

Abstract
Resistance to trimethoprim (TMP) resulting from point mutations in the enzyme drug target dihydrofolate reductase (DHFR) drives the development of new antifolate inhibitors effective against methicillin-resistant Staphlococcus aureus (MRSA). For the past several years we have used structure-based design to create propargyl-linked antifolates that are highly potent antibacterial agents. In order to focus priority on the development of lead compounds with a low propensity to induce resistance, we prospectively evaluated resistance profiles for two of these inhibitors in an MRSA strain. By selection with the lead inhibitors, we generated resistant strains that contain single point mutations F98Y and H30N associated with TMP resistance and one novel mutation, F98I, in DHFR. Encouragingly, the pyridyl propargyl-linked inhibitor selects mutants at low frequency (6.85 × 10−10 to 1.65 × 10−9) and maintains a low MIC (2.5 μg/ml) and a low mutant prevention concentration (1.25 μg/ml), strongly supporting its position as a lead compound. Results from this prospective screening method inform the continued design of antifolates effective against mutations at the Phe 98 position. Furthermore, the method can be used broadly to incorporate ideas for overcoming resistance early in the development process.
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) is a Gram-positive, highly pathogenic bacterium prevalent in hospital environments (16). In recent years, the transition from hospital-acquired (HA) to community-acquired (CA) infections (2, 22, 24) and the emergence of multidrug-resistant MRSA have presented major problems in the treatment of these infections. Several diverse MRSA strains have emerged and now account for more than 60% of clinical S. aureus strains isolated from intensive care units in the United States (31). In addition to methicillin resistance, clinically isolated S. aureus strains can also be resistant to aminoglycosides, macrolides, tetracycline, or several disinfectants (23). Although vancomycin is often used to treat hospital-acquired infections, several MRSA strains were identified with vancomycin resistance transferable from Enterococcus (4, 23).
Dihydrofolate reductase (DHFR) is a highly conserved enzyme required for reducing folate cofactors involved in the biosynthesis of deoxythymidine monophosphate (dTMP) and several amino acids. Because DHFR is an essential enzyme, it is a validated drug target for bacterial and protozoal infections as well as malignancies (12, 21). Trimethoprim (TMP) is a successful broad-spectrum inhibitor of DHFR frequently used in combination with sulfamethoxazole (Bactrim) to treat bacterial infections. In fact, sulfamethoxazole has been successfully used to treat serious CA MRSA infections (20). However, point mutations in DHFR associated with reduced TMP sensitivity, primarily H30N/F98Y and F98Y/H149R, have been identified; presently, 28% of MRSA strains are TMP resistant (15).
We have developed a series of propargyl-based DHFR inhibitors to inhibit wild-type and TMP-resistant MRSA strains (10). In previous work, we used crystal structures to understand trimethoprim resistance in S. aureus DHFR with point mutations F98Y and H30N/F98Y (10, 11). From these structural studies, we found that our leading first-generation compounds, characterized by a diaminopyrimidine ring, propargyl linker, and meta-biphenyl ring system (Fig. 1), retain potency for the F98Y DHFR mutant with 50% inhibition concentration (IC50) values in the low nanomolar range by promoting favorable interactions with the standard, extended conformation of cofactor NADPH (10). Additional studies for the H30N/F98Y DHFR mutant revealed potency losses of up to 95-fold compared to wild-type DHFR, most likely owing to the loss of a water-mediated hydrogen bond between the pyrimidine ring and Thr111 (11).
Fig 1.
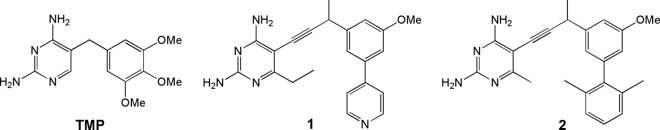
Chemical structures of trimethoprim and compounds 1 and 2.
From these discoveries, a new generation of propargyl-linked compounds was developed with improved solubility and potency for both wild-type and mutant DHFR. The new series replaces the distal biphenyl with a pyridine ring (Fig. 1). Several representative compounds from this series are potent inhibitors of the wild-type DHFR enzyme (IC50s range from 12 to 26 nM) and several clinically isolated MRSA strains (MIC values between 0.02 and 2.8 μg/ml) (36). Four of the pyridyl-containing compounds have been shown to retain potency against a TMP-resistant strain, with an MIC value of 0.09 μg/ml (36).
Given the emergence of TMP-resistant MRSA strains, we decided to prospectively determine resistance profiles for the leading propargyl-linked compounds. In this work, we generated spontaneous mutants resistant to propargyl-linked compounds with meta-biphenyl or pyridine moieties (Fig. 1) from progenitor MRSA strain Staphylococcus aureus subsp. aureus ATCC 44300. For each of the resistant strains, we characterized the genotypic sequence of dfrA, the gene encoding DHFR, for analysis of point mutations in the target enzyme. Selected DHFR mutants were characterized using mutation frequencies, fitness, MICs, and mutant prevention concentrations (MPCs). To explore any correlation of bacterial fitness with enzyme fitness, we constructed the mutant DHFR enzymes and determined kinetic parameters and inhibition constants.
Interestingly, selection with our propargyl-linked pyridyl compound reveals two mutations in DHFR: the F98Y mutation associated with TMP resistance and a novel mutation, F98I. Similarly, selection with the meta-biphenyl compound reveals the single mutations H30N and F98Y in DHFR; these two mutations are often found together in clinically isolated TMP-resistant strains (8, 10). While resistance mutations emerge in response to the propargyl-linked inhibitors, the mutational frequencies are similar to or lower than those observed for TMP. Also encouraging, the MPC for the lead pyridyl compound is only 1.25 μg/ml. The pyridyl compound also maintains potency for the mutant enzymes; there are losses in potency of only 4-fold for the Sa (F98Y) DHFR enzyme and 2-fold for the Sa (H30N) enzyme. There do not appear to be any significant penalties in enzyme fitness imparted by the inhibitor-specific point mutations.
In determining a resistance profile for the lead propargyl-linked antifolate, we have developed a prospective study, experimentally investigating resistance genotypes during the early stages of drug design. This investigation differs from previous retrospective studies that analyzed resistance genotypes emerging in MRSA strains selected by clinically used antibiotics (26, 34, 35). Results from the resistance profile determination suggest that the pyridine-based compound may be useful in preventing the emergence of resistant strains, as it possesses a low MPC value and maintains a low MIC value for the resistant bacteria. These results emphasize that the experimental prescreening of new inhibitors for resistance is a worthy effort during the early stages of drug design, as it yields a more detailed understanding of the lead series and informs the continued design of the compounds.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and antimicrobial agents.
All experiments were performed using Staphylococcus aureus subsp. aureus ATCC 44300 as the progenitor strain. Bacterial cultures were grown with aeration at 37°C in Iso-sensitest broth (ISB) from Oxoid. Compounds 1 and 2 were synthesized as described previously (1, 36). Trimethoprim (TMP) was purchased from Sigma-Aldrich.
Minimum inhibition concentrations (MICs) were determined using the broth microdilution method (39) according to CLSI standards (6), using a final inoculum of 5 × 105 CFU/ml. The MIC was defined as the lowest concentration of antibiotic or inhibitor preventing visible growth after monitoring cell turbidity at an optical density at 600 nm (OD600) following an incubation period of 18 h at 37°C. Mutant prevention concentrations (MPCs) were determined using methods similar to those described in reference 9. The MRSA progenitor strain was cultured in ISB until the stationary phase and was then resuspended in fresh media. An inoculum of 1010 CFU/ml, containing various concentrations of TMP and the inhibitors, was plated onto ISA plates and then subsequently grown for 30 h at 37°C. TMP concentrations used for MPC determinations were several magnitudes above and below the determined MIC value, ranging from 0.039 to 20 μg/ml, where the concentration in each plate increased by successive 2-fold increments. Similarly, a range of concentrations for compound 1 (0.0098 to 5 μg/ml), increasing by 2-fold increments, was applied to ISA plates. The MPC was defined as the MIC of the least susceptible single-step mutant, as determined by the prevention of mutant growth on ISA plates containing TMP or inhibitor (33).
Selection of TMP-resistant and inhibitor-resistant DHFR mutants.
TMP-resistant and inhibitor-resistant DHFR mutants were selected by plating 100 μl of saturated overnight cultures (approximately 1011 CFU/ml) in triplicate onto selective ISA plates containing 5× the MIC for TMP and 10× the MIC for inhibitors 1 and 2 (35). Selective plates were grown for 30 h at 37°C. An estimate of mutation frequency (prior to DNA sequencing) for each inhibitor was calculated by averaging the number of TMP/inhibitor-resistant mutants recovered on triplicate plates and then expressing the results as a fraction of the viable count plate.
Detection of point mutations in DHFR.
All TMP-resistant and inhibitor-resistant mutants were subjected to repassage on selective ISA plates. Whole colonies were lysed with stapholysin (Sigma Aldrich) to release genomic DNA and serve as a template for PCR. Specific primers designed previously (10) were used to isolate and amplify the dfrA gene from resistant strains by the use of methods described in reference 19 for the detection of the mecA gene. All PCR products were screened for point mutations in DHFR by high-quality DNA sequencing (Genewiz). Primers used to detect the dfrA gene contained restriction enzyme sites for NdeI and XhoI in order to ligate PCR products into the pET41 vector for further validation using DNA sequencing with the T7 promoter sequencing primer. Mutation frequencies for each point mutation were recalculated based on the sequencing information provided for each mutant colony.
Determination of fitness.
Pairwise competition assays as described by Lenski (18, 35) were used to determine the fitness for wild-type and mutant strains. Bacterial cultures were prepared in triplicate by inoculating ISB with a mixture of the susceptible progenitor strain Staphylococcus aureus subsp. aureus ATCC 44300 and each mutant strain in a 10:1 ratio. Cultures were diluted and plated on nonselective and selective ISA plates (with or without inhibitors) at 0 h and 24 h time points to determine colony counts. All plates were incubated for 24 h at 37°C. Relative competitive fitness data were calculated according to the relative fitness equation determined by Lenski (18, 35).
Effux pump evaluation.
Resistant colonies that showed increased MIC values but did not contain point mutations in the dfrA gene were alternatively tested for efflux pump activity using methods described in reference 13. MIC values were tested using the microdilution method described above with and without the known efflux pump inhibitors (EPIs) at concentrations corresponding to half the MIC value determined for each inhibitor: reserpine (at 20 μg/mL), thioridazine (at 12.5 μg/mL), and verapamil (at 200 μg/mL) (7, 15). All MIC value determinations for susceptible and resistant strains were performed in triplicate on the same day.
Cloning, expression, and purification of DHFR mutant enzymes.
Sa (F98Y), Sa (F98I), and Sa (H30N) DHFR PCR sequences recovered from the selection studies were ligated into the pET41 vector containing a His8 tag at the C-terminal end. Recombinant clones were verified by ABI DNA sequencing. All constructs were propagated in E. coli BL21(DE3) cells via transformation. The mutant DHFR enzymes were overexpressed at high yields (∼60 mg/liter of cells) by induction with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and growth for an additional 6 h at 30°C. Expression and purification conditions for the wild-type DHFR enzyme (Sa DHFR) were the same as previously described, using nickel affinity chromatography followed by size exclusion chromatography (10, 11).
Enzyme kinetic and inhibition assays for DHFR mutants.
Enzyme activity and inhibition assays were performed in triplicate for each mutant by monitoring the rate of NADPH oxidation by the DHFR enzyme at an absorbance of 340 nm (10, 11). Using enzyme inhibition assays, IC50s were calculated for TMP and inhibitors 1 and 2. Kinetic parameters were measured by performing triplicate enzyme activity assays at various substrate concentrations of dihydrofolate (12.5, 25, 50, 75, and 100 μM) and analyzed with Lineweaver-Burk plots to calculate Km, Vmax, kcat, and catalytic efficiency (kcat/Km). Km values, in addition to the obtained IC50s, were used to calculate Ki values for each enzyme and inhibitor (5). Km values for NADPH were determined by adjusting the NADPH concentration (20, 40, 60, 80, and 100 μM) while keeping the DHF concentration constant at 100 μM and then analyzing the data with Lineweaver-Burk and Eadie-Hofstee double-reciprocal plots.
RESULTS
Characterization of DHFR mutants from selection with TMP and inhibitors.
Several independent cultures were used to generate TMP-resistant mutants (TMP was used as a positive control in these experiments) and inhibitor-resistant mutants from the progenitor strain 43300. Similar to previous reports (28, 35), we observed breakthrough colonies that were difficult to distinguish from mutants when bacteria were plated at concentrations below the MIC values obtained in culture. Therefore, mutants were selected on plates with 5 times (TMP) or 10 times (compounds 1 and 2) the concentration above the MIC values determined (35). All of the DHFR mutants identified were prepared as single-step mutants. Sequencing results reveal that all point mutations are conferred by a single nucleotide base change (Table 1), where the replacement of thymine with adenine is most prevalent for mutations at the F98 position.
Table 1.
Resistance profiles for strains with DHFR mutations
| Inhibitor | DHFR mutation [no. of colonies containing mutations/total no. of colonies recovered] | Single nucleotide polymorphisma | Mutation frequency (± SD) | Wild-type MIC (μg/ml) | Mutant MIC (μg/ml) | Fold potency lossb | Fitness ratio (± SD)c |
|---|---|---|---|---|---|---|---|
| TMP | F98Y [5/7] | TTT to TAT | 1.65 × 10−9 (± 0.39) | 0.625 | 10 | 16 | 0.982 (± 0.097) |
| 1 | F98I [4/14] | TTT to ATT | 6.87 × 10−10 (± 2.3) | 0.078 | 2.5 | 32 | 1.04 (± 0.23) |
| F98Y [7/14] | TTT to TAT | 1.20 × 10−9 (± 0.19) | 0.078 | 2.5 | 32 | 0.993 (± 0.21) | |
| 2 | H30N [5/11] | CAT to AAT | 8.88 × 10−10 (± 0.50) | 1.25 | 20 | 16 | 0.974 (± 0.26) |
| F98Y [4/11] | TTT to TAT | 7.10 × 10−10 (± 0.53) | 1.25 | 20 | 16 | 0.993 (± 0.21) |
The single nucleotide polymorphisms are indicated in bold.
Fold potency loss = (MICMutant/MICParent).
Fitness ratios represent the relative fitness of mutants compared to the wild type determined using pair-wise competition assays as described by Lenski et al. (18).
In the control experiment, TMP-resistant mutants with the F98Y mutation in DHFR were selected with a frequency of 1.65 × 10−9 (Table 1), which is consistent with earlier studies, where the frequency ranged between 1.06 × 10−10 and 1.18 × 10−9 (35). Mutants generated from selection with inhibitor 1 were identified with F98Y and F98I mutations and had frequencies similar to or slightly lower than those of the F98Y mutants selected with TMP, with frequencies of 1.20 × 10−9 and 6.87 × 10−10, respectively (Table 1). Specific to inhibitor 1, the F98I DHFR mutation is a novel mutation generated at a lower frequency than the F98Y mutation. Finally, mutants selected with compound 2 have single F98Y and H30N DHFR mutations, both associated with TMP-resistant strains and often observed as double mutations in clinically isolated strains. Despite the generation of resistance genotypes in dfrA, the mutational frequencies observed for inhibitors 1 and 2 were lower than those observed for members of other classes of antibiotics that include norfloxacin, rifampin, and fusidic acid, for which point mutations accumulate in the enzyme target at frequencies near 2.87 (± 0.27) × 10−8 (34), 2.0 × 10−7 (25), and 8.0 × 10−7 (25), respectively.
Susceptibility testing for all mutants using the microdilution method (39) revealed a significant increase in MIC values compared to the wild-type progenitor strain, indicating resistance to the inhibitor. As observed for TMP and inhibitors 1 and 2, strains containing the F98Y mutation showed 16-fold and 32-fold losses in potency (Table 1). Similarly, strains with the F98I and H30N mutations revealed a 32-fold loss and 16-fold loss in potency for the inhibitor, respectively. Since the F98I mutation is a novel mutation and was selected only with high concentrations of inhibitor 1, these strains were also tested for cross-resistance to TMP. The MIC values confirm that the F98I mutation also confers resistance to TMP, with an 8-fold difference (changing from 0.625 to 5 μg/ml). While a significant increase in the MIC value was observed for all of the inhibitors, pyridine compound 1 maintained considerable potency for strains containing the F98Y and F98I mutations, with MIC values increasing only to 2.5 μg/ml. For comparison, several MRSA strains without point mutations in DHFR have an MIC range for trimethoprim of 1 to 4 μg/ml (19, 35).
Previous work emphasizes the importance of evaluating the fitness costs associated with antibacterial resistance (25, 26, 35). Compelled by the selection of novel mutation F98I in DHFR, we evaluated the bacterial fitness for all of the mutant strains by the use of pairwise competition assays (18, 35). All bacterial strains containing point mutations in dfrA were relatively fit compared to the progenitor strain, with fitness ratios close to 1.00 (Table 1), suggesting that the mutations imparted no significant fitness penalties to the bacterial strains. Our overall results are similar to those of an earlier study, in which MRSA strains containing DHFR mutations L41R, F98Y, F98S, and H150R were also relatively fit compared to the progenitor strain (35).
Confirming the resistance profiles for pyridyl compounds using the MPC.
In order to validate that the pyridyl compound (compound 1) retains potency for MRSA strains containing mutations in DHFR, we determined mutant prevention concentration (MPC) values for TMP and compound 1 using methods described previously for fluoroquinolones (9). An inoculum of 1.0 × 1010 CFU/ml was used to determine the MPC for both inhibitors; therefore, the MPC value is more specifically called the MPC1010. Similar to the fluoroquinolone experiment (9), two sharp declines in cell recovery were observed as the inhibitor concentration increased successively by 2-fold increments (Fig. 2). The first decline was observed near the MIC values for both TMP and compound 1, at nearly 0.625 μg/ml and 0.078 to 0.156 μg/ml, respectively. The second decline was observed before the MPC was reached and can be described as representing the concentration range where single-step mutants are recovered. The MPC emerges as the curve begins to plateau, suggesting that this concentration is sufficient to block the growth of single-step mutants. The MPC values for TMP and compound 1 were 10 μg/ml and 1.25 μg/ml, respectively.
Fig 2.

Effect of TMP and compound 1 concentration on selection of resistant mutants. After growth on selective plates, cells were counted and expressed as the fraction of the cells recovered over the 1010 CFU/ml inoculum. Approximate MIC and MPC values for TMP and compound 1 are indicated on the graph.
The MPC values for TMP and compound 1 reveal a 16-fold change compared to their MIC values for the progenitor strain. Consistent with our hypothesis from the evaluation of susceptibility, the pyridyl compound (compound 1) had a low MPC value of 1.25 μg/ml; this value is still within the range of MIC values of TMP for the wild-type strain. Hypothetical treatment with compound 1 using a concentration within the range of the MPC (1.25 to 2.50 μg/ml) might be able to limit or minimize the development of resistant strains that rely on mechanisms involving point mutations in DHFR.
Evaluation of resistant strains lacking point mutations in DHFR.
DNA sequencing of the dfrA gene for some resistant colonies isolated from selection plates showed that they lacked point mutations in the DHFR gene. These colonies were present at a low frequency for selection with inhibitors 1 and 2 and TMP. On average, of the approximately 11 colonies that are recovered upon selection with each inhibitor, 2 to 3 colonies did not have any point mutations in DHFR (Table 1). Susceptibility testing of these resistant colonies with wild-type DHFR sequences (referred to as F98* strains) revealed a significant reduction in inhibitor potency, with MIC values comparable to those seen with strains containing DHFR point mutations. It is well known that there can be multiple mechanisms of resistance to the archetypical inhibitor, TMP, including alteration of cell wall permeability, overexpression of the wild-type DHFR gene, acquisition of a second, plasmid-encoded DHFR, and metabolic bypass (14). Although drug efflux is not considered a major resistance pathway for TMP, we felt that it was important to ascertain whether our series of compounds were susceptible to extrusion by the multiple drug resistance (MDR) efflux pumps that are often overexpressed in MRSA. MIC values were determined in the presence of the known efflux inhibitors reserpine, thioridazine, and verapamil by the use of concentrations (Table 2) that correspond to half the MIC value reported for each efflux pump inhibitor (EPI). Based on the criteria set forth for efflux experiments in the literature (17, 27, 29, 30), an 8-fold or higher reduction of the MIC value for the EPI is considered indicative of efflux activity. Our results (Table 2) reveal that there were no significant reductions in MIC values for TMP or compound 1 with any of the EPIs. In the presence of compound 2 and thioridazine, there was an 8-fold reduction in the MIC value, suggesting that there might be a potential for the involvement of efflux with this more hydrophobic lead series. We were pleased to see that our most promising and less hydrophobic lead series, represented by compound 1, more closely mirrored the results seen with TMP, which is not susceptible to these mechanisms.
Table 2.
Efflux pump inhibitor evaluation
| Compound | MIC in μg/ml [fold potency loss] for strainF98* |
|||
|---|---|---|---|---|
| No EPI (F98*/wild type) | Reserpine | Thioridazine | Verapamil | |
| TMP | 20 [32] | 20 [1.0] | 10 [2.0] | 5 [4.0] |
| 1 | 10 [128] | 5.0 [2.0] | 2.5 [4.0] | 2.5 [4.0] |
| 2 | 20 [16] | 20 [1.0] | 2.5 [8.0] | 10 [2.0] |
Enzyme inhibition assays to assess enzyme resistance.
In order to validate that mutations in DHFR confer resistance to TMP and inhibitors 1 and 2, we prepared the Sa (F98Y), Sa (F98I), and Sa (H30N) DHFR enzymes for inhibition assays and compared the Ki values with those for the wild-type enzyme, Sa DHFR (Table 3). As shown in previous work (11), the F98Y mutation reduces the potency of TMP by 30-fold. Clinically relevant DHFR mutants Sa (H30N) and Sa (F98Y) have minimal effects on the affinity of the pyridyl compound 1, with only 2-fold and 4-fold losses in potency, respectively. While the F98Y mutation has minimal effects on the potency of meta-biphenyl compound 2, the H30N mutation significantly affects its potency, as observed by its 50-fold reduction in Ki value. The loss of potency for compound 2 toward the Sa (H30N) DHFR enzyme is consistent with previous work showing that the meta-biphenyl compounds maintain potency for the Sa (F98Y) DHFR but lose significant potency (at levels as great as 95-fold) for the Sa (H30N, F98Y) DHFR enzyme (10). Structural analysis of the clinically isolated double mutant Sa (H30N, F98Y) DHFR (11) reveals that the H30N mutation prevents a water-mediated hydrogen bond between the 2-amino group and Thr111. From the extension of this analysis, it is likely that the single H30N mutation also impacts compound 2 by preventing the formation of a key hydrogen bond in the active site.
Table 3.
Ki values for wild-type and mutant DHFR enzymes
| Compound | Ki (± SD) for Sa | Ki (± SD) for Sa (F98Y) | Fold loss (F98Y/Sa) | Ki (± SD) for Sa (F98I) | Fold loss (F98I/Sa) | Ki (± SD) for Sa (H30N) | Fold loss (H30N/Sa) |
|---|---|---|---|---|---|---|---|
| TMP | 0.0033 (± 0.00048) | 0.10 (± 0.0073) | 30 | 0.13 (± 0.051) | 39 | 0.0066 (± 0.0012) | 2.0 |
| 1 | 0.0028 (± 0.00041) | 0.012 (± 0.00088) | 4.3 | 0.055 (± 0.022) | 20 | 0.0059 (± 0.0011) | 2.1 |
| 2 | 0.011 (± 0.0016) | 0.023 (± 0.0017) | 2.1 | 0.17 (± 0.067) | 16 | 0.55 (± 0.10) | 50 |
The novel F98I mutation reduces the potency of TMP and compounds 1 and 2 as reflected by the 39-fold, 20-fold, and 16-fold losses, respectively. These losses in potency are quite dramatic compared to the minimal losses observed for the Sa (F98Y) DHFR mutant and compound 1. In order to understand the greater effect imparted by F98I, we examined the structure of the wild-type enzyme (Protein Data Bank [PDB] code 3SGY) (36). Figure 3 represents the active site of the wild-type enzyme bound to compound 1 and NADPH, with residues His30 and Phe98 highlighted in orange. While a crystal structure has not yet been determined for the Sa (F98I) DHFR enzyme or inhibitors 1 and 2, we can infer that the Ile98 mutation would reduce interactions with NADPH and the propargyl linker of the ligand.
Fig 3.
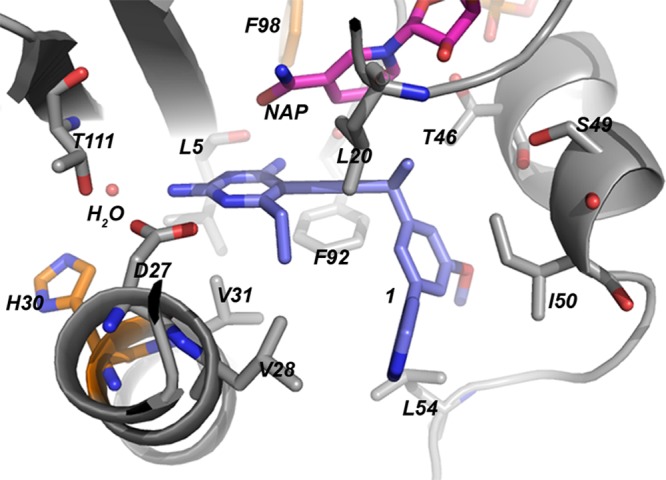
Sa DHFR active site residues (gray) bound to NADPH (pink) and compound 1 (lavender). Mutating residues are highlighted in orange.
Enzyme kinetic parameters to correlate with bacterial fitness.
Since the resistant bacterial strains exhibit no significant loss in fitness compared to the progenitor strain, we investigated enzyme fitness by assessing kinetic properties for both the wild-type and mutant DHFR enzymes (Table 4). The Sa (F98Y) and Sa (H30N) DHFR enzymes maintain activity with catalytic efficiency (kcat/Km) values close to those of Sa DHFR, ranging from 1.4 to 2.4 μM/s. While the Sa (F98I) DHFR enzyme has an approximately 2.7-fold reduction in the Km value for dihydrofolate (DHF), the kcat/Km ratio is nearly identical to the values observed for the wild-type and Sa (F98Y) and Sa (H30N) DHFR enzymes. Based on these observations, enzyme fitness may not necessarily be compromised by the resistance mutations.
Table 4.
Kinetic parameters for wild-type and mutant DHFR enzymesa
| Enzyme | DHF Km (μM ± SD) | DHF Km fold loss (SaMut/SaWT) | NAP Km (μM ± SD) | NAP Km fold loss (SaMut/SaWT) | DHF kcat (s−1) | DHF kcat/KM |
|---|---|---|---|---|---|---|
| Sa DHFR | 14.5 ± 0.35 | 1.0 | 38.0 ± 2.3 | 1.0 | 31 | 2.1 |
| Sa (F98Y) DHFR | 7.3 ± 0.40 | 1.0 | 45.1 ± 2.2 | 1.2 | 11.1 | 1.5 |
| Sa (F98I) DHFR | 39.4 ± 3.2 | 2.7 | 58.1 ± 0.45 | 1.5 | 17.5 | 2.3 |
| Sa (H30N) DHFR | 18.9 ± 0.75 | 1.3 | 32.9 ± 2.0 | 1.0 | 13.7 | 1.4 |
Mut, mutant; WT, wild type.
In previous studies, we found a correlation between an alternative NADPH conformation identified in the Sa (F98Y) DHFR crystal structure and the reduction of potency for the propargyl-linked inhibitors (10). Because of this observation and to distinguish whether any of the point mutations affect the cofactor, we measured Km for NADPH. All of the DHFR mutants had Km values that are nearly identical to the value observed for the Sa DHFR enzyme. Consistent with earlier studies (8), the assay data here show that the F98Y, F98I, and H30N mutations have minimal effects on catalysis.
DISCUSSION
Trimethoprim resistance associated with point mutations in DHFR is emerging in MRSA strains at an alarming rate, thus necessitating the development of new chemotherapies to overcome resistant strains. While we have successfully designed potent inhibitors for TMP-resistant MRSA strains, we were compelled to investigate potential resistance to our lead propargyl-linked inhibitors during the development process. Here, we have developed a prospective method to prescreen the most potent compounds for potential resistance in a MRSA strain during the early stages of drug design. By screening our lead compounds for a resistance profile, we have identified that the same mutations associated with trimethoprim resistance also emerge for the propargyl-based pyridine and meta-biphenyl compounds. These mutations in DHFR include the clinically observed F98Y and H30N mutations, in addition to the novel F98I mutation, all of which are observed at a low frequency for inhibitors 1 and 2. As identified in earlier studies, F98Y is the most prevalent DHFR mutation that emerges for TMP and also inhibitors 1 and 2.
Resistance mutations in DHFR are observed at a low frequency upon selection with compound 1. In addition to low mutational frequencies, compound 1 retains potency for resistant strains, maintaining an MIC value of 2.5 μg/ml and MPC values within the range of MIC values for trimethoprim (within 1 to 4 μg/ml) against the wild-type strain. In fact, other clinically used antibiotics have MPC values that are much higher than the MPC value observed for compound 1. For example, the MPC value for norfloxacin is 7 μg/ml (S. aureus MT5), an approximate 23-fold difference from the MIC value, observed as 0.3 μg/ml (9). In addition, MPC values have been determined for S. aureus RN450 and several antibiotics that include erythromycin, for which the MPC value is 32 μg/ml, with an MPC/MIC ratio of almost 64-fold. Based on the current literature regarding MPC values, it is recommended that concentrations above the MPC should be administered to prevent the development of resistant strains (33). Low MPC values are desirable, since they provide for the reduction of resistance while remaining in the therapeutic window below dose-limiting toxicity. The specific value of determining the MPC in early studies is to rank preliminary candidates and to assess their relative MPC values against those of other clinically used drugs. In the long term, however, it should be noted that drugs used over a longer, clinically relevant time span may have altered resistance profiles.
Previous studies suggest that resistance mutations may affect bacterial fitness by affecting growth rates, survival rates, or catalysis of the enzyme drug target (3, 25, 37). In this study, we investigated the bacterial fitness of resistant strains containing the F98Y, F98I, and H30N genotypes. Results from the pairwise competition assays confirm that all of the resistance genotypes are relatively fit compared to the wild-type strain. In an attempt to correlate bacterial fitness with enzyme fitness, we evaluated kinetic parameters for the mutant enzymes and found that the Sa (F98Y), Sa (F98I), and Sa (H30N) DHFR enzymes have Km values similar to those of the Sa DHFR enzyme. In addition, these mutant enzymes are all catalytically efficient, with kcat/Km values close to those of the wild type. Previous work aimed at understanding pyrimethamine resistance mutations in DHFR suggests that resistance is selected without jeopardizing the overall fitness of the enzyme (3). Other DHFR mutants from Plasmodium falciparum, which include sets of 3 to 4 point mutations associated with pyrimethamine resistance, also show kcat/Km values very similar to those of the wild type (32). Similarly, the results here also show a correlation between bacterial fitness and enzyme catalytic efficiency for all of the mutant enzymes generated from inhibitor-induced selection pressure.
Sa (F98I) DHFR emerges as an enzyme that is highly resistant to TMP as well as compounds 1 and 2 that retains affinity for the DHF substrate and cofactor NADPH and overall catalytic efficiency compared to Sa DHFR. Bacterial strains with the novel F98I resistance genotype are relatively fit and resistant to inhibitor 1. These strains are also cross-resistant to TMP, as observed by the increase in MIC values. Although the F98I resistance genotype is identified at a low frequency in vitro (for compound 1), the inhibitor Ki values reveal that Sa (F98I) DHFR is unusually resistant to all three compounds, especially compared to the clinically relevant Sa (F98Y) and Sa (H30N) DHFR enzymes. Given that Sa (F98I) confers 34-fold resistance for TMP and remains fit while not impairing catalytic efficiency, we were puzzled as to why the F98I resistance genotype does not evolve in MRSA strains in response to TMP. As observed in the literature describing resistance mutants, there are other factors in addition to kinetic properties that can describe enzyme fitness and its correlation with bacterial fitness, such as enzyme stability, function, and aggregation and degradation within the cell (38). These factors should be investigated in the future to understand why the F98I resistance genotype does not emerge either in vitro or in TMP-resistant strains of MRSA in the clinic.
Mutational resistance is a major problem in treating serious bacterial infections and in designing new therapeutics to overcome resistant strains. In order to effectively design inhibitors that can overcome these limitations, we have incorporated an experimental screening method to evaluate resistance genotypes in dfrA during the early stages of drug design. The positive results for the pyridyl compound (compound 1) suggest that this compound is worth continued investigation, including assessing toxicity in vivo, and that future optimization should be tailored to continue to avoid the effects of the F98Y and F98I mutations.
ACKNOWLEDGMENTS
We gratefully acknowledge funding from the NIH (AI065143 and GM067542).
Footnotes
Published ahead of print 9 April 2012
REFERENCES
- 1. Bolstad D, Bolstad E, Frey K, Wright D, Anderson A. 2008. A structure-based approach to the development of potent and selective inhibitors of dihydrofolate reductase from Cryptosporidium. J. Med. Chem. 51:6839–6852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boucher H, Corey G. 2008. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 46(Suppl. 5):S344–S349 [DOI] [PubMed] [Google Scholar]
- 3. Brown K, et al. 2010. Compensatory mutations restore fitness during the evolution of dihydrofolate reductase. Mol. Biol. Evol. 27:2682–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang S, et al. 2003. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 348:1342–1347 [DOI] [PubMed] [Google Scholar]
- 5. Cheng Y, Prusoff W. 1973. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- 6. CLSI 2006. Performance standards for antimicrobial susceptibility testing; sixteenth informational supplement, vol M100-S16. CLSI, Wayne, PA [Google Scholar]
- 7. Couto I, Costa S, Viveiros M, Martins M, Amaral L. 2008. Efflux-mediated response of Staphylococcus aureus exposed to ethidium bromide. J. Antimicrob. Chemother. 62:504–513 [DOI] [PubMed] [Google Scholar]
- 8. Dale G, et al. 1997. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J. Mol. Biol. 266:23–30 [DOI] [PubMed] [Google Scholar]
- 9. Dong Y, Zhao X, Domagala J, Drlica K. 1999. Effect of fluoroquinolone concentration on selection of resistant mutants of Mycobacterium bovis BCG and Staphylococcus aureus. Antimicrob. Agents Chemother. 43:1756–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frey K, et al. 2009. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternative conformation of NADPH that may be linked to trimethoprim resistance. J. Mol. Biol. 387:1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frey K, Lombardo M, Wright D, Anderson A. 2010. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J. Struct. Biol. 170:93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hawser S, Lociuro S, Islam K. 2006. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharmacol. 71:941–948 [DOI] [PubMed] [Google Scholar]
- 13. Huet A, Raygada J, Mendiratta K, Seo S, Kaatz G. 2008. Multidrug efflux pump overexpression in Staphylococcus aureus after single and multiple in vitro exposures to biocides and dyes. Microbiology 154:3144–3153 [DOI] [PubMed] [Google Scholar]
- 14. Huovinen P. 1987. Trimethoprim resistance. Antimicrob. Agents Chemother. 31:1451–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huovinen P, Sundstrom L, Swedberg G, Skold O. 1995. Trimethoprim and sulfonamide resistance. Antimicrob. Agents Chemother. 39:279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kluytmans-Vandenberg M, Kluytmans J. 2006. Community-acquired methicillin-resistant Staphylococcus aureus: current perspectives. Clin. Microbiol. Infect. 12(Suppl. 1):9–15 [DOI] [PubMed] [Google Scholar]
- 17. Kriengkauykiat J, Porter E, Lomovskaya O, Wong-Beringer A. 2005. Use of an efflux pump inhibitor to determine the prevalence of efflux pump-mediated fluoroquinolone resistance and multidrug resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lenski R. 1988. Experimental studies of pleiotropy and epistasis in Escherichia coli. I. Variation in competitive fitness among mutants resistant to virus T4. Evolution 42:425–432 [DOI] [PubMed] [Google Scholar]
- 19. Lorian V. 2005. Antibiotics in laboratory medicine, 5th ed Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 20. Markowitz N, Quinn E, Saravolatz L. 1992. Trimethoprim-sulfamethoxazole compared with vancomycin for the treatment of Staphylococcus aureus infection. Ann. Intern. Med. 117:390–398 [DOI] [PubMed] [Google Scholar]
- 21. McGuire J. 2003. Anticancer antifolates: current status and future directions. Curr. Pharm. Des. 9:2593–2613 [DOI] [PubMed] [Google Scholar]
- 22. Naimi T, et al. 2003. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 290:2976–2984 [DOI] [PubMed] [Google Scholar]
- 23. Nikaido H. 2009. Multidrug resistance in bacteria. Annu. Rev. Biochem. 78:119–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. NNIS 2004. National Nosocomial Infections Surveillance (NNIS) System report, data summary from January 1992 through June 2004, issued October 2004. Am. J. Infect. Control 32:470–485 [DOI] [PubMed] [Google Scholar]
- 25. O'Neill A, Chopra I. 2004. Preclinical evaluation of novel antibacterial agents by microbiological and molecular techniques. Expert Opin. Invest. Drugs 13:1045–1063 [DOI] [PubMed] [Google Scholar]
- 26. O'Neill A, Cove J, Chopra I. 2001. Mutation frequencies for resistance to fusidic acid and rifampicin in Staphylococcus aureus. J. Antimicrob. Chemother. 47:647–650 [DOI] [PubMed] [Google Scholar]
- 27. Pages J-M, et al. 2009. Efflux pump, the masked side of ß-lactam resistance in Klebsiella pneumoniae clinical isolates. PLoS One 4:e4817 doi:10.1371/journal.pone.0004817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phillips I, Warren C. 1976. Activity of sulfamethoxazole and trimethoprim against Bacteroides fragilis. Antimicrob. Agents Chemother. 9:736–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rajendran R, et al. 2010. Efflux pumps may play a role in tigecycline resistance in Burkholderia species. Int. J. Antimicrob. Agents 36:151–154 [DOI] [PubMed] [Google Scholar]
- 30. Ribera A, Ruiz J, Jimenez de Anta T, Vila J. 2002. Effect of an efflux pump inhibitor on the MIC of nalidixic acid for Acinetobacter baumannii and Stenotrophomonas maltophilia clinical isolates. J. Antimicrob. Chemother. 49:697–702 [DOI] [PubMed] [Google Scholar]
- 31. Sakoulas G, Moellering R. 2008. Increasing antibiotic resistance among methicillin-resistant Staphylococcus aureus strains. Clin. Infect. Dis. 46(Suppl. 5):S360–S367 [DOI] [PubMed] [Google Scholar]
- 32. Sandefur CI, Wooden J, Quaye I, Sirawaraporn W, Sibley C. 2007. Pyrimethamine-resistant dihydrofolate reductase enzymes of Plasmodium falciparum are not enzymatically compromised in vitro. Mol. Biochem. Parasitol. 154:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith H, Nichol K, Hoban D, Zhanel G. 2003. Stretching the mutant prevention concentration (MPC) beyond its limits. J. Antimicrob. Chemother. 51:1323–1325 [DOI] [PubMed] [Google Scholar]
- 34. Vickers A, O'Neill A, Chopra I. 2007. Emergence and maintenance of resistance to fluoroquinolones and coumarins in Staphylococcus aureus: predictions from in vitro studies. J. Antimicrob. Chemother. 60:269–273 [DOI] [PubMed] [Google Scholar]
- 35. Vickers A, Potter N, Fishwick C, Chopra I, O'Neill A. 2009. Analysis of mutational resistance to trimethoprim in Staphylococcus aureus by genetic and structural modelling techniques. J. Antimicrob. Chemother. 63:1112–1117 [DOI] [PubMed] [Google Scholar]
- 36. Viswanathan K, et al. 2012. Toward new therapeutics for skin and soft tissue infections: propargyl-linked antifolates are potent inhibitors of MRSA and Streptococcus pyogenes. PLoS One 7:e29434 doi:10.1371/journal.pone.0029434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang J, Bambara R, Demeter L, Dykes C. 2010. Reduced fitness in cell culture of HIV-1 with nonnucleoside reverse transcriptase inhibitor-resistant mutations correlates with relative levels of reverse transcriptase content and RNase H activity in virions. J. Virol. 84:9377–9389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang X, Minasov G, Shoichet B. 2002. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320:85–95 [DOI] [PubMed] [Google Scholar]
- 39. Wiegand I, Hilpert K, Hancock R. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163–175 [DOI] [PubMed] [Google Scholar]