

Abstract
PSI-352938 is a novel cyclic phosphate prodrug of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine-5′-monophosphate with potent anti-HCV activity. In order to inhibit the NS5B RNA-dependent RNA polymerase, PSI-352938 must be metabolized to the active triphosphate form, PSI-352666. During in vitro incubations with PSI-352938, significantly larger amounts of PSI-352666 were formed in primary hepatocytes than in clone A hepatitis C virus (HCV) replicon cells. Metabolism and biochemical assays were performed to define the molecular mechanism of PSI-352938 activation. The first step, removal of the isopropyl group on the 3′,5′-cyclic phosphate moiety, was found to be cytochrome P450 (CYP) 3A4 dependent, with other CYP isoforms unable to catalyze the reaction. The second step, opening of the cyclic phosphate ring, was catalyzed by phosphodiesterases (PDEs) 2A1, 5A, 9A, and 11A4, all known to be expressed in the liver. The role of these enzymes in the activation of PSI-352938 was confirmed in primary human hepatocytes, where prodrug activation was reduced by inhibitors of CYP3A4 and PDEs. The third step, removal of the O6-ethyl group on the nucleobase, was shown to be catalyzed by adenosine deaminase-like protein 1. The resulting monophosphate was consecutively phosphorylated to the diphosphate and to the triphosphate PSI-352666 by guanylate kinase 1 and nucleoside diphosphate kinase, respectively. In addition, formation of nucleoside metabolites was observed in primary hepatocytes, and ecto-5′-nucleotidase was able to dephosphorylate the monophosphate metabolites. Since CYP3A4 is highly expressed in the liver, the CYP3A4-dependent metabolism of PSI-352938 makes it an effective liver-targeted prodrug, in part accounting for the potent antiviral activity observed clinically.
INTRODUCTION
Hepatitis C virus (HCV), a member of the Flaviviridae family of viruses, is a major health problem affecting approximately 170 million people worldwide. In the United States, where it is the leading cause of liver transplantation, nearly 2% of the U.S. population are HCV carriers (1). Recently, the protease inhibitors boceprevir and telaprevir were approved to be used in combination with pegylated-alpha interferon (IFN-α) and ribavirin (PEG-RBV) for the treatment of chronic HCV infection. While these treatments show improved response rates and the potential for shorter duration treatment over PEG-RBV alone, they lack efficacy against genotypes other than genotype 1 and require thrice-daily dosing, and patients may develop additional side effects on top of the already challenging tolerability profile of PEG-RBV. Therefore, there is a need for more potent anti-HCV compounds with improved clinical efficacy and greater tolerability to more broadly address the unmet medical needs of those with chronic HCV. Combinations of antiviral agents targeting viral proteins essential to HCV replication have the potential to achieve increased clinical efficacy across HCV genotypes, have fewer adverse effects, and shorten treatment duration compared to the current standard of care.
Nucleoside/nucleotide analogs, which inhibit virally encoded polymerases, have a proven track record as therapies for viral infections caused by other viruses, including herpesviruses, HIV, and hepatitis B virus (5). The HCV RNA-dependent RNA polymerase NS5B protein is essential for HCV replication and therefore is an ideal therapeutic target (15, 20, 21, 29). Recent clinical studies suggest that nucleoside/nucleotide analogs have potent pangenotype antiviral activity and a high barrier to resistance development (11, 17, 19, 27).
A number of 2′-C-methylpurine analogs, including 2′-fluoro-2′-C-methylguanosine, are reported to have only weak activity in the replicon assay (4, 6, 22, 26). Because the 5′-triphosphates of these compounds are potent inhibitors of the NS5B polymerase, the weak activity in the replicon assay appears to be due to inefficient phosphorylation of the compound to the corresponding 5′-monophosphate. Since cyclic monophosphate prodrugs of the 2′-C-methyl ribonucleosides have been shown to have greater stability and improved cellular permeability (12), cyclic prodrugs of the 2′-deoxy-2′-fluoro-2′-C-methylguanosine monophosphate nucleoside were investigated (26). Among the compounds synthesized and tested, PSI-352938, a novel cyclic phosphate prodrug of β-d-2′-fluoro-2′-C-methylguanosine 5′-monophosphate with pangenotypic anti-HCV activity, showed the best profile in terms of anti-HCV activity, cytotoxicity, stability, and intracellular triphosphate production (26). Furthermore, PSI-352938 lacked cross-resistance with other modified nucleoside/nucleotide analogs targeting HCV NS5B (18), suggesting that a complementary effect could be achieved when combined with other classes of nucleoside/nucleotide inhibitors.
Since we previously demonstrated that the metabolism of PSI-7851, a phosphoramidate prodrug of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine-5′-monophosphate, and the metabolism of its single isomer, PSI-7977, were different in Huh 7-derived clone A replicon cells and primary human hepatocytes due to differences in the expression of certain enzymes involved in the metabolism of these compounds, we compared the metabolisms of PSI-352938 in these two cell types (24). Finally, the activation pathway of PSI-352938 in primary human hepatocytes is proposed (Fig. 1).
Fig 1.

Chemical structures of PSI-352938 and its potential metabolites and the proposed metabolic pathway of PSI-352938 activation. Thick arrows represent primary pathway, and dotted arrows represent slow processes. *, PK and 3PGK can also catalyze this reaction, but NDPK is the primary enzyme.
MATERIALS AND METHODS
Materials.
High-throughput cytochrome P450 (CYP) screening kits were purchased from BD Biosciences (Woburn, MA). Recombinant human cathepsin A, carboxylesterases 1 and 2, aminopeptidases P1 and P2, ectonucleotide pyrophosphatase/phosphodiesterase 1 (PDE1), and ecto-5′-nucleotidase/CD73 (5′-NTase CD73) were purchased from R & D Systems (Minneapolis, MN), human trypsin, chymase, and neutrophil elastase were obtained from Calbiochem (San Diego, CA), chymotrypsin was obtained from MP Biomedicals (Solon, OH), cathepsins B, D, and L and lipase were obtained from Sigma (St. Louis, MO), cathepsin H, calpain 1, and caspases 1 to 10 were obtained from BioVision (Mountain View, CA), HCV NS3 protease was obtained from AnaSpec (Fremont, CA), human phosphodiesterases 2A1, 5A, 9A, and 11A4 were obtained from BPS Biosciences (San Diego, CA), and human cytosolic 5′-nucleotidase II (5′-NTase cN-II) was obtained from NovoCIB (Lyon, France). PSI-352938, M1, M2, M3, M4, M5, M6, M7, M8, and PSI-352666 (whose structures are shown in Fig. 1) were synthesized at Pharmasset with a purity of >95%, and the identities of these compounds were confirmed by liquid chromatography coupled to triple-quadropole mass spectrometery (LC/MS/MS) and nuclear magnetic resonance (NMR). 3H-labeled PSI-352938, M1, M2, and M3 were synthesized by Moravek Biochemicals (Brea, CA). Ketoconazole, aminobenzotriazole, rolipram, sildenafil, and ibudilast were purchased from Sigma (St. Louis, MO). Ritonavir was purchased from Toronto Research Chemical (Ontario, Canada).
Cellular metabolism study.
Clone A HCV genotype 1b (Con1 strain, GenBank accession no. AJ238799.1) replicon cells (Apath LLC, Brooklyn, NY) in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) were seeded into a 6-well plate at about 500,000 cells/well, and human primary hepatocytes in cell plating medium (CellzDirect, Inc.) were seeded at about 1,000,000 cells/well in a 6-well collagen I-coated plate. After overnight incubation to allow the cells to attach, the cells were incubated for up to 72 h for clone A and 48 h for primary hepatocytes at 37°C in a 5% CO2 atmosphere with DMEM–10% FBS for clone A and cell maintenance medium (CellzDirect, Inc.) for human hepatocytes containing 5 μM [3H]PSI-352938 (4,000 dpm/pmol). At selected times, the extracellular medium was removed, and the cell layer was washed with cold phosphate-buffered saline (PBS). After trypsinization, cells were counted and centrifuged at 1,200 rpm for 5 min. The cell pellets were suspended in 1 ml cold 60% methanol and incubated overnight at −20°C. The samples were centrifuged at 14,000 rpm for 5 min, and the supernatants were dried using a SpeedVac concentrator (Thermo Electron Corporation, MA) and then stored at −20°C until they were analyzed by high-performance liquid chromatography (HPLC). Residues were suspended in 100 μl of water, and 50-μl aliquots were injected into HPLC. PSI-352938 and metabolites were separated by ion-exchange HPLC with a Whatman 10 μm SAX column (Whatman, Maidstone, England) using a Series 200 HPLC system (PerkinElmer, Wellesley, MA). The mobile phase consisted of buffer A (0.02 M KH2PO4, pH 3.5) and buffer B (1 M KH2PO4 [pH 3.5]). Elution was performed using a linear gradient of buffer B from 0 to 100% for 100 min. Radioactivity was analyzed using a 610TR radiometric flow scintillation analyzer (PerkinElmer, Wellesley, MA). PSI-352938 and its respective metabolites were identified based on the retention time of synthesized standards.
Effects of PDE and CYP inhibitors on triphosphate formation in primary human hepatocytes.
Primary human hepatocytes were purchased from Life Technology (Grand Island, NY) in 12-well plates seeded at confluence (0.888 × 106 cells/well). PSI-352938 was incubated at 10 μM either alone or in combination (including a 30-min preincubation with inhibitor) with various phosphodiesterase (PDE) or cytochrome P450 (CYP) inhibitors for 4 h. The inhibitors tested were 5 μM ritonavir, 10 μM ketoconazole, 5 μM montelukast, 500 μM 1-aminobenzotriazole, 10 μM rolipram, 10 μM sildenafil, and 100 μM ibudilast. Following removal of extracellular media, cells were washed with 2 ml of ice-cold 0.9% normal saline and scraped into 0.5 ml ice-cold 70% methanol containing 100 nM 2-chloro-adenosine-5′-triphosphate (Sigma-Aldrich, St. Louis, MO) as an internal standard. Samples were stored overnight at −20°C to facilitate nucleotide extraction and centrifuged at 15,000 × g for 15 min, and then supernatant was transferred to clean tubes for drying in a MiVac Duo concentrator (Genevac, Gardiner, NY). Dried samples were then reconstituted in 3 mM ammonium formate with 10 mM dimethylhexylamine (DMH) for analysis by LC/MS/MS. Briefly, analytes were separated using a 50- by 2-mm and 2.5-μm-particle-size Luna C18 (2) HST column (Phenomenex, Torrance, CA) connected to an LC-20ADXR ternary pump system (Shimadzu, Columbia, MD) and HTS PAL autosampler (LEAP Technologies, Carrboro, NC). A multistage linear gradient from 10% to 50% acetonitrile in a mobile phase containing 3 mM ammonium formate (pH 5.0) with 10 mM DMH at a flow rate of 150 μl/min was used to separate analytes. Detection was performed on an API4000 (Applied Biosystems, Foster City, CA) MS/MS operating in the positive-ion and multiple-reaction-monitoring modes. The MS/MS parameters used to detect PSI-352666 include parent and daughter mass-to-charge ratios (m/z) of 540.2 and 152.1, respectively, a declustering potential of 165 V, and a collision energy of 48 eV. PSI-352666 was quantified using a 7-point standard curve of authentic PSI-352666 ranging in concentration from 0.1 to 100 pmol/million cells prepared in cell extract from untreated primary human hepatocytes. To ensure accuracy and precision within 20% over the course of the analysis, quality control samples of known concentration were included at the beginning and end of each sample set.
PSI-352938 hydrolysis by various enzymes.
Hydrolysis of PSI-352938 was tested using various human recombinant enzymes in a 100-μl reaction volume containing 100 μM PSI-352938. The amounts of protein in the reaction mixtures were 1 μg for trypsin, chymotrypsin chymase, elastase, cathepsins B, D, and L, calpain 1, lipase, and HCV NS3 protease, 0.6 μg for cathepsin H, 0.5 μg for ectonucleotide pyrophosphatase/phosphodiesterase 1, carboxylesterases 1 and 2, and aminopeptidase P1, and 0.2 μg for cathepsin A and aminopeptidase P2; 1 U of enzyme (i.e., enzyme activity that cleaves 1 nmol of the individual caspase substrate per hour at 37°C) for caspases 1 to 10 was used in the reaction. Different buffer systems were used, depending on the enzyme: 200 mM Tris-HCl (pH 7.5) plus 20 mM CaCl2 for trypsin, chymotrypsin, and chymase; 100 mM Tris-HCl (pH 7.5) and 500 mM NaCl for elastase; 50 mM Tris-HCl (pH 8.0), 250 mM NaCl, and 0.5 mM MnCl2 for aminopeptidase P1; 100 mM HEPES (pH 8.0) and 0.5 mM MnCl2 for aminopeptidase P2; 50 mM HEPES (pH 7.0), 100 mM NaCl, and 0.5% NP-40 for cathepsin A; 100 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 5 mM dithiothreitol (DTT) for cathepsins B and L; 400 mM sodium acetate (pH 5.2), 10 mM DTT, and 5 mM EDTA for cathepsin D; 50 mM Tris-HCl (pH 9.5) and 250 mM NaCl for ectonucleotide pyrophosphatase/phosphodiesterase 1; 50 mM Tris-HCl (pH 7.5) for carboxylesterases 1 and 2 and lipase; 100 mM Tris-HCl (pH 7.5), 5 mM CaCl2, and 5 mM DTT for calpain 1; 50 mM HEPES (pH 7.2), 50 mM NaCl, 0.1% CHAPS {3-[(cholamidopropyl)-dimethylammonio]-1-propanesulfonate}, 10 mM DTT, 10 mM EDTA, and 5% glycerol for cathepsin H and caspases 1 to 10; and AnaSpec assay buffer for HCV NS3 protease. After incubation at 37°C for 3 h, the reaction mixture was applied to a YM-10 Microcon filter (Millipore, Billerica, MA) to remove the protein. The flowthrough from the filter was collected and analyzed by HPLC with a Gemini 5-μm-particle-size C18 column (Phenomenex, Torrance, CA) using a Series 200 HPLC system (PerkinElmer, Wellesley, MA). The mobile phase consisted of buffer A (water plus 0.1% formic acid) and buffer B (acetonitrile plus 0.1% formic acid). Elution was performed using a linear gradient of buffer B from 0 to 50% for 40 min.
CYP assay.
The cytochrome P450 (CYP) assays were conducted in a 50-μl reaction mixture containing 100 mM potassium phosphate buffer (pH 7.4), 3.3 mM MgCl2, 1.3 mM NADPH, 3.3 mM glucose-6-phosphate, 0.4 μg/ml glucose-6-phosphate dehydrogenase, and 5 μM [3H]PSI-352938, and the reaction was started by adding 0.5 μM CYP1A2, 1.5 μM CYP2C8, 1 μM CYP2C9, 1 μM CYP2C19, 1 μM CYP2D6, or 0.5 μM CYP3A4 and incubation at 37°C for 24 h, except for CYP3A4, which was incubated for 2 h. All of the CYP preparations contained P450 reductase and cytochrome b5. The samples were centrifuged at 14,000 rpm for 20 min, and the supernatants were used for HPLC analysis. PSI-352938 and metabolites were separated by reverse-phase HPLC with a Gemini 5-μm-particle-size C18 column (Phenomenex, Torrance, CA) using a Series 200 HPLC system (PerkinElmer, MA). The mobile phase consisted of buffer A (water plus 0.1% formic acid) and buffer B (acetonitrile plus 0.1% formic acid). Elution was performed using a linear gradient of buffer B from 0 to 50% for 40 min. Radioactivity was analyzed using a 610TR radiometric flow scintillation analyzer (PerkinElmer, Wellesley, MA).
PDE assay.
The assay to determine phosphodiesterase (PDE) activity for PSI-352938 was performed with a 100-μl reaction volume containing 2.6 μg/ml of PDE2A, 0.68 μg/ml of PDE5A, 4 μg/ml of PDE9A, or 4.8 μg/ml of PDE11A, 200 μM PSI-352938, 20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, and 0.5 mM MnCl2. After incubation at 37°C for 90 min, the reaction mixture was applied to a YM-10 Microcon filter (Millipore) to remove the protein. The flowthrough from the filter was collected and loaded onto a Gemini 5-μm-particle-size C18 110A column (Phenomenex) for HPLC. The substrates and products were separated by a 35-ml linear gradient of 0 to 50% acetonitrile at a flow rate of 1 ml/min.
Steady-state kinetic assays were performed with M1 and M3 at 37°C. A 20-μl reaction mixture contained 4 μg/ml PDE and various concentrations of 3H-labeled M1 or M3 in 20 mM Tris-HCl (pH 7.5) buffer supplemented with 5 mM MgCl2, and 0.5 mM MnCl2. Reaction incubation times ranged from 10 to 120 min, depending on the substrate and the enzyme being investigated. A 5-μl sample of the reaction mixture was spotted on a sheet of DE81 paper to stop the reaction. After drying, the paper was washed three times for 5 min with 5 mM Tris-HCl (pH 7.8). The paper was dried, the spots were cut out of the paper, and the radioactivity was counted in a liquid scintillation counter. The nucleoside 5′-monophosphate product was retained on the DE81 paper under these washing conditions. A 5-μl aliquot of the reaction mixture was also spotted on DE81 paper, and the radioactivity was directly counted without washing the paper to determine the total radioactivity in the reaction. Based on the total radioactivity and the radioactivity from the cyclic phosphate product, the amount of product was quantified.
5′-Nucleotidase assays.
The ecto-5′-nucleotidase (5′-NTase CD73) reactions for GMP and O6-MeGMP were performed in a 96-well plate with 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.8 nM CD73, and various concentrations of the tested compounds at 37°C for 0, 2, 4, 6, 8, and 10 min. The product formation was quantified by measuring a releasing phosphate group, which was detected by BIOMOL Green reagent (Enzo Life Sciences, Farmingdale, NY). For the rest of the compounds, the assays were conducted in a 50-μl reaction mixture containing 25 mM Tris-HCl (pH 7.5) and 5 mM MgCl2 and various concentrations of the tested compounds. The reactions were started by addition of the appropriate amount of CD73 (0.8 nM for 2′-C-Me-O6-MeGMP, 4 nM for 2′-C-MeGMP, 4 nM for 2′-F-2′-C-Me-O6-MeGMP, 4 nM for M4, and 8 nM for M5) and incubated at 37°C for 0, 2, 4, 6, 8, and 10 min and then stopped by adding 100 μl of 1 N HCl. The samples were analyzed by HPLC using a Whatman 10-μm SAX column (Whatman, Maidstone, England).
The cytosolic 5′-NTase II (cN-II) reactions were performed in a 96-well plate with 50 mM Tris-HCl (pH 7.5), 100 mM KCl, 20 mM MgCl2, 5 mM DTT, and 200 nM cN-II and various concentrations of the test compounds at 37°C for 0, 6, 12, 18, 24, and 30 min. The product formation was quantified by measuring a releasing phosphate group, which was detected by BIOMOL Green reagent (Enzo Life Sciences).
RESULTS
Intracellular metabolism of PSI-352938.
In order to be an active inhibitor of HCV replication, PSI-352938 must be metabolized to its corresponding 5′-triphosphate (PSI-352666). Metabolism studies were therefore performed to characterize the metabolic pathway of PSI-352938 by incubating primary human hepatocytes with [3H]PSI-352938 and analyzing the intracellular metabolites by HPLC. A total of nine potential metabolites were predicted, and eight of them were identified in primary human hepatocytes by comparing the retention times with those of the synthesized compound standards. The structures of PSI-352938 and the nine potential metabolites are shown in Fig. 1. Among the metabolites, three different 3′,5′-cyclic phosphate derivatives, PSI-354091 (M1), PSI-354713 (M2), and PSI-354519 (M3), were detected. Also identified were the two 5′-nucleoside monophosphate derivatives PSI-353221 (M4), which contains an O6-ethyl substitution, and PSI-353222 (M5). Two nucleoside analogs were identified: PSI-352700 (M6), which contains the O6-ethyl substitution, and PSI-6567 (M7). PSI-353579 (M8) and PSI-352666 are the 5′-diphosphate and 5′-triphosphate metabolites, respectively.
In order to elucidate the temporal sequence of events in the metabolic pathway, a time course experiment following formation of the metabolites was conducted using clone A replicon cells or primary human hepatocytes incubated with 5 μM [3H]PSI-352938 for up to 72 h or 48 h, respectively. These two different cell types were used since clone A cells have been used in our antiviral activity assays in vitro and primary human hepatocytes are a more physiologically relevant model. The HPLC chromatograms after incubation of clone A or primary human hepatocytes with [3H]PSI-352938 for 8 h are shown in Fig. 2A and B, respectively, and the time courses of formation of the metabolites in clone A cells and primary human hepatocytes are shown in Fig. 2C and D, respectively. In clone A, the metabolites detected were M2, M7, M8, and PSI-352666 (Fig. 2C). The predominant metabolite formed throughout the 72-h time course was M2, which accumulated rapidly and remained at a concentration between 2 and 3 μM for up to 72 h. The level of the active triphosphate PSI-352666 increased linearly over the time course, and at 72 h, the concentration was 1.7 μM. The nucleoside metabolite M7 and the diphosphate M8 were formed at low levels over the time course, and they were undetectable at the early time points (Fig. 2A and C). Other metabolites (M1, M3, M4, M5, and M6) were not detected.
Fig 2.

Metabolism of PSI-352938 in clone A and primary human hepatocytes. (A and B) HPLC chromatogram of the cell extract collected from clone A cells (A) or primary human hepatocytes (B) after 8 h of incubation with 5 μM radiolabeled PSI-352938. (C and D) Dependence of intracellular concentrations of PSI-352938 metabolites on incubation time in clone A cells (C) or primary human hepatocytes (D). The time points were defined as the times when cells were trypsinized.
In primary human hepatocytes, all of the metabolites with the exception of M3 were detected at 8 h (Fig. 2B). Most of the metabolites were able to be separated using an anion-exchange column; however, M1 and M4 eluted together (Fig. 2B). The predominant metabolite was the active triphosphate PSI-352666, which formed after 2 h of incubation with PSI-352938 (Fig. 2D). The maximum intracellular concentration (∼11 μM) was reached at about 8 h followed by a gradual decline to ∼4 μM at the 48-h time point. M2 was formed early in the time course and peaked at 2 h with a maximum concentration of ∼6 μM, which then gradually declined. The intracellular levels of the monophosphate M5 were maintained a significant level (2 to 3 μM) for 24 h and declined to 1 μM at 48 h. The concentration of the diphosphate metabolite, M8, was initially lower than M5 at the earlier time points but increased after 4 h of incubation. The concentration of M1 and M4 combined remained low (below 2 μM) throughout the time course. Among the two nucleoside metabolites, a significant level of M6 (∼4 μM) was formed by 2 h and slowly declined and the intracellular levels of M7 remained low throughout the time course (∼1.5 μM).
This metabolism study suggested that the first step of PSI-352938 metabolism is either removal of the isopropyl group in the cyclic phosphate moiety to form M1 or removal of the O6-ethyl group in the nucleobase to form M2. In order to determine which reaction is the primary step in the metabolic pathway for PSI-352938, metabolism studies using the 3′,5′-cyclic phosphate derivatives (PSI-352938, M1, M2, and M3) were performed using clone A cells and primary human hepatocytes. As shown in Fig. 3A, when clone A cells were incubated with M1 for 24 h, a significantly larger amount of PSI-352666 was formed than the levels of PSI-352666 formed when the cells were incubated with PSI-352938, M2, or M3. These results indicated that the removal of the isopropyl group is highly inefficient. In primary hepatocytes, the highest level of PSI-352666 was formed when the cells were incubated with M3 for 8 h. Incubation with PSI-352938 or M1 resulted in similar levels of PSI-352666. The lowest level of PSI-352666 was formed when primary human hepatocytes were incubated with M2 (Fig. 3B). Since a significantly smaller amount of PSI-352666 was formed when the cells were incubated with M2 than with PSI-352938, it is unlikely that M2 is a major metabolite in PSI-352938 activation, although a contribution from reduced cellular permeability of M2 cannot be ruled out in these whole-cell assays.
Fig 3.

PSI-352666 formation upon incubation of cells with PSI-352938, M1, M2, or M3. Clone A cells (A) or primary human hepatocytes (B) were incubated with 5 μM 3H-labeled PSI-352938, M1, M2, or M3 for 24 or 8 h, respectively, and the cellular concentration of PSI-352666 was determined.
Removal of the isopropyl group.
We explored enzymes that could remove the isopropyl group in the cyclic phosphate moiety of PSI-352938. Since the isopropyl group is attached to the phosphate via a phosphoester bond, we tested a panel of hydrolases, including trypsin, chymotrypsin chymase, elastase, cathepsins A, B, D, H, and L, carboxylesterases 1 and 2, calpain 1, caspases 1 to 10, HCV NS3 protease, lipase, aminopeptidases 1 and 2, and ectonucleotide pyrophosphatase/phosphodiesterase 1, for their ability to hydrolyze the bond. However, none of these enzymes was able to hydrolyze the phosphoester (data not shown). It has been reported that the benzyl triesters of cyclic AMP (cAMP) or cyclic GMP (cGMP) can be hydrolyzed spontaneously in Krebs-Henseleit buffer (16). Therefore, we examined chemical degradation PSI-352938 in Krebs-Henseleit buffer. Even after 24 h of incubation at 37°C, the isopropyl group was not removed (data not shown). Since PSI-352938 was actively metabolized in primary human hepatocytes, we next investigated the possibility that removal of the isopropyl group was being catalyzed by cytochrome P450 (CYP)-dependent oxidation. To identify which isoforms of CYP are responsible for the reaction, PSI-352938 was incubated with different CYP preparations in a reaction buffer containing an NADPH regeneration system as described in Materials and Methods. As shown in Fig. 4, small amounts of guanine base and M1 were observed in the no-CYP control, probably due to chemical degradation. CYP1A2, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 failed to convert PSI-352938 to M1. However, when PSI-352938 was incubated with CYP3A4, there was a disappearance of the PSI-352938 peak and the subsequent appearance of M1 (Fig. 4). This result indicates that CYP3A4 is capable of removing the isopropyl group from PSI-352938. In addition, an increase in the guanine peak and the appearance of the O6-ethylguanine peak were detected (Fig. 4). The ability of CYP3A4 to remove the isopropyl group from M2 to form M3 was also tested. Our results indicated that M2 was estimated to be a >20-fold less-efficient substrate for CYP3A4 than PSI-352938 (data not shown).
Fig 4.
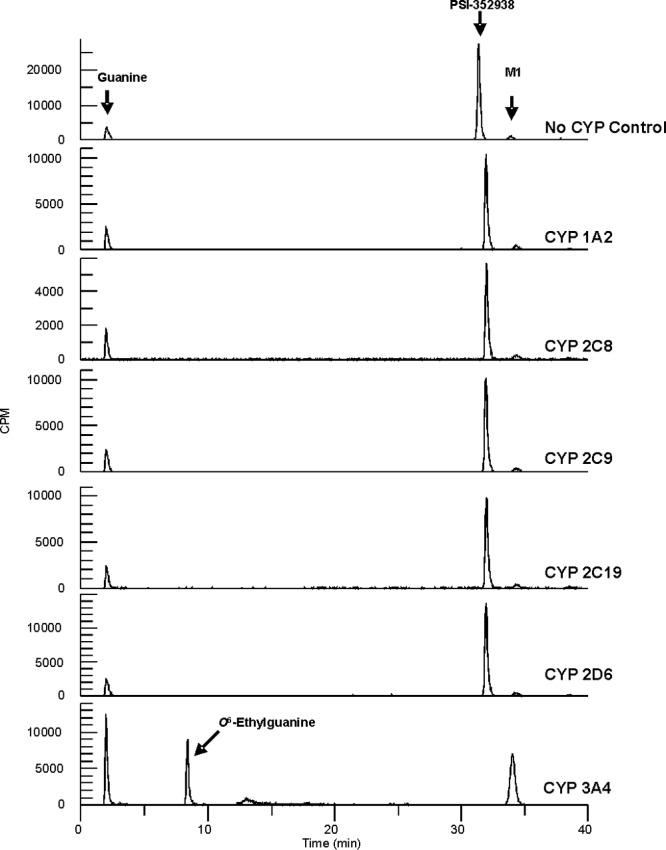
Conversion of PSI-352938 to M1. PSI-352938 was incubated with 0.5 μM CYP1A2, 1.5 μM CYP2C8, 1 μM CYP2C9, 1 μM CYP2C19, 1 μM CYP2D6, or 0.5 μM CYP3A4, and the product was separated by HPLC. All reaction mixtures were incubated for 24 h, except for the CYP3A4 reaction mixture, which was incubated for 2 h.
Conversion of M1 to M4 by PDEs.
The cyclic nucleotide phosphodiesterases (PDEs) are a family of related phosphohydrolases that selectively catalyze the hydrolysis of the adenosine and/or guanosine-3′,5′-cyclic monophosphate (cAMP and/or cGMP). In this study, we tested the intracellular metabolism of PSI-352938, M1, and M3 with four PDEs (PDE2A1, PDE5A, PDE9A, and PDE11A4) that are known to be expressed in the liver and able to hydrolyze cGMP (2). PSI-352938 was not hydrolyzed by any of the PDEs tested after incubation at 37°C for 90 min (data not shown). Both M1 and M3 were substrates for all four PDEs, and the steady-state kinetic parameters were determined. As shown in Table 1, PDE9A demonstrated the highest activity and PDE2A1 was the least active enzyme in hydrolyzing M1. PDE5A and 11A4 showed intermediate activities. The kcat/Km values for M1 were significantly higher than those for M3 for all four enzymes, mostly due to tighter substrate binding (lower Km values).
Table 1.
Hydrolysis of M1 and M3 by phosphodiesterasesa
| Enzyme | M1 |
M3 |
||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | |
| PDE2A1 | 34 ± 13 | 0.15 ± 0.02 | 0.0044 | 373 ± 103 | 0.62 ± 0.10 | 0.0017 |
| PDE5A | 23 ± 5 | 1.49 ± 0.09 | 0.065 | 117 ± 12 | 1.81 ± 0.07 | 0.015 |
| PDE9A | 17 ± 10 | 1.21 ± 0.19 | 0.071 | 194 ± 42 | 1.37 ± 0.13 | 0.0071 |
| PDE11A4 | 46 ± 10 | 0.46 ± 0.03 | 0.01 | 215 ± 21 | 0.84 ± 0.04 | 0.0039 |
Results are reported as average values (±standard errors, where shown) based on three and two independent experiments for M1 and M3, respectively.
Effect of CYP and PDE inhibitors on the activation of PSI-352938 in primary human hepatocytes.
Cellular metabolism studies in primary human hepatocytes were performed in the presence of a CYP3A4 inhibitor, ketoconazole, to confirm that CYP3A4 is responsible for converting PSI-352938 to M1. Primary human hepatocytes from two different donors were treated with 5 μM [3H]PSI-352938 in the presence of different concentrations of ketoconazole, and the intracellular concentrations of M1 plus M4, M2, M5, M7, M8, and PSI-352666 were determined (Fig. 5A and B). Formation of all of the metabolites was inhibited by ketoconazole in a dose-dependent manner, indicating that CYP3A4 is likely to be the major enzyme that removes the isopropyl group.
Fig 5.

Effect of ketoconazole on PSI-352938 metabolism. Primary human hepatocytes from two different donors (A and B) were treated with 5 μM [3H]PSI-352938 for 4 h in the presence of different concentrations of ketoconazole, and the amounts of metabolites were quantified. The effects of inhibitors of CYPs and PDEs on the intracellular activation of PSI-352938 to PSI-352666 are shown in panel C. Primary human hepatocytes were incubated with 10 μM PSI-352938 for 4 h either alone or in the presence of 5 μM ritonavir (CYP3A4), 10 μM ketoconazole (CYP3A4), 5 μM montelukast (CYP2C8 and -9), 500 μM aminobenzotriazole (pan-CYP), 10 μM rilopram (PDE4), 10 μM sidenafil (PDE5), and 100 μM ibudilast (pan-PDE). Values are the means ± standard deviations from at least three independent experiments done in duplicate with cells from separate hepatocyte donors. Statistical significance was assessed by Student's unpaired two-tailed t test, assuming equal variance (*, P < 0.05; **, P < 0.01).
In order to further establish the role of CYP3A4 and PDEs in the intracellular activation of PSI-352938, the effects of inhibitors of CYPs and PDEs on the formation of PSI-352666 in primary human hepatocytes were studied (Fig. 5C). Consistent with results described above, the CYP3A4 inhibitors ritonavir and ketoconazole were able to almost completely block the formation of PSI-352666. While the nonspecific CYP inhibitor aminobenzotriazole also significantly inhibited PSI-352666 formation, the inhibitor of CYP2C8 and 2C9, montelukast, did not show a significant effect on activation. The PDE4 inhibitor rilopram did not show an effect on PSI-352938 activation, suggesting either a reduced role for PDE4 or inhibitor concentrations were not sufficient. In contrast, the PDE5 inhibitor sildenafil and the pan-PDE inhibitor ibudilast significantly reduced triphosphate formation. Combined, these results further establish the specific role of CYP3A4 and the involvement of multiple PDEs in the hepatic activation of PSI-352938.
Dephosphorylation by 5′-NTase.
The family of 5′-nucleotidases (5′-NTases) catalyzes the dephosphorylation of ribo- and deoxyribonucleoside monophosphates to nucleosides and organic phosphate. Two 5′-nucleotidases, ecto-5′-nucleotidase (5′-NTase CD73; low-Km 5′-nucleotidase) and cytosolic-5′-nucleotidase II (cN-II; high-Km 5′-nucleotidase) have been extensively studied and are known to catalyze the dephosphorylation of purine nucleoside-5′-monophosphates (3, 14, 28). Since nucleoside metabolites M6 and M7 were formed in our metabolic study, we examined the ability of both enzymes to catalyze the dephosphorylation of 5′-phosphate metabolites of PSI-352938 and other related compounds.
In order to understand structure-activity relationships (SAR), compounds shown in Fig. 6 were tested against both 5′-NTase CD73 and cN-II. As shown in Table 2, all of the compounds were dephosphorylated by 5′-NTase CD73, but when the activity was tested with cN-II, kinetic parameters could not be determined because the Km values were too high (>5 mM), except for the natural nucleotide GMP. Therefore, 5′-NTase CD73 is likely to be primarily responsible for the formation of the nucleoside metabolites. Compounds with different substitutions at the 2′-α position (R1) were compared. The kcat/Km values for the compounds with 2′-α-OH (compounds 3 and 4) were 4- and 2.5-fold higher than those with 2′-α-F (compounds 7 and 5), respectively. Addition of a 2′-C-methyl group (R2 position) significantly reduced the 5′-NTase activity: compounds 3 and 4 exhibited a 26- or 17-fold-lower kcat/Km value than compound 1 or 2, respectively. The reduction in the activity is mostly due to weaker binding as the Km values were significantly increased by addition of a 2′-C-methyl group. We also examined the effects of different substitutions at the O6 position (R3) on the 5′-NTase CD73. The kcat/Km values for the compounds with O6-methylguanine (compounds 2, 4, and 5) were 2-, 3-, and 5-fold higher than those with the guanine base (compounds 1, 3, and 7), respectively.
Fig 6.
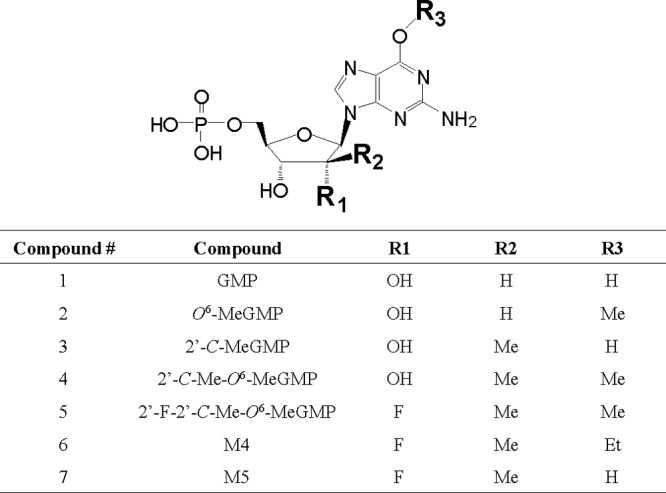
Structures of compounds tested with 5′-NTases CD73 and cN-II.
Table 2.
Dephosphorylation of GMP and GMP analogs by 5′-NTases CD73 and cN-IIa
| Compound no. | CD73 |
cN-II |
||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) | |
| 1 | 15.9 ± 0.8 | 5.0 ± 0.9 | 0.31 | 838 ± 78 | 0.010 ± 0.001 | 1.2 × 10−5 |
| 2 | 11.4 ± 0.2 | 7.3 ± 1.5 | 0.64 | >5,000 | NDb | ND |
| 3 | 273 ± 29 | 3.3 ± 0.4 | 0.012 | >5,000 | ND | ND |
| 4 | 255 ± 39 | 9.6 ± 0.3 | 0.038 | >5,000 | ND | ND |
| 5 | 261 ± 9 | 4.0 ± 1.3 | 0.015 | >5,000 | ND | ND |
| 6 | 316 ± 8 | 4.8 ± 1.1 | 0.015 | >5,000 | ND | ND |
| 7 | 346 ± 16 | 1.1 ± 0.4 | 0.003 | >5,000 | ND | ND |
Results are reported as average values (±standard deviation where shown) based on at least three independent experiments.
ND, not determined.
DISCUSSION
PSI-352938 is a cyclic phosphate prodrug of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine 5′-monophosphate with potent anti-HCV activity both in vitro and in vivo. This prodrug must be metabolized to the active 5′-triphosphate form to inhibit HCV NS5B RNA polymerase. In this study, cell-based metabolism studies and enzyme assays were performed to understand the metabolic pathway of PSI-352938 and to identify the human enzymes that could play a role in metabolizing the prodrug. Figure 1 provides a proposed metabolic pathway of PSI-352938 in primary human hepatocytes. Eight of the nine metabolites shown in Fig. 1 were detected in primary human hepatocytes. The results shown in Fig. 3 suggested that M3 may be metabolized efficiently. Therefore, it is possible that even if M3 was formed in the cells, it may not be detectable due to rapid metabolism.
The first step of PSI-352938 metabolism could be either removal of the isopropyl group in the cyclic phosphate moiety of PSI-352938 to form M1 or removal of the O6-ethyl group to form M2. The enzyme (if any) that is responsible for the removal of the O6-ethyl group is unknown. Our metabolism studies described in Fig. 3B demonstrated that conversion of M2 to M3 was highly inefficient since incubation of primary hepatocytes with M3 resulted in formation of a large amount of PSI-352666, whereas very little PSI-362666 was formed from M2. In addition, a significantly smaller amount of PSI-352666 was formed when cells were incubated with M2 than with PSI-352938, indicating that M2 is not a primary metabolite. Therefore, the first step of the primary pathway most likely involves the removal of the isopropyl group to form M1 metabolite.
We found that the first step of the metabolism appears to be CYP dependent, and among the CYP isozymes tested, CYP3A4 was the only enzyme of the panel tested that was able to catalyze this reaction. This was also confirmed by our metabolism results, which showed that ketoconazole, ritonavir, and aminobenzotriazole, inhibitors of CYP3A4, inhibited the metabolism of PSI-352938. This CYP3A4-dependent activation of PSI-352938 could explain the difference in metabolic profiles in primary human hepatocytes and clone A cells, which are Huh 7 cells containing subgenomic HCV replicons, as shown in Fig. 2. It has been reported that CYP3A4 was highly expressed in primary hepatocytes, but it was not detected in Huh 7 cells (13). Therefore, in clone A cells, CYP3A4-dependent steps (PSI-352938 to M1 and M2 to M3) cannot take place, resulting in accumulation of the M2 metabolite. Slow formation of PSI-352666 in clone A is likely due to a trace amount of CYP3A4 expressed in the cells or some other unidentified enzyme that can slowly catalyze the reaction. In primary human hepatocytes, on the other hand, a significant level of PSI-352666 was formed since CYP3A4 is highly expressed in these cells. CYP3A4 is predominantly expressed in liver and intestine (25). It is important that the activation of PSI-352938 requires CYP3A4 because HCV is a chronic liver disease.
The reaction catalyzed by CYPs is a monooxygenase reaction, and it has been reported that organophosphates could be substrates for CYP3A4, including nucleotide prodrugs using HepDirect technology, which contain cyclic 1,3-propanyl esters (7, 8). Here we have demonstrated for the first time that a 3′,5′-cyclic phosphate prodrug can be activated by CYP3A4. We propose the reaction mechanism to be oxidation of the secondary carbon of the isopropyl group to form a hemiketal intermediate followed by rapid hydrolysis of the hemiketal to release the cyclic phosphate and the by-product, acetone.
The second step in the metabolic pathway involves either hydrolysis of the 3′,5′-cyclic phosphate moiety of the nucleoside analogs to their corresponding monophosphates or removal of the O6-ethyl group in the nucleobase. We have reported previously that adenosine deaminase-like protein 1 (ADAL1) is able to hydrolytically remove the O6-ethyl group from M4, but not from M1. Therefore, the second step is likely to be hydrolysis of the 3′,5′-cyclic phosphate. Our results showed the cellular enzymes responsible for this step include a number of phosphodiesterases (PDEs). The PDEs are grouped into 11 families (PDE1 through PDE11) according to differences in their amino acid sequences, kinetic properties, modes of allosteric regulation, and sensitivity to chemical inhibitors (9). Some PDE families specifically hydrolyze cAMP (e.g., PDE4, PDE7, and PDE8), some specifically hydrolyze cGMP (e.g., PDE5, PDE6, and PDE9), and others can hydrolyze both substrates (e.g., PDE1, PDE2, PDE3, PDE10, and PDE11). In particular, we tested PDE2A, PDE9A, PDE5A1, and PDE11A4 because they are known to be expressed in the liver and are able to hydrolyze cGMP (2). All four PDEs were able to catalyze the hydrolysis of M1 or M4 but not PSI-352938. Interestingly, the kcat/Km values for compounds with O6-ethylguanine were significantly higher than those with the natural guanine base, mostly due to a lower Km, suggesting that the O6-ethyl group is contributing to the tighter binding. Furthermore, the activation of PSI-352938 was observed to be sensitive to the PDE5 inhibitor sildenafil and the pan-PDE inhibitor ibudilast in primary human hepatoctyes. Therefore, the second step of the primary pathway for PSI-352938 metabolism should be hydrolysis of M1 to form M4. Since multiple PDEs can catalyze this reaction, selective PDE inhibitors are unlikely to affect metabolism of PSI-352938.
The final three steps leading to the formation of the active 5′-triphosphate are removal of the O6-ethyl group followed by phosphorylation events catalyzed by cellular kinases. We demonstrated previously that ADAL1 can efficiently remove the O6-ethyl group to form M5 (23). M5 is then phosphorylated to M8 by guanylate kinase (GUK1) (10). The diphosphate metabolite M8 is subsequently metabolized to PSI-352666 by nucleoside diphosphate kinase (NDPK), pyruvate kinase (PK), and 3-phosphoglycerate kinase (3PGK) (10). Among the three enzymes, NDPK is the primary enzyme that phosphorylates M8. PSI-352666 was shown to be a potent inhibitor of HCV NS5B RNA polymerase (10, 18).
In our metabolism studies, formation of the nucleoside metabolites M6 and M7 was observed. Our results demonstrated that the 5′-monophosphate metabolites (M4 and M5) can be dephosphorylated to the corresponding nucleoside metabolites (M6 and M7) by 5′-NTase. We examined two 5′-NTases, ecto-5′-NTase CD73 (low-Km 5′-nucleotidase) and cytosolic 5′-NTase II (high-Km 5′-nucleotidase), which are known to catalyze the dephosphorylation of purine nucleoside-5′-monophosphates. The kinetic parameters for M6 and M7 with 5′-NTase cN-II could not be determined because the Km values for those compounds were too high (>5 mM). The parameters for 5′-NTase CD73 were determined, but the catalytic efficiencies (kcat/Km) for M6 and M7 were approximately 20- and 100-fold lower, respectively, than that of the natural substrate, GMP. Our SAR study further demonstrated that the low activities with M6 and M7 are primarily due to the 2′-C-methyl substitution. Nevertheless, 5′-nucleotidase CD73 is at least one of the enzymes responsible for formation of the nucleoside metabolites.
The nucleoside metabolites could be further processed by cellular enzymes. M6 can be converted to M7 by adenosine deaminase (ADA) but much less efficiently than its O6-methylated counterpart, 2′-F-2′-C-Me-O6-methylguanosine. M7 can act as a poor substrate for 2′-deoxyguanosine kinase (dGK) to be phosphorylated back to M5, but M6 cannot be phosphorylated to M4 by dGK (unpublished results). Since these nucleoside metabolites were poorly phosphorylated, they may be degraded to the nucleobase either chemically or enzymatically. Indeed, such nucleobase metabolites were observed in the extracellular fractions collected after incubation of primary human hepatocytes with PSI-352938 (data not shown).
In summary, our studies have determined the primary metabolic pathway and identified enzymes involved in activating PSI-352938. The first step, removal of the isopropyl group in the cyclic phosphate moiety of PSI-352938 to form M1, is catalyzed by CYP3A4. The second step, hydrolysis of the cyclic phosphate of M1 to form the 5′-monophosphate intermediate M4, can be catalyzed by multiple PDEs, including PDEs 2A1, 5A, 9A, and 11A4. The third step can be catalyzed by ADAL1, which removes O6-ethyl group from M4 to form M5 (23). The fourth and fifth steps are phosphorylation reactions mediated by GUK1 and NDPK, which convert M5 to M8 and M8 to PSI-352666, respectively (10). Overall the results from this study characterize the intracellular events involved in activating PSI-352938 and provide a better understanding of how this cyclic phosphate nucleotide prodrug is metabolized to its active form that ultimately inhibits HCV replication.
Footnotes
Published ahead of print 23 April 2012
REFERENCES
- 1. Alter HJ. 2005. HCV natural history: the retrospective and prospective in perspective. J. Hepatol. 43:550–552 [DOI] [PubMed] [Google Scholar]
- 2. Bender AT, Beavo JA. 2006. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58:488–520 [DOI] [PubMed] [Google Scholar]
- 3. Bianchi V, Spychala J. 2003. Mammalian 5′-nucleotidases. J. Biol. Chem. 278:46195–46198 [DOI] [PubMed] [Google Scholar]
- 4. Clark JL, et al. 2006. Synthesis and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methyl purine nucleosides as inhibitors of hepatitis C virus RNA replication. Bioorg. Med. Chem. Lett. 16:1712–1715 [DOI] [PubMed] [Google Scholar]
- 5. De Clercq E. 2004. Antiviral drugs in current clinical use. J. Clin. Virol. 30:115–133 [DOI] [PubMed] [Google Scholar]
- 6. Eldrup AB, et al. 2004. Structure-activity relationship of purine ribonucleosides for inhibition of hepatitis C virus RNA-dependent RNA polymerase. J. Med. Chem. 47:2283–2295 [DOI] [PubMed] [Google Scholar]
- 7. Erion MD, et al. 2004. Design, synthesis, and characterization of a series of cytochrome P(450) 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J. Am. Chem. Soc. 126:5154–5163 [DOI] [PubMed] [Google Scholar]
- 8. Erion MD, et al. 2005. Liver-targeted drug delivery using HepDirect prodrugs. J. Pharmacol. Exp. Ther. 312:554–560 [DOI] [PubMed] [Google Scholar]
- 9. Francis SH, Turko IV, Corbin JD. 2001. Cyclic nucleotide phosphodiesterases: relating structure and function. Prog. Nucleic Acid Res. Mol. Biol. 65:1–52 [DOI] [PubMed] [Google Scholar]
- 10. Furman PA, et al. 2011. Activity and the metabolic activation pathway of the potent and selective hepatitis C virus pronucleotide inhibitor PSI-353661. Antiviral Res. 91:120–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gane E, et al. 2009. Combination therapy with a nucleoside polymerase (R7128) and protease (R7227/ITMN-191) inhibitor in HCV: safety, pharmacokinetics, and virologic results from INFORM-1, abstr 193. Abstr. 60th Annu. Meet. Am. Assoc. Study Liver Dis., Boston, MA [Google Scholar]
- 12. Gunic E, et al. 2007. Cyclic monophosphate prodrugs of base-modified 2′-C-methyl ribonucleosides as potent inhibitors of hepatitis C virus RNA replication. Bioorg. Med. Chem. Lett. 17:2452–2455 [DOI] [PubMed] [Google Scholar]
- 13. Guo L, et al. 2011. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab. Dispos. 39:528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ipata PL, Tozzi MG. 2006. Recent advances in structure and function of cytosolic IMP-GMP specific 5′-nucleotidase II (cN-II). Purinergic Signal. 2:669–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishii K, et al. 1999. Expression of hepatitis C virus NS5B protein: characterization of its RNA polymerase activity and RNA binding. Hepatology 29:1227–1235 [DOI] [PubMed] [Google Scholar]
- 16. Korth M, Engels J. 1979. The effects of adenosine- and guanosine 3′,5′-phosphoric acid benzyl esters on guinea-pig ventricular myocardium. Naunyn Schmiedebergs Arch. Pharmacol. 310:103–111 [DOI] [PubMed] [Google Scholar]
- 17. Lalezari J, et al. 2008. Potent antiviral activity of the HCV nucleoside polymerase inhibitor R7128 with PEG-IFN and ribavirin: interim results of R7128 500mg BID for 28 days, abstr 66. Abstr. 43rd Annu. Meet. Eur. Assoc. Study Liver, Milan, Italy [Google Scholar]
- 18. Lam AM, et al. 2011. Inhibition of hepatitis C virus replicon RNA synthesis by PSI-352938, a cyclic phosphate prodrug of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine. Antimicrob. Agents Chemother. 55:2566–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawitz E, et al. 2009. Potent antiviral activity observed with PSI-7851, a novel nucleotide polymerase inhibitor for HCV, following multiple ascending oral doses for 3 days in patients with chronic HCV infection, abstr 103. HEP DART, Kohala Coast, HI, 6 to 10 December 2010 [Google Scholar]
- 20. Lohmann V, Korner F, Herian U, Bartenschlager R. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71:8416–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lohmann V, Roos A, Korner F, Koch JO, Bartenschlager R. 1998. Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology 249:108–118 [DOI] [PubMed] [Google Scholar]
- 22. Migliaccio G, et al. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278:49164–49170 [DOI] [PubMed] [Google Scholar]
- 23. Murakami E, et al. 2011. Adenosine deaminase-like protein 1 (ADAL1): characterization and substrate specificity in the hydrolysis of N(6)- or O(6)-substituted purine or 2-aminopurine nucleoside monophosphates. J. Med. Chem. 54:5902–5914 [DOI] [PubMed] [Google Scholar]
- 24. Murakami E, et al. 2010. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 285:34337–34347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paine MF, et al. 2006. The human intestinal cytochrome P450 “pie.” Drug Metab. Dispos. 34:880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reddy PG, et al. 2010. 2′-Deoxy-2′-alpha-fluoro-2′-beta-C-methyl 3′,5′-cyclic phosphate nucleotide prodrug analogs as inhibitors of HCV NS5B polymerase: discovery of PSI-352938. Bioorg. Med. Chem. Lett. 20:7376–7380 [DOI] [PubMed] [Google Scholar]
- 27. Reddy R, et al. 2007. Antiviral activity, pharmacokinetics, safety, and tolerability of R7128, a novel nucleoside HCV polymerase inhibitor, following multiple, ascending, oral doses in patients with HCV genotype 1 infection who have failed prior interferon therapy, abstr LB9. Abstr. 58th Annu. Meet. Am. Assoc. Study Liver Dis., Boston, MA [Google Scholar]
- 28. Resta R, Yamashita Y, Thompson LF. 1998. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 161:95–109 [DOI] [PubMed] [Google Scholar]
- 29. Yamashita T, et al. 1998. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J. Biol. Chem. 273:15479–15486 [DOI] [PubMed] [Google Scholar]