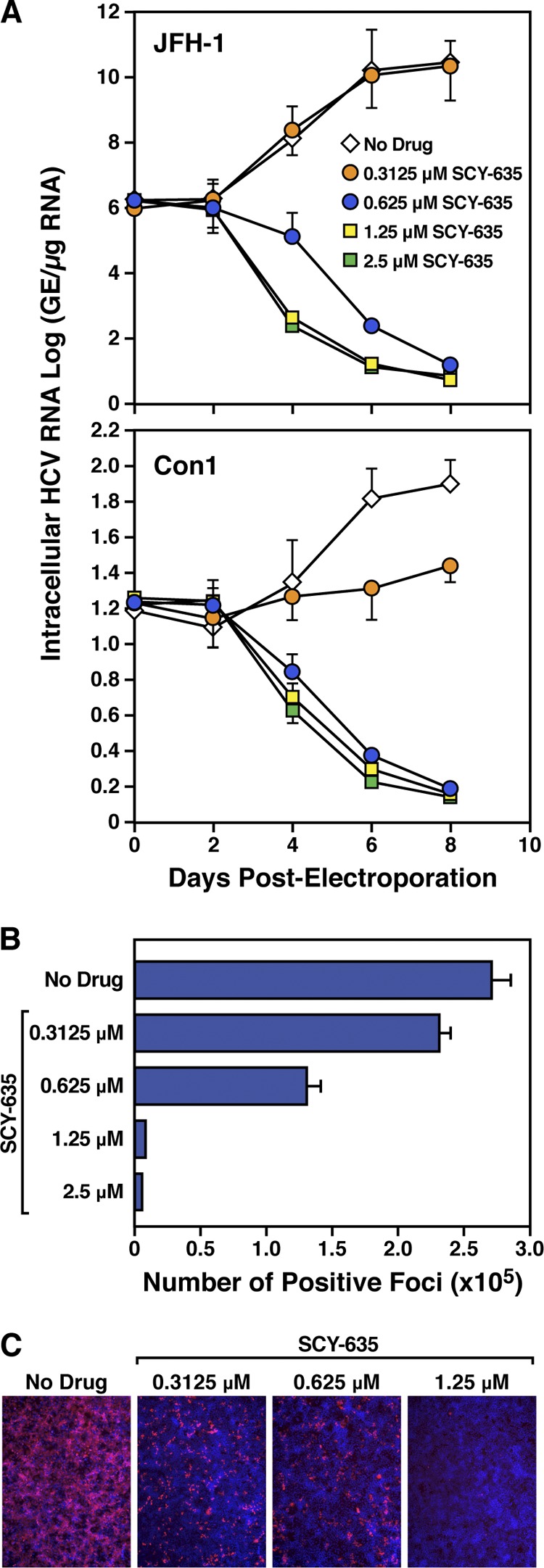
(A) Ten micrograms of in vitro-transcribed subgenomic Con1 or genomic JFH-1 or RNA was electroporated into Huh7 cells in the presence of increasing concentrations of SCY-635 (0.3125, 0.625, 1.25, and 2.5 μM). At the indicated time points, intracellular HCV RNA was analyzed via reverse transcription-quantitative PCR; the results are presented as genome equivalents (GE) per microgram of total RNA. (B) After day 7, JFH-1-electroporated cells were fixed and stained with anti-NS5A human IgG (E1) (1:1,000) for 2 h at room temperature. After 3 washes, cells were incubated for 1 h at room temperature with secondary Alexa Fluor 555-conjugated goat anti-human IgG (1:1,000). Cells were washed 3 times; cells and plates were stored in the dark at 4°C until pictures were taken. The number of JFH-1-positive foci was counted by fluorescence microscopy. Panel C is the same as panel B, except that typical fluorescence microscopy immunostainings of JFH-1-positive foci are presented. These results are representative of 2 independent experiments. Error bars represent standard errors of triplicate determinations.
SCY-635 clears HCV replicon-containing cells.
We then analyzed the possibility that SCY-635 clears HCV from hepatoma cells harboring the Con1 replicon (GT1b) and prevents its rebound upon drug removal. Specifically, Huh7-Con1 cells were passaged eight consecutive times without G418 in the presence or absence of two concentrations of CsA or SCY-635 (0.5 and 1 μM). We used telaprevir as a control. At each passage, five clones were collected and RNA was extracted and analyzed by RT-qPCR for replicon content. As expected, both CsA and SCY-635 drastically reduced HCV replication (Fig. 2). Under drug selection, the presence of viral RNA was dramatically diminished already after the first passage, and it remained undetectable throughout the eight consecutive passages (Fig. 2). After eight passages, drugs were removed and cells were analyzed for viral RNA content by qPCR to determine if a viral rebound occurs or not. Importantly, no viral rebound was observed when the virus was placed under SCY-635 selection pressure (Fig. 2). In contrast, rapid viral rebound occurred when HCV was initially placed under CsA pressure (Fig. 2). These data suggest that SCY-635 has the potency to clear hepatoma cells from their HCV replicon even when used alone.
Fig 2.
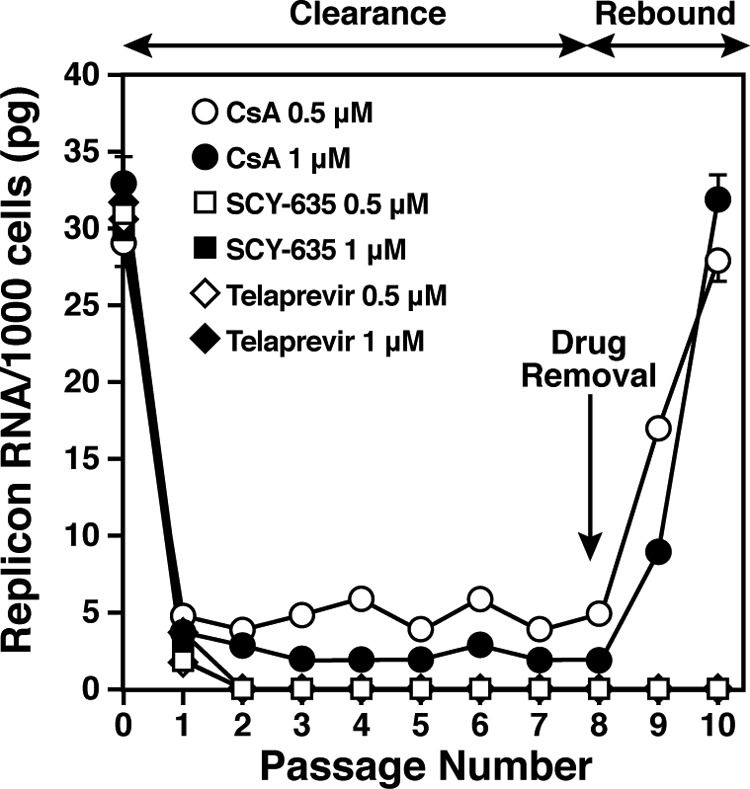
Huh7-Con1 cells were passaged eight consecutive times without G418 in the presence or absence of two concentrations of CsA, SCY-635, or telaprevir (0.5 and 1 μM). At each passage, five clones were collected, and RNA was extracted and analyzed by RT-qPCR for replicon content. After eight passages, drugs were removed and cells were analyzed for viral RNA content by qPCR for two additional passages. Data are representative of two independent experiments. Error bars represent standard errors of duplicate determinations.
SCY-635 disrupts NS5A-CypA interactions.
We and others demonstrated that HCV highly relies on CypA to replicate in hepatocytes and that the enzymatic active pocket of CypA is absolutely required for HCV replication (5, 28, 31, 50). Moreover, we and others showed that CypA binds NS5A directly and that the enzymatic active pocket of CypA contains the NS5A-binding site (5, 9, 31). Together, these findings suggested that there is a direct correlation between CypA assistance to HCV replication, CypA expression, and CypA binding to NS5A. One can envision that CypA, by binding to NS5A, enhances or triggers an activity of NS5A that is vital to HCV replication. If this model is correct, one could envision that SCY-635, which binds to the hydrophobic pocket of CypA, would interfere with the contact between CypA and NS5A. We thus tested this hypothesis, by examining the effect of SCY-635 on CypA-NS5A interactions using a GST-CypA capture assay recently developed in our laboratory (6). Briefly, recombinant GST-CypA and full-length NS5A Con1 were produced and purified as previously described (6). Glutathione beads were blocked for 2 h with BSA (5 mg/ml) and washed twice. Meanwhile, GST-CypA was incubated with NS5A-His for 3 h at 4°C. Glutathione beads were then added to the GST-CypA–NS5A mixture for 30 min at 4°C and washed 3 times. Bound material was eluted and analyzed by Western blotting using anti-His or anti-GST antibodies. Since previous work suggested that NS5A binds to CypB as well (21), we also examined the effect of SCY-635 on GST-CypB–NS5A interactions.
In the absence of SCY-635, NS5A binds efficiently GST-CypA (Fig. 3A, left panels). Importantly, SCY-635 inhibits the CypA-NS5A interaction in a dose-dependent manner (Fig. 3A, left panels). This suggests that SCY-635, by binding to the hydrophobic active pocket of CypA, prevents the contact between viral NS5A and host CypA. We also found that GST-CypB, like GST-CypA, very efficiently captures NS5A in the absence of SCY-635 (Fig. 3A, right panels). This is in accordance with previous studies that showed that both recombinant CypA and CypB proteins bind to recombinant NS5A (9, 21). Importantly, as demonstrated for GST-CypA, SCY-635 inhibits GST-CypB–NS5A interactions in a dose-dependent manner (Fig. 3A, right panels).
Fig 3.

(A) GST-CypA or GST-CypB (1 μg) was mixed with Con1 NS5A-His (200 ng) for 3 h at 4°C in the presence or absence of increasing concentrations of SCY-635. Glutathione beads were added to the GST-Cyp–NS5A mixture for 30 min at 4°C and washed. Bound material was eluted and analyzed by Western blotting using anti-His and anti-GST antibodies. (B) Plates were coated with GST-CypA or GST-CypB (10 μg/ml) for 16 h at 4°C. Recombinant Con1 NS5A-His (1 ng/ml) was added to wells for 16 h at 4°C. Captured NS5A-His was detected using mouse anti-His antibodies (1 μg/ml) followed by anti-mouse HRP-conjugated antibodies (1:1,000 dilution). Adsorbed levels of GST-CypA or GST-CypB were monitored using mouse anti-GST IgG (1 μg/ml) and rabbit anti-CypA or mouse anti-CypB antibodies (1 μg/ml) followed by anti-mouse and anti-rabbit HRP-conjugated antibodies (1:1,000 dilution). These results are representative of two independent experiments. Error bars represent standard errors of triplicate determinations.
To further confirm that SCY-635 prevents NS5A-CypA and NS5A-CypB complex formation, we used a different binding assay (6). Briefly, recombinant GST-CypA or GST-CypB proteins were immobilized onto 8-well-strip plates for 16 h at 4°C. Wells were blocked with BSA for 16 h at 4°C. Recombinant NS5A-His was added to wells for 16 h at 4°C. Captured NS5A-His was subsequently detected using mouse anti-His antibodies. Similar to the GST-CypA pulldown assay (Fig. 3A), we found that SCY-635 inhibits in a dose-dependent manner the binding of NS5A to immobilized GST-CypA (Fig. 3B, top panels) or GST-CypB (Fig. 3B, bottom panels). Thus, both pulldown and ELISA data reveal that there is a direct correlation between the SCY-635-mediated inhibition of HCV replication (Fig. 1) and inhibition of CypA-NS5A and/or CypB-NS5B interactions (Fig. 3). We verified that similar amounts of GST-CypA and GST-CypB were immobilized onto plates using anti-GST and anti-Cyp antibodies (Fig. 3B). Our demonstration that SCY-635 blocks CypA-NS5A interactions in a dose-dependent manner, further suggests that the interaction is specific. Our observation that SCY-635 blocks the CypA-NS5A interaction (Fig. 3) is perfectly in accordance with our previous finding that the enzymatic hydrophobic pocket of CypA, where Cyp inhibitors bind, is vital for both HCV replication (5) and NS5A binding (6).
SCY-635 disrupts the contact between CypA and NS5A derived from various HCV genotypes.
We demonstrated that SCY-635 efficiently and specifically disrupts the contact between CypA and NS5A derived from the HCV Con1 strain, which belongs to genotype 1b (GT1b). We then asked whether SCY-635 disrupts the contact between CypA and NS5A derived from other genotypes, such as H77 (genotype 1a), JFH-1 (genotype 2a), and MD2b-1 (genotype 2b). To address this issue, we cloned NS5A from H77, JFH-1, and MD2b-1 into the bacterial expression pET-Ub vector. The four recombinant NS5A H77, Con1, JFH-1, and MD2b-1 proteins were expressed, purified, and tested for GST-CypA binding as described above. All four 1a, 1b, 2a, and 2b NS5A proteins efficiently bound CypA in the absence of SCY-635 (Fig. 4A), further suggesting that CypA-NS5A interactions are conserved among various HCV genotypes. No significant difference in CypA binding was observed between the NS5A proteins derived from the different genotypes (Fig. 4A). Importantly, SCY-635 prevents all CypA-NS5A interactions in a dose-dependent manner (Fig. 4A).
Fig 4.

(A) Same as Fig. 2A, except that GST-CypA (1 μg) was mixed with recombinant NS5A-His (200 ng) derived from genotypes 1a, 1b, 2a, and 2b with increasing concentrations of SCY-635 (from 0.3125 to 0.625 μM) for 3 h at 4°C. Glutathione beads were added to the GST-CypA–NS5A mixture for 30 min at 4°C and washed. Bound material was eluted and analyzed by Western blotting using anti-His and anti-GST antibodies. (B) Same as Fig. 2B. These results are representative of two independent experiments. (C) To calculate SCY-635 and CsA IC50s, NS5A-His (1 ng/ml) was added to wells in the presence of increasing concentrations of SCY-635 or CsA (0.078, 0.156, 0.3125, 0.625, 1.25, and 2.5 μM) for 16 h at 4°C. Captured NS5A-His was subsequently detected using mouse anti-His and rabbit anti-mouse HRP-conjugated antibodies. The amounts of SCY-635 necessary to inhibit 50% of NS5A binding in the absence of drug were calculated (IC50). Results (duplicate determinations) are representative of two independent experiments. Error bars represent standard errors of triplicate determinations.
We confirmed the GST-CypA pulldown data by ELISA. Specifically, we found that SCY-635 blocks all CypA-NS5A interactions, independently of the genotype origin of NS5A (Fig. 3B). Because the ELISA allows a more accurate analysis of the Cyp-NS5A interaction block than Western blotting, we measured by ELISA the concentration of SCY-635 necessary to block 50% of the CypA-NS5A interaction (50% inhibitory concentration [IC50]). We found that lower concentrations of SCY-635 were required to prevent the interaction between CypA and H77 and Con1 NS5A than between CypA and JFH-1 and MD2b-1 NS5A (Fig. 4C). This is in accordance with our recent data, which showed that the affinities of JFH-1 and MD2b-1 NS5A to CypA are slightly superior to those of H77 and Con1 NS5A (6). Indeed, we calculated KD (equilibrium dissociation constant) values of 131, 126, 108, and 112 μM for wild-type H77, Con1, JFH-1, and MD2b-1 NS5A, respectively (6). In addition, we calculated the concentration of CsA necessary to block 50% of the CypA-NS5A interaction (IC50) in order to compare SCY-635 and CsA inhibitory efficacies (Fig. 4C). We found that IC50s for CsA were greater than the corresponding values calculated for SCY-635 (Fig. 4C). This is consistent with previous observations that SCY-635 is more potent inhibitor of HCV RNA replication and exhibits higher-affinity binding to CypA in comparison to CsA (24).
Development of HCV replicons resistant to SCY-635.
To better understand the mechanisms of antiviral action of SCY-635, we generated HCV replicons that replicate in hepatoma cells even in the presence of high concentrations of SCY-635. Specifically, Huh7 cells expressing the subgenomic Con1 (GT1b) replicon were incubated with 0.5 μM SCY-635 and 100 μg/ml of G418 in 10-cm CellBIND plates. Cells were split every 3 days at 1:6, and the G418 concentration was increased sequentially to 200 and 300 μg/ml. Most of the cells were dying 3 weeks after drug incubation. At this point, medium was changed every 3 to 4 days until the emergence of nine SCY-635-resistant clones. Clones were individually transferred to 24-well CellBIND using cloning disks. Of the nine clones, three survived after the transfer process and were expanded to 6-well plates. Each clone was split into 2 identical plates and was separately maintained under 0.5 or 1.0 μM SCY-635 and 100 μg/ml of G418. Once the colonies were established, G418 concentration was increased to 300 μg/ml. Clones resistant to 1 μM SCY-635 were further screened for resistance to 2 μM SCY-635 as described above.
The three clones selected above were analyzed for mutations in NS5A and NS5B. All three clones examined contain mutations in NS5A, but not in NS5B. Specifically, we found that two clones contained a single D320E mutation, whereas the third clone contained a single Y321N mutation. To truly demonstrate that these mutations are responsible for the resistance of HCV to SCY-635, D320E and Y321N mutations were engineered into the HCV Con1 subgenomic replicon and the resulting mutant viral genomes were electroporated into Huh7.5.1 cells in the presence or absence of increasing concentrations of SCY-635. Viral RNA replication was monitored by luciferase activity at various time points. First, we confirmed that SCY-635 efficiently blocks RNA replication of wild-type virus in a dose-dependent manner (Fig. 5). Second, we found that in the absence of SCY-635, both D320E and Y321N NS5A mutant viruses replicate similarly to wild-type virus, suggesting that these mutations do not alter the fitness of the virus. This also suggests that the SCY-635-resistant replicons do not require the drug to replicate. Importantly, both D320E and Y321N mutations render HCV significantly more resistant to SCY-635 than wild-type virus (Fig. 5). These data strongly suggest that these mutations are indeed responsible for the resistance of HCV to SCY-635.
Fig 5.

Wild-type and D320E and Y321N mutant Con1 RNAs were electroporated into Huh7.5.1 cells, and the HCV RNA replication was monitored over time by measuring luciferase activity in cell lysates. SCY-635 was added to cells at the time of the cell seeding, immediately after the electroporation. Results are representative of two independent experiments. Error bars represent standard errors of triplicate determinations.
Interactions Between CypA and NS5A derived from SCY-635-resistant HCV replicons.
We then asked whether the D320E and Y321N mutations found in the SCY-635-resistant viruses influence either the interaction between CypA and NS5A or the sensitivity of this interaction to SCY-635. To address this issue, we cloned D320E and Y321N mutants into the context of the bacterial vector encoding full-length Con1 NS5A. We produced and purified wild-type and D320E and Y321N NS5A mutant proteins and tested them for their capacities to bind CypA using the GST-CypA pulldown assay. We first found that in the absence of SCY-635, wild-type, D320E and Y321N NS5A mutant proteins bind similarly to GST-CypA (Fig. 6A). Importantly, SCY-635 prevents all CypA-NS5A interactions in a dose-dependent manner (Fig. 6A). We confirmed the GST-CypA pulldown data by ELISA. Specifically, we found that SCY-635 blocks the interaction between CypA and wild-type, D320E, and Y321N NS5A mutant proteins (Fig. 6B). This finding is important because it suggests that the D320E and Y321N NS5A mutations, which arose in SCY-635-resistant HCV replicons, do not require correspondingly high concentrations of SCY-635 to cause dissociation of the CypA-mutant NS5A complexes.
Fig 6.
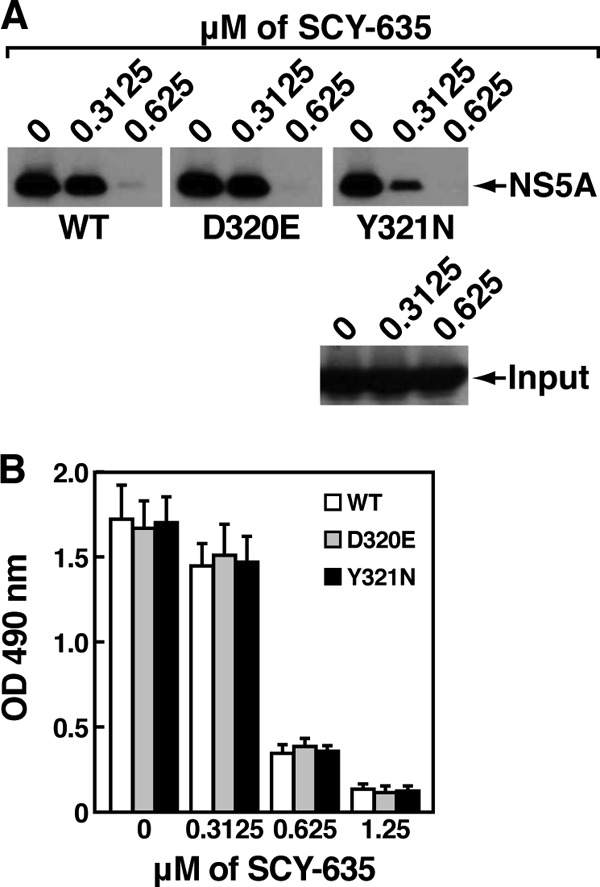
(A) Same as Fig. 4A, except that GST-CypA (1 μg) was mixed with recombinant wild-type or D320E or Y321N mutant NS5A-His (200 ng) with increasing concentrations of SCY-635 (from 0.3125 to 0.625 μM) for 3 h at 4°C. Glutathione beads were added to the GST-CypA–NS5A mixture for 30 min at 4°C and washed. Bound material was eluted and analyzed by Western blotting using anti-His and anti-GST antibodies. (B) Same as Fig. 4B. These results are representative of two independent experiments. Error bars represent standard errors of triplicate determinations.
HCV resistance to SCY-635 correlates with CypA independence.
One possibility to explain how HCV replicons replicate even in the presence of SCY-635 is that the D320E and Y321N NS5A mutations render the viruses less dependent on CypA. The high concentration of SCY-635 (>1 μM) added to cells should suffice to completely saturate the PPIase active site and inhibit enzymatic activity of the total intracellular pool of CypA molecules, therefore preventing their association with NS5A. To test this hypothesis, wild-type and D320E and Y321N mutant viral RNAs were electroporated into parental or CypA-knockdown (CypA-KD) Huh7.5.1 cells. The establishment of CypA-KD cells was described previously (5). Wild-type subgenomic Con1 replicates poorly in CypA-KD cells compared to parental or CypB-KD cells (Fig. 7), consistent with the absolute requirement for CypA expression, but not CypB expression, to support efficient viral RNA replication (5, 15, 17, 28, 34, 50). Importantly, both D320E and Y321N mutations rescue viral RNA replication in CypA-KD cells treated or not with SCY-635 (1 μM) (Fig. 7). These data strongly suggest that the D320E and Y321N mutations not only confer resistance to SCY-635, but also render HCV CypA independent.
Fig 7.

Wild-type and D320E and Y321N mutant Con1 RNAs were electroporated into parental, CypB-KD, or CypA-KD Huh7.5.1 cells treated with or without SCY-635 (1 μM), and HCV RNA replication was monitored over time by measuring luciferase activity in cell lysates. Results are representative of two independent experiments. Error bars represent standard errors of triplicate determinations.
DISCUSSION
In this study, we showed in vitro that the Cyp inhibitor SCY-635 is a very potent inhibitor of both HCV RNA replication and HCV infection. We also showed that SCY-635 alone is capable of clearing HCV RNA from replicon-containing cells. Since a viral rebound was observed with the other Cyp inhibitor, CsA, these data further demonstrate the superior antiviral efficacy of SCY-635 to that of CsA. Importantly, we demonstrated that SCY-635 prevents the formation of NS5A-CypA complexes in a dose-dependent manner. There is thus a direct correlation between blocking NS5A-CypA interactions and inhibiting HCV replication, suggesting that NS5A-CypA interactions are vital for viral replication. It has been proposed in vitro that CypA, via its isomerase pocket, accelerates the rotation of peptidyl-prolyl bonds in recombinant NS5A (21, 44, 49). However, the biologically relevant action of CypA on NS5A in a physiological cellular context remains to be elucidated. More importantly, the actual role of NS5A in HCV RNA replication also remains to be fully investigated. Interestingly, a recent study nicely showed that CypA, by binding to domain II of NS5A, enhances the capacity of NS5A to bind RNA (13). One can envision that CypA, by affecting the conformational structure of NS5A, promotes the binding of NS5A to HCV RNA and subsequently enhances HCV RNA replication.
We showed that CypB binds NS5A similarly to CypA and that SCY-635 prevents NS5A-CypB and NS5A-CypA interactions equally. This could indicate that NS5A-CypB interactions are also important for HCV replication as previously suggested (9, 15, 21). However, several CypA-KD studies clearly showed that CypB is dispensable and/or not sufficient for HCV replication (5, 28, 50). This apparent lack of an essential function for CypB in HCV replication is in accordance with the fact that CypB, which resides in the lumen of the endoplasmic reticulum (ER) (4), is not appropriately located to interact with the HCV replication complex, which is exposed to the cytosolic side of the ER. In contrast, CypA, which resides in the cytosol (20), is properly positioned to interact with the HCV replication complex, further supporting an important role for CypA, but not CypB, in HCV replication.
We found that SCY-635 prevents the direct contact between CypA and NS5A derived from various genotypes (GT1a, GT1b, GT2a, and GT2b). While SCY-635 has only been tested in GT1a and 1b patients, our data are in accordance with previous studies that showed that other Cyp inhibitors, such as CsA, NIM811, and alisporivir, like SCY-635, exhibit pangenotypic anti-HCV activities (10, 18, 25, 26, 32, 33, 35, 40).
We found that HCV replicons escape SCY-635 selection pressure only after >10 weeks. This extended period of time for the development of resistance suggests a high barrier to resistance to SCY-635. This is in agreement with recent studies that showed that in contrast to direct-acting antivirals (DAAs), such as protease and NS5A inhibitors, which exhibit a low barrier to resistance, Cyp inhibitors such as alisporivir have a high barrier to resistance (7). It is important to emphasize that although HCV replicons could escape SCY-635 selection pressure in vitro, no evidence of viral rebound was observed and no selection of resistant virus was detected following 15 days of SCY-635 monotherapy at a dose of 900 mg/day in patients with chronic GT1 infection (23). Nonetheless, future phase II and III studies with SCY-635 should address this possibility. To date, no viral breakthrough has been observed in alisporivir-treated patients until week 12 of treatment (B. Li et al., presented at the 62nd Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, 2011). In 3 of 6 treated patients, viral breakthrough occurred after pIFN-α/RBV dose adjustment/stoppage; in 2 of the other 3 viral breakthroughs, pharmacokinetics analysis revealed suboptimal alisporivir exposure (B. Li et al., presented at the 62nd Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, 2011). Sequencing of the HCV genome did not identify any genotypic changes (at least in NS5A), which were consistently associated with viral breakthrough (B. Li et al., presented at the 62nd Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, 2011). Altogether, these data suggest a low potential for development of resistance to Cyp inhibitors such as SCY-635 in treated patients.
We identified two mutations in NS5A that emerge under in vitro SCY-635 selection—D320E and Y321N. The D320E mutation seems to be a common resistance change for Cyp inhibitor resistance. Specifically, the D320E NS5A mutation was identified in CsA-, NIM811-, and alisporivir-resistant HCV Con1 replicons (GT1b) (6, 7, 9, 17, 39). The D320E NS5A mutation was also identified in CsA-resistant HCV JFH1 (GT2a) (D320E corresponds to D316E in JFH-1 NS5A) (49). The Y321N mutation was previously reported for HCV Con1 (GT1b) resistance to CsA and NIM811 (10, 17, 39), as well as for HCV JFH1 (GT2a) (49) (Y321N corresponds to Y317N in JFH-1 NS5A). We demonstrated in this study that the D320E and Y321N mutations render HCV Con1 both SCY-635 resistant and CypA independent. This is perfectly in accordance with a recent study that nicely showed that D316E and Y317N NS5A mutations render JFH-1 more resistant to CsA and more independent of CypA (49). These data suggest that the D320E and Y321N mutations confer low-level resistance of HCV (Con1) to Cyp inhibitors, possibly by reducing the need for CypA-dependent isomerization of NS5A. It will be interesting to determine whether the D320E and Y321N mutations confer “universal” cross-resistance among Cyp inhibitors, including SCY-635, NIM811, alisporivir, CsA, sanglifehrins A to D, and sangamides (amide derivatives of sanglifehrin A) (19). It is important to note that the RNA binding of domain II of NS5A containing the D320E mutation was unaffected by CypA (14). Further studies are required to determine first if NS5A isomerization truly occurs in a cell and, if it occurs, what the impact is on NS5A function in HCV replication. NS5A isomerization may affect any of several features of NS5A: (i) binding to the viral RNA, (ii) binding to the HCV polymerase since NS5A and NS5B interact (42), (iii) binding to host ligands given that NS5A binds to multiple host proteins (22); and (iv) the IFN pathway since NS5A modulates the innate response (22). Nevertheless, our data strongly suggest that the main initial mechanism of antiviral action of SCY-635 is the drug-mediated competitive dissociation of NS5A-CypA complexes.
It is important to mention that SCY-635 and alisporivir behave very similarly both in vitro and in vivo. Indeed, alisporivir, like SCY-635, inhibits in vitro HCV replication at a nanomolar concentration range (7, 35), efficiently blocks the peptidyl-prolyl isomerase activity of CypA (35), abrogates in a dose-dependent manner NS5A-CypA interactions (7), and exhibits high efficacy in HCV patients (12; R. Flisiak et al., presented at the 43rd Annual Meeting of the European Association for the Study of the Liver, Milan, Italy, 2008; R. Flisiak et al., presented at the 46th Annual Meeting of the European Association for the Study of the Liver, Berlin, Germany, 2011). To our knowledge, NIM811 has never been tested for its ability to prevent NS5A-CypA interactions, but it is likely that like parental CsA and the CsA derivates SCY-635 and alisporivir, NIM811, which is also a CsA derivate, will block NS5A-CypA interactions. Interestingly, in contrast to SCY-635 and alisporivir, NIM811, although potent in vitro (32), did not exert antiviral activity as monotherapy (29). These results indicate that although the in vitro efficacies of the three nonimmunosuppressive CsA derivates are similar, their in vivo efficacies differ, likely due to dissimilar cellular pharmacologies and/or in vivo biodistributions.
In conclusion, several findings suggest that SCY-635 represents a promising novel anti-HCV agent. (i) It blocks HCV RNA replication and infection in vitro. (ii) It clears HCV-containing cells in vitro. (iii) It prevents the contact between CypA and NS5A derived from various genotypes. (iv) It targets a host protein that has been shown to be optional for cell growth and survival, validating the potential of CypA as a therapeutic target. (v) It provides (at least in vitro) a high barrier to the development of resistance. (vi) It exhibits broad genotypic anti-HCV activities. (vii) It efficiently decreases the plasma viral loads in SCY-635-treated patients. (viii) Finally, it exhibits an acceptable safety profile in SCY-635-treated patients (23).
ACKNOWLEDGMENTS
We thank J. Kuhns for secretarial assistance, C. Cameron for the pET26Ub-NS5A-His Con1 plasmid, C. Rice for Huh7.5.1 cells, R. Bartenschlager for Con1 plasmid and Huh7-Luc/Neo ET cells, F. Chisari for JFH-1-Huh7.5.1 cells, T. Wakita, R. Bartenschlager, and T. Pietschmann for the Luc-JFH1 plasmid, and Ann Sluder and Katyna Katyna Borroto-Esoda for careful reading of the manuscript.
We acknowledge financial support from U.S. Public Health Service grant no. AI087470 (P.A.G.).
Footnotes
Published ahead of print 14 May 2012
This article is publication no. 21719 from the Department of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, California, USA.
REFERENCES
- 1. Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 13:2436–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Armstrong GL, et al. 2006. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann. Intern. Med. 144:705–714 [DOI] [PubMed] [Google Scholar]
- 3. Asselah T, Benhamou Y, Marcellin P. 2009. Protease and polymerase inhibitors for the treatment of hepatitis C. Liver Int. 29(Suppl 1):57–67 [DOI] [PubMed] [Google Scholar]
- 4. Bose S, Mucke M, Freedman RB. 1994. The characterization of a cyclophilin-type peptidyl prolyl cis-trans-isomerase from the endoplasmic-reticulum lumen. Biochem. J. 300:871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chatterji U, et al. 2009. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 284:16998–17005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chatterji U, et al. 2010. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 53:50–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coelmont L, et al. 2010. DEB025 (alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 5:e13687 doi:10.1371/journal.pone.0013687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dienstag JL, McHutchison JG. 2006. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 130:231–264 [DOI] [PubMed] [Google Scholar]
- 9. Fernandes F, Ansari IU, Striker R. 2010. Cyclosporine inhibits a direct interaction between cyclophilins and hepatitis C NS5A. PLoS One 5:e9815 doi:10.1371/journal.pone.0009815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandes F, et al. 2007. Sensitivity of hepatitis C virus to cyclosporine A depends on nonstructural proteins NS5A and NS5B. Hepatology 46:1026–1033 [DOI] [PubMed] [Google Scholar]
- 11. Fischer G, Gallay P, Hopkins S. 2010. Cyclophilin inhibitors for the treatment of HCV infection. Curr. Opin. Investig. Drugs 11:911–918 [PubMed] [Google Scholar]
- 12. Flisiak R, et al. 2008. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 47:817–826 [DOI] [PubMed] [Google Scholar]
- 13. Foster GR, et al. 2011. Telaprevir alone or with peginterferon and ribavirin reduces HCV RNA in patients with chronic genotype 2 but not genotype 3 infections. Gastroenterology 141:881.e1–889.e1 doi:10.1053/j.gastro.2011.05.046 [DOI] [PubMed] [Google Scholar]
- 14. Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 85:7460–7464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gaither LA, et al. 2010. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology 397:43–55 [DOI] [PubMed] [Google Scholar]
- 16. Gallay PA. 15 December 2011. Cyclophilin inhibitors: a novel class of promising host-targeting anti-HCV agents. Immunol. Res. [Epub ahead of print.] doi:10.1007/s12026-011-8263-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goto K, Watashi K, Inoue D, Hijikata M, Shimotohno K. 2009. Identification of cellular and viral factors related to anti-hepatitis C virus activity of cyclophilin inhibitor. Cancer Sci. 100:1943–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goto K, et al. 2006. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 343:879–884 [DOI] [PubMed] [Google Scholar]
- 19. Gregory MA, et al. 2011. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrobial Agents Chemother. 55:1975–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. 1984. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226:544–547 [DOI] [PubMed] [Google Scholar]
- 21. Hanoulle X, et al. 2009. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 284:13589–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He Y, Staschke KA, Tan SL. 2006. HCV NS5A: a multifunctional regulator of cellular pathways and virus replication. In Tan SL. (ed), Hepatitis C viruses: genomes and molecular biology. Horizon Bioscience, Norfolk, United Kingdom: [PubMed] [Google Scholar]
- 23. Hopkins S, et al. 13 March 2012. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J. Hepatol. [Epub ahead of print.] doi:10.1016/jhep.2012.02.024 [DOI] [PubMed] [Google Scholar]
- 24. Hopkins S, et al. 2010. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 54:660–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Houck DR, Hopkins S. 2006. Preclinical evaluation of SCY-635, a cyclophilin inhibitor with potent anti-HCV activity. Hepatology 44(Suppl 1):S534–S535 [Google Scholar]
- 26. Ishii N, et al. 2006. Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 80:4510–4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kapadia SB, Brideau-Andersen A, Chisari FV. 2003. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. U. S. A. 100:2014–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaul A, et al. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546 doi:10.1371/journal.ppat.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lawitz E, et al. 2011. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral Res. 89:238–245 [DOI] [PubMed] [Google Scholar]
- 30. Lin K. 2010. Development of novel antiviral therapies for hepatitis C virus. Virol. Sin. 25:246–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Z, Yang F, Robotham JM, Tang H. 2009. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 83:6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma S, et al. 2006. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 50:2976–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakagawa M, et al. 2004. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 313:42–47 [DOI] [PubMed] [Google Scholar]
- 34. Nakagawa M, et al. 2005. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology 129:1031–1041 [DOI] [PubMed] [Google Scholar]
- 35. Paeshuyse J, et al. 2006. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 43:761–770 [DOI] [PubMed] [Google Scholar]
- 36. Pawlotsky JM. 2012. New antiviral agents for hepatitis C. F1000 Biol. Rep. 4:5 doi:10.3410/B4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pawlotsky JM. 2011. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53:1742–1751 [DOI] [PubMed] [Google Scholar]
- 38. Pockros PJ. 2010. New direct-acting antivirals in the development for hepatitis C virus infection. Ther. Adv. Gastroenterol. 3:191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Puyang X, et al. 2010. Mechanism of resistance of hepatitis C virus replicons to structurally distinct cyclophilin inhibitors. Antimicrob. Agents Chemother. 54:1981–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robida JM, Nelson HB, Liu Z, Tang H. 2007. Characterization of hepatitis C virus subgenomic replicon resistance to cyclosporine in vitro. J. Virol. 81:5829–5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567 [DOI] [PubMed] [Google Scholar]
- 42. Shirota Y, et al. 2002. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 277:11149–11155 [DOI] [PubMed] [Google Scholar]
- 43. Soriano V, et al. 2008. Emerging drugs for hepatitis C. Expert Opin. Emerg. Drugs 13:1–19 [DOI] [PubMed] [Google Scholar]
- 44. Verdegem D, et al. 2011. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J. Biol. Chem. 286:20441–20454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vermehren J, Sarrazin C. 2011. New hepatitis C therapies in clinical development. Eur. J. Med. Res. 16:303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. von Hahn T, Ciesek S, Manns MP. 2011. Arrest all accessories—inhibition of hepatitis C virus by compounds that target host factors. Discov. Med. 12:237–244 [PubMed] [Google Scholar]
- 47. Waller H, Chatterji U, Gallay P, Parkinson T, Targett-Adams P. 2010. The use of AlphaLISA technology to detect interaction between hepatitis C virus-encoded NS5A and cyclophilin A. J. Virol. Methods 165:202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. 2003. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38:1282–1288 [DOI] [PubMed] [Google Scholar]
- 49. Yang F, et al. 2010. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 6:e1001118 doi:10.1371/journal.ppat.1001118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang F, et al. 2008. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 82:5269–5278 [DOI] [PMC free article] [PubMed] [Google Scholar]