

Abstract
Legionella pneumophila, the causative agent of Legionnaires' disease, is a ubiquitous freshwater bacterium whose virulence phenotypes require a type IV secretion system (T4SS). L. pneumophila strain JR32 contains two virulence-associated T4SSs, the Dot/Icm and Lvh T4SSs. Defective entry and phagosome acidification phenotypes of dot/icm mutants are conditional and reversed by incubating broth-grown stationary-phase cultures in water (WS treatment) prior to infection, as a mimic of the aquatic environment of Legionella. Reversal of dot/icm virulence defects requires the Lvh T4SS and is associated with a >10-fold induction of LpnE, a tetratricopeptide repeat (TPR)-containing protein. In the current study, we demonstrated that defective entry and phagosome acidification phenotypes of mutants with changes in LpnE and EnhC, another TPR-containing protein, were similarly reversed by WS treatment. In contrast to dot/icm mutants for which the Lvh T4SS was required, reversal for the ΔlpnE or the ΔenhC mutant required that the other TPR-containing protein be present. The single and double ΔlpnE and ΔenhC mutants showed a hypersensitivity to sodium ion, a phenotype associated with dysfunction of the Dot/Icm T4SS. The ΔlpnE single and the ΔlpnE ΔenhC double mutant showed 3- to 9-fold increases in translocation of Dot/Icm T4SS substrates, LegS2/SplY and LepB. Taken together, these data identify TPR-containing proteins in a second mechanism by which the WS mimic of a Legionella environmental niche can reverse virulence defects of broth-grown cultures and implicate LpnE and EnhC directly or indirectly in translocation of Dot/Icm T4SS protein substrates.
INTRODUCTION
Legionella pneumophila is a ubiquitous freshwater bacterium and the causative agent of Legionnaires' disease, a potentially fatal community- and hospital-derived pneumonia. Infection and replication in freshwater amoebae are proposed to constitute an initiating event in transition of the bacterium from a free-living aquatic organism into a pathogen capable of intracellular multiplication in pulmonary macrophages. Inhalation of Legionella-laden amoebae following aerosolization from contaminated domestic water supplies is the route by which Legionnaires' disease is spread (20, 22, 23, 28, 33, 40, 54).
A functional type IV secretion system (T4SS) is required for infection and replication of L. pneumophila in amoeba and macrophage hosts (20, 23, 31, 36). The Philadelphia-1 strain of Legionella pneumophila, isolated following the 1976 disease outbreak that gave the organism its name (22), contains two virulence-associated T4SSs: the Dot/Icm and the Lvh T4SSs (13). Strains with null mutations of many dot/icm genes are defective in entry, phagosome acidification, and intracellular replication phenotypes when eukaryotic hosts are infected by broth-grown stationary-phase bacteria. Studies from our laboratory demonstrated that those virulence defects of dot/icm mutants are reversed when broth stationary-phase bacteria are exposed to water prior to infection. This exposure, termed water stress (WS) treatment, is a mimic of the L. pneumophila aquatic environmental niche (2, 3). Following WS treatment, dotA and dotB mutants, encoding a membrane protein and an ATPase essential for Dot/Icm T4SS function, respectively, showed entry and phagosome acidification phenotypes equal to those of parental strain JR32, a derivative of the Philadelphia-1 strain. Reversal of these virulence defects by WS treatment was shown to require the lvh locus, encoding the Lvh T4SS (2). These studies established that the Legionella Dot/Icm T4SS is conditionally required for virulence phenotypes and implicated the Lvh locus in the environmental lifestyle and transition of Legionella pneumophila from aquatic bacterium to intracellular pathogen mimicked by WS treatment.
To implicate proteins associated with WS reversal, broth-grown stationary-phase cultures of JR32 were compared by proteomics with JR32 cultures exposed to conditions that reverse virulence defects of dot/icm mutants. This comparison identified a >10-fold induction of protein Lpg2222 (3). Subsequently, open reading frame (ORF) Lpg2222 was implicated in Legionella virulence phenotypes by two independent approaches. Subtractive genomic hybridization of L. pneumophila strain 130b and the less pathogenic Legionella micdadei identified the corresponding ORF in strain 130b, also known as the Wadsworth strain. The gene was named lpnE when a mutant of strain 130b was shown to be defective in entry and phagosome acidification phenotypes following infection of macrophages with broth stationary-phase cultures (41). In strain JR32, LpnE was identified in an affinity chromatography screen for proteins that bind to OCRL1, an inositol polyphosphate 5-phosphatase whose Dictyostelium discoideum homolog, Dd5P4, restricts the intracellular replication of L. pneumophila in D. discoideum (59).
LpnE is one of 10 proteins in the Philadelphia-1 genome that contain tetratricopeptide (TPR) repeats. The TPR repeat is a motif of ∼34 amino acid residues frequently associated with functional protein-protein interactions in eukaryotes and prokaryotes (6, 16, 18, 24). EnhC, another L. pneumophila TPR-containing protein, was initially identified in strain AA100, a derivative of strain 130b, in a screen for mutants with reduced uptake by nonphagocytic HEp-2 human epidermoid carcinoma cells (14). EnhC was also identified in a screen in a dotA mutant of strain Lp02, a Philadelphia-1 derivative, for mutants with reduced plating efficiency on laboratory media in the presence of a plasmid overexpressing dotA+ (15). In this report, we describe data on virulence phenotypes of lpnE, enhC, and lpnE enhC double mutants that implicate the LpnE and EnhC directly or indirectly in T4SS function of L. pneumophila strain JR32.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Parental strain JR32, serotype 1, a derivative of L. pneumophila strain Philadelphia-1 isolated from the 1976 outbreak of Legionnaires' disease (46, 60), and mutants constructed from it were plated on charcoal–N-(2-acetamido)-2-aminoethanesulfonic acid (ACES)-buffered (pH 6.9) yeast extract agar (CYE) plates and cultured in ACES-buffered yeast extract (AYE) broth (21, 26). When required, chloramphenicol and hygromycin were present at 5 and 100 μg/ml, respectively. Escherichia coli strain DH5α, cultured in Luria-Bertani broth, was used for recombinant constructions with chloramphenicol and hygromycin at 15 and 150 μg/ml, respectively. All bacteria were cultured at 37°C. WS-treated cultures were prepared as previously described (2), resuspending 22- to 24-h-old AYE broth cultures in water and incubating them for an additional 18 to 19 h in 16- by 150-mm glass culture tubes on a rotary drum.
Amoeba and macrophage cell line and culture conditions.
Acanthamoeba castellanii trophozoites (ATCC 30234) were cultured at 28°C in peptone yeast extract glucose (PYG) medium as described previously (25, 49). The BALB/c mouse peritoneal macrophage line J774 was maintained in RPMI 1640 medium with 2 mM l-glutamine, 10% heat-inactivated fetal bovine serum, and penicillin-streptomycin solution (PenStrep, 5,000 U/ml) at 37°C in humidified 5% CO2 (2, 12).
Construction of an lpnE deletion mutant and lpnE complementing plasmid.
The entire lpnE ORF, from 21 nucleotides (nt) upstream of the start codon to 95 nt downstream of the stop codon, was deleted using the sacB-containing allelic exchange vector pNPTS138 used previously (1, 44). A PstI-EcoRI PCR fragment containing 1.14 kbp of upstream sequence was digested with EcoRI and ligated to an EcoRI-digested 1.17-kbp downstream BspEI-EcoRI PCR fragment. The gel-purified ligation product was digested with BspEI and PstI and ligated into similarly digested pNPTS138. The resultant allelic exchange plasmid was electroporated into strain JR32 and Kmr colonies streaked on CYE plates with 5% sucrose. PCR was used to screen Kms Sucr colonies for the ΔlpnE mutation. For complementation, the lpnE ORF, from 194 nt upstream of the ATG start codon to 100 nt downstream of the UAA stop codon, was amplified by PCR. The BamHI sites from the primers were cleaved and the product ligated to BamHI-digested, calf intestinal phosphatase (CIP)-treated complementation vector pMMB207αB-Km-14.
Construction of enhC deletion mutants.
An in-frame deletion of the enhC ORF was constructed in which the first five enhC codons were fused to the last four codons by a PstI site. An upstream BamHI-PstI PCR fragment beginning 1.16 kp upstream of the enhC start codon was digested with PstI and then annealed and ligated to a PstI-digested downstream ApaI-PstI PCR fragment which began 1.14 kbp downstream of the enhC stop codon. After BamHI-ApaI, the product was ligated into the similarly digested allelic exchange vector pNPTS138. The ΔenhC mutant of strain JR32 and the ΔlpnE ΔenhC double mutant were constructed using sucrose counterselection as described above.
Immunofluorescence assay for entry of Legionella into A. castellanii amoebae and J774 macrophages.
Amoeba trophozoites (5 × 106 cells in A. castellanii buffer) or macrophages (1.5 × 106 cells in tissue culture medium) were applied in 0.5-ml amounts to 12-mm-diameter glass coverslips in 24-well microtiter dishes, as described previously (3, 25). Legionella strains containing plasmid green fluorescent protein (GFP), pJN105-hygro-GFP (3), were constructed by natural competence (34, 55) or electroporation. Bacteria, grown as indicated below, were resuspended in A. castellanii buffer or tissue culture medium, added at the multiplicity of infection (MOI) indicated below, incubated for 30 min at 28°C for amoebae and 1 h at 37°C for macrophages, and then fixed with formaldehyde and stained with rabbit anti-L. pneumophila serotype 1 antibody (m-Tech, Atlanta, GA) and then Cy3-conjugated donkey anti-rabbit immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) as previously described (3, 25). Immunofluorescence samples were observed at a magnification of ×60 with a Zeiss Axioskop epifluorescence microscope. Scoring of internalized bacteria, data acquisition, and statistical analyses to calculate means ± standard deviations and P values from a chi-square test were performed as described previously (2, 3).
Immunofluorescence assay for acidification of Legionella-containing vacuoles.
J774 macrophages were resuspended at 2 × 106 cells/ml in tissue culture medium lacking penicillin and streptomycin and containing 50 nM LysoTracker red DND 99 (Molecular Probes Invitrogen, Carlsbad, CA), adhered in duplicate to glass coverslips, and then infected with L. pneumophila containing plasmid GFP as described previously (2). After 1 h at 37°C, monolayers were fixed with formaldehyde, prepared, and observed by fluorescence microscopy as described above. Colocalization was scored as the percentage of internalized legionellae in which bacterial GFP fluorescence localized with LysoTracker red fluorescence inside the macrophage. As previously described (2, 3), means ± standard deviations and P values from a chi-square test were calculated, scoring each internalized bacterium as colocalized or not colocalized with LysoTracker.
Assay of intracellular multiplication by titer of released L. pneumophila organisms.
Monolayers of A. castellanii trophozoites at 28°C or 37°C or J774 macrophages at 37°C were infected with L. pneumophila as described previously (2–4), and titers of bacteria in the infection medium were determined on CYE plates. In any given experiment, data from titer determinations in replicate wells were in good agreement.
Construction of fusions to adenylate cyclase.
Plasmids expressing fusions between B. pertussis CyaA and L. pneumophila effector proteins were constructed from pcya-ralF, a derivative of pMMB207 containing the M45 epitope fused upstream of a cyaA fusion to the amino terminus of L. pneumophila RalF (38). The ralF portion was liberated from pcya-ralF by digestion with BamHI and PstI and the remaining plasmid ligated to a PCR product containing the ORF of the desired effector, following BamHI-PstI digestion of the PCR-amplified effector ORF. These constructions fused the carboxyl terminus of CyaA to the amino terminus of L. pneumophila effector proteins. In each plasmid construct, the start codon of the effector ORF was replaced by a Leu codon and ∼100 nt of sequence downstream of the translational stop included.
Quantifying translocation of T4SS substrates into macrophages.
Translocation was assayed by immunoassay of cyclic AMP (cAMP) in lysates of macrophages, following infection with Legionella strains harboring plasmid fusions of adenylate cyclase to L. pneumophila effector proteins (12, 38). J774 macrophages were resuspended at 3 × 106 cells per ml in RPMI medium lacking penicillin and streptomycin, and 0.5 ml was added per well of a 24-well plate. After 2 h at 37°C, monolayers were washed 3 times with RPMI medium lacking penicillin, streptomycin, and fetal bovine serum, and then 0.5 ml of the same medium was added per well. Stationary AYE-chloramphenicol (AYE-Cm) cultures of strains with cyclase fusion plasmids were diluted to 1.25 × 109 CFU per ml in RPMI lacking penicillin, streptomycin, and fetal bovine serum, and 100 μl was added to each well of J774 cells (MOI, 83). The plate was centrifuged for 15 min at 37°C and then immediately placed in a water bath at 37°C for 1 h. Monolayers were washed 3 times with phosphate-buffered saline (PBS), and then 0.25 ml of water and 0.25 ml of 0.2 N HCl or 0.25 ml of 0.1 N HCl were added directly. After 20 min at room temperature (RT), the extract was incubated at 95°C for 5 min and supernatants stored at −20°C until assayed for cAMP using the Direct Cyclic AMP Correlate-EIA kit (Assay Designs, Ann Arbor, MI). Absorbance at 405 nm was measured and the pmoles of cAMP computed with a SpectraMax microplate reader and software (Molecular Devices, Sunnyvale, CA). The ratio of cAMP in ΔlpnE and JR32 parental strains was corrected for differences in the relative expression of the cyclase fusion in parental strain JR32 and the ΔlpnE mutant, measured by Western blotting, as described below. Correction factors by which experimental values were multiplied were small and did not change trends or conclusions: RalF, 1.0; SidB, 1.2; SdeA, 0.82; LegS2, 1.1; and LepB cyclase fusion, 1.3.
Western blotting for expression of adenylate cyclase fusion proteins.
Cell pellets from strain JR32 and its ΔlpnE mutant were passed through four cycles of freezing and thawing, resuspended in a solution containing 4 M urea, 4% SDS, 100 mM Tris-HCl (pH 6.8), 10% glycerol, 2 mM EDTA, 1× mini-EDTA-free protease inhibitor (Boehringer, Indianapolis, IN), and 5 mM dithioerythritol, and then disrupted by sonication and heated at 95°C for 5 min (38). Supernatants were electrophoresed by SDS-PAGE and analyzed by Western blotting to Hybond ECL (pore size, 0.45 μm; Amersham Biosciences, Piscataway, NJ). Blots were blocked for 1 h in 150 mM NaCl–10 mM Tris-HCl (pH 8.3) with 5% skim-milk powder, then incubated with a 1:100 dilution of mouse monoclonal antiserum against the M45 epitope and then with a 1:1,000 dilution of horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (Amersham Biosciences Piscataway, NJ), and developed with the peroxidase ECL detection system (Pierce Protein Research, Rockford, IL). X-ray films were scanned and bands quantified using ImageJ software, using a calibration curve established with extracts of E. coli strain DH5α harboring the cyaA∷ralF plasmid. Relative protein levels of cyclase fusions in JR32 and in the ΔlpnE strain were interpolated from the calibration curve, and experimentally determined pmoles of cAMP from translocation experiments were corrected for the relative protein levels.
Stress tests.
Resistance of stationary-phase AYE cultures to sodium ion stress was quantified by comparing the titers on CYE plates containing 100 mM NaCl to titers on CYE plates without added NaCl (4, 10). Resistance of stationary-phase AYE cultures to H2O2 was quantified by the zone-of-inhibition method, loading 7-mm Whatmann 3MM disks with 10 μl of 0.5 or 1 M H2O2 onto a bacterial lawn in 1.5% top agar (1).
RESULTS
Defective entry of a Legionella ΔlpnE mutant into macrophage and amoeba hosts is reversed by WS treatment.
An unmarked ΔlpnE mutant of strain JR32 was defective for entry into J774 macrophages infected with broth-grown stationary-phase cultures (Fig. 1A, left). These data, from microscopic examination of internalized GFP-expressing bacteria, are in excellent agreement with defective entry reported for an lpg2222∷Km mutant of Legionella strain 130b. In that study, entry into A549 human alveolar epithelial cells and macrophages from human THP-1 cells, infected by broth stationary-phase cultures, was analyzed by gentamicin protection (41). Entry of our ΔlpnE mutant was 8% that of the parental strain JR32, compared to 18% that of parental strain 130b for the lpnE∷Km mutant. Complementation with plasmid lpnE in vector pKm14 restored entry to 45% that of strain JR32 (Fig. 1A, right), compared to 53% for the lpg2222∷Km mutant of strain 130b. Partial complementation may be attributable to differences in expression or regulation of chromosomal versus plasmid lpnE.
Fig 1.

Analysis of Legionella entry into macrophage and amoeba hosts. (A) WS treatment reverses defective entry of ΔlpnE Legionella into macrophages. J774 macrophages were infected at 37°C and an MOI of 100 with stationary-phase (Stat) or water stress (WS)-treated bacteria expressing GFP. Means and standard deviations are plotted. On the left, it can be seen that WS treatment of the ΔlpnE strain restores entry to that of parental JR32. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE mutant. For WS bacteria, P = 0.31. On the right, it can be seen that complementation with plasmid pKm14 reverses defective entry of Stat bacteria. For ΔlpnE pKm14 Stat versus ΔlpnE pKm14∷lpnE Stat bacteria, P < 0.001. (B) WS treatment reverses defective entry of ΔlpnE Legionella into amoebae. A. castellanii trophozoites were infected at 28°C and an MOI of 5 with Stat or WS-treated bacteria expressing GFP. Means and standard deviations are plotted. On the left, it can be seen that WS treatment of the ΔlpnE strain restores entry to that of parental JR32. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE mutant. For WS bacteria, P = 0.25 for JR32 versus the ΔlpnE mutant. On the right, it can be seen that complementation with plasmid pKm14 restores parental entry. For JR32 pKm14 Stat versus ΔlpnE pKm14 Stat, P < 0.001. For JR32 pKm14 Stat versus ΔlpnE pKm14∷lpnE Stat, P = 0.73. (C) The lvh locus is not required for WS reversal of defective ΔlpnE mutant entry into macrophages. J774 macrophages were infected at 37°C and an MOI of 100 with Stat or WS-treated bacteria expressing GFP. Means and standard deviations are plotted. WS treatment of the ΔlpnE and ΔlpnE Δlvh strains restores entry to that of parental JR32. For WS-treated bacteria, P = 0.60 for JR32 versus the ΔlpnE mutant and P = 0.65 for JR32 versus the ΔlpnE Δlvh mutant. WS reverses the entry defect in Stat bacteria. P < 0.001 for Stat versus WS-treated ΔlpnE mutant and P < 0.001 for the ΔlpnE Δlvh mutant. (D) The lvh locus is not required for WS reversal of defective ΔlpnE mutant entry into amoebae. A. castellanii trophozoites were infected at 28°C and an MOI of 5 with Stat or WS-treated bacteria expressing GFP. Means and standard deviations are plotted. WS treatment of ΔlpnE and ΔlpnE Δlvh strains restores entry to that of parental JR32. For WS-treated bacteria, P = 0.61 for JR32 versus the ΔlpnE mutant and P = 0.99 for JR32 versus the ΔlpnE Δlvh mutant. WS reverses the entry defect in Stat bacteria. P < 0.001 for Stat versus WS-treated ΔlpnE mutant and P < 0.001 for the ΔlpnE Δlvh mutant. (E) EnhC is required for WS reversal of defective ΔlpnE mutant entry into macrophages. J774 macrophages were infected at 37°C and an MOI of 50 with Stat or WS-treated bacteria expressing GFP. Means and standard deviations are plotted. WS treatment of ΔlpnE and ΔenhC strains reverses the entry defect in Stat bacteria. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE mutant and for JR32 versus the ΔenhC mutant. For Stat versus WS bacteria, P < 0.001 for the ΔlpnE and ΔenhC mutants. WS treatment of the ΔlpnE ΔenhC double mutant fails to reverse the entry defect. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE ΔenhC mutant. For WS bacteria, P < 0.001 for JR32 versus the ΔlpnE ΔenhC mutant. (F) EnhC is required for WS reversal of defective entry of ΔlpnE Legionella into amoebae. A. castellanii trophozoites were infected at 28°C and an MOI of 5 with Stat or WS-treated bacteria expressing GFP. Means and standard deviations are plotted. WS treatment of ΔlpnE and ΔenhC strains restores entry to that of parental JR32. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE mutant and for JR32 versus the ΔenhC mutant. For WS bacteria, P < 0.75 for JR32 versus the ΔlpnE mutant and P = 0.82 for JR32 versus the ΔenhC mutant. WS treatment of the ΔlpnE ΔenhC double mutant fails to reverse the entry defect. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE ΔenhC mutant. For WS bacteria, P < 0.001 for JR32 versus the ΔlpnE ΔenhC mutant.
Entry of the JR32 ΔlpnE mutant into Acanthamoeba castellanii was analyzed, since the lpg2222∷Km mutant of strain 130b was not tested in an amoeba host (41). Similar to results with J774 macrophages, entry of the ΔlpnE mutant was defective when amoebae were infected with broth-grown stationary-phase cultures (Fig. 1B, left) and was restored to that of parental strain JR32 with plasmid lpnE complementation (Fig. 1B, right). These data established that defective entry was attributable to deletion of lpnE and that LpnE is required for efficient entry into an environmental amoeba host of Legionella pneumophila, as well as cell lines of human and mouse macrophages and human alveolar epithelial cells, all of these being cell types encountered by L. pneumophila as a pulmonary pathogen.
Defective entry of stationary-phase ΔlpnE Legionella into amoeba and macrophage hosts is reversed by WS treatment.
We previously reported that the defective entry of broth-grown stationary-phase cultures of dot/icm mutants into macrophages and amoebae was reversed by water stress (WS treatment), a mimic of the freshwater ponds, streams, and lakes in which L. pneumophila cycles through growth and stasis as an environmental bacterium (22, 40, 54). A similar reversal of defective entry was observed with the ΔlpnE mutant. After WS treatment of broth stationary-phase cultures, entry of the ΔlpnE mutant into macrophages (Fig. 1A) and amoeba hosts (Fig. 1B) was comparable to that of parental strain JR32. These results demonstrated that the defective entry phenotype was conditional and dependent on culture conditions relevant to the Legionella lifestyle.
Reversal of defective entry into macrophages and amoebae by WS treatment does not require the Lvh T4SS.
Reversal of virulence defects indicated a WS-dependent expression of compensatory virulence factors. For WS reversal of defective entry in broth-grown stationary-phase dot/icm mutants, we implicated compensatory expression of the Lvh T4SS by demonstrating that the lvh locus was required (2). Those data suggested that the Lvh T4SS could functionally compensate for the Dot/Icm T4SS in WS-treated bacteria. Therefore, we tested if the lvh locus was required for WS reversal of defective entry in the ΔlpnE mutant, comparing WS reversal of ΔlpnE and Δlvh single mutants with that of a ΔlpnE Δlvh double mutant. Entry of the Δlvh single mutant was identical to that of parental strain JR32 for broth stationary-phase and WS-treated cultures in macrophage (Fig. 1C) and amoeba (Fig. 1D) hosts. The ΔlpnE Δlvh double mutant showed defective entry for broth stationary-phase cultures, as expected, and showed WS reversal with macrophage (Fig. 1C) and amoeba (Fig. 1D) hosts. These results established that the lvh locus and the Lvh T4SS it encodes were not required for WS reversal of defective entry in the ΔlpnE mutant. This contrasts with the lvh requirement for WS reversal of defective entry in dot/icm mutants.
EnhC is required for WS reversal of defective entry in the ΔlpnE mutant.
Deletion of the TPR-containing protein EnhC in strain AA100 resulted in defective entry when broth-grown stationary-phase cultures were used to infect HEp-2 human epithelial cells and THP-1-derived macrophages (14). Consistent with those studies, broth stationary-phase cultures of an ΔenhC mutant of strain JR32 were defective for entry in J774 macrophages (Fig. 1E) and A. castellanii amoebae (Fig. 1F). In contrast, a ΔenhC mutant of strain Lp02 was not defective for entry into HEp-2 cells or macrophages derived from bone marrow of A/J mice (30). Differences in entry phenotypes between L. pneumophila strains AA100, JR32, and Lp02 may be attributable to genomic differences, including the absence of an Lvh T4SS in strain Lp02.
Similar to WS reversal in the JR32 ΔlpnE mutant, the entry defect of the ΔenhC mutant was reversed by WS treatment in both hosts (Fig. 1E and F). In contrast to the case with the single ΔlpnE and ΔenhC mutants, the entry defect of the double mutant was not reversed by WS treatment for either host (Fig. 1E and F). These data implicated enhC in the WS reversal of defective entry of the ΔlpnE mutant and conversely implicated lpnE in WS reversal of the ΔenhC mutant phenotype. Since entry is dependent on T4SS function, these results suggest direct or indirect involvement of LpnE and EnhC in L. pneumophila T4SS function under WS culture conditions.
Defective vacuole acidification in the ΔlpnE mutant is reversed by WS treatment.
Immediately after entry into macrophage hosts, Legionella resides in a specialized vacuole which is not acidified (23, 28, 40, 43, 53, 56). This acidification phenotype was quantified as the percentage of internalized bacteria that colocalized with LysoTracker red, a fluorescent marker of acidic vacuoles. For parental strain JR32, ∼20% of internalized stationary-phase bacteria colocalized with LysoTracker at 1 h postinfection. For stationary-phase ΔlpnE bacteria, colocalization was nearly 80%, comparable to that of a dotA mutant defective in the Dot/Icm T4SS (Fig. 2A, left). After transformation with plasmid lpnE, colocalization of the △lpnE mutant was comparable to that of the parental strain with the empty complementing vector (Fig. 2A, right), thus attributing the acidification defect to deletion of lpnE.
Fig 2.
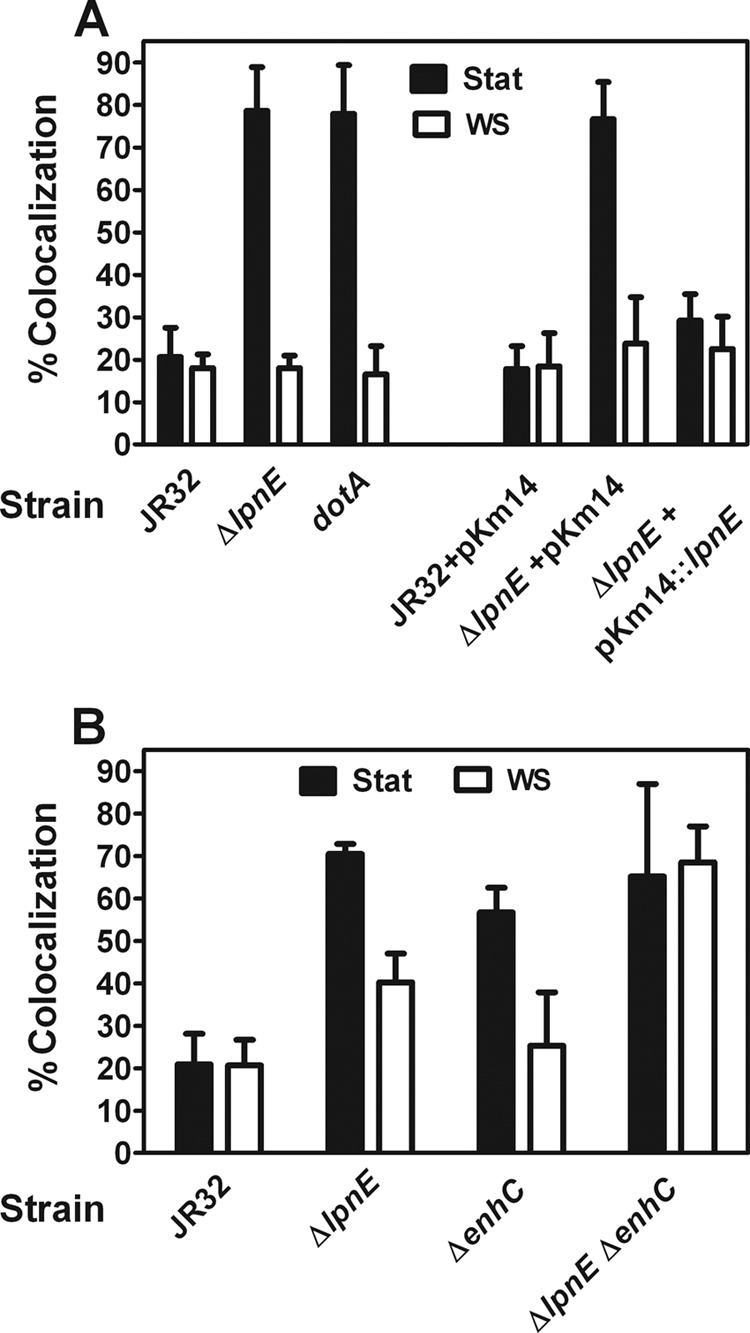
Analysis of vacuole acidification phenotype in macrophages. (A) WS treatment reverses defective ΔlpnE strain vacuole acidification. J774 macrophages were infected at 37°C and an MOI of 50 with stationary-phase (Stat) or WS-treated bacteria expressing GFP. Colocalization with LysoTracker red was calculated as a percentage of internalized bacteria. On the left, it can be seen that colocalization is increased for Stat bacteria and reversed by WS treatment. P < 0.001 for JR32 versus the ΔlpnE mutant. Colocalization is no different from that of a dotA mutant. P = 0.90 for the dotA versus the ΔlpnE mutant. WS treatment reverses the colocalization defect. For Stat versus WS-treated bacteria, P < 0.001 for the ΔlpnE mutant and P < 0.001 for the dotA mutant. On the right, it can be seen that complementation with plasmid pKm14 restores parental colocalization. For Stat bacteria, P < 0.001 for ΔlpnE pKm14 versus ΔlpnE pKm14∷lpnE. (B) EnhC is required for WS reversal of defective ΔlpnE strain vacuole acidification. J774 macrophages were infected at 37°C and an MOI of 50 with Stat or WS-treated bacteria expressing GFP. Colocalization with LysoTracker red was calculated as a percentage of internalized bacteria. WS treatment of ΔlpnE and ΔenhC strains reverses defective colocalization. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE mutant and for JR32 versus the ΔenhC mutant. For Stat versus WS-treated bacteria, P < 0.001 for the ΔlpnE mutant and P = 0.001 for the ΔenhC mutant. WS treatment of the ΔlpnE ΔenhC double mutant fails to reverse the entry defect. For Stat bacteria, P < 0.001 for JR32 versus the ΔlpnE ΔenhC mutant. For WS-treated bacteria, P < 0.001 for JR32 versus the ΔlpnE ΔenhC mutant.
Our results in Fig. 2A are in good agreement with those for defective acidification of an lpnE∷Km insertion mutant of L. pneumophila strain 130b, using macrophages derived from THP-1 human monocytes and LAMP-1 as a marker of late endosomes. Following infection with broth stationary-phase cultures, levels of colocalization of Legionella bacteria with the LysoTracker were 21, 79, and 78% for strain JR32 and its ΔlpnE and dotA mutants, respectively (Fig. 2A). These values compare favorably with colocalization levels of 37, 66, and 75% for strain 130b and its ΔlpnE and dotA mutants, based on the LAMP-1 marker (42). The phenotype of the ΔlpnE mutant in L. pneumophila strain 130b was complemented by plasmid lpnE under the control of the constitutively active Legionella mip promoter (42).
We previously reported that defective acidification of broth-grown stationary-phase dot/icm mutants was reversed by WS treatment, similar to reversal of defective entry (2). WS treatment similarly reversed defective acidification of broth stationary-phase ΔlpnE bacteria, with restoration to values of parental strain JR32 (Fig. 2A). These results established that the role of LpnE in vacuole acidification was conditional, dependent on culture conditions, and indicated WS-dependent expression of virulence-related functions redundant with or overlapping those of LpnE.
The doubling time of the ΔlpnE mutant was identical to that of parental strain JR32 in AYE broth, 1.5 ± 0.09 and 1.5 ± 0.05 h, respectively. Thus, defective entry and acidification of stationary-phase cultures were not attributable to a general growth defect of the ΔlpnE mutant in broth medium. The LysoTracker method could not be used to quantify acidification in A. castellanii because LysoTracker was uniformly dispersed in the amoeba cell and not sequestered in acidic vesicles.
EnhC is required for WS reversal of the defective acidification phenotype in the ΔlpnE mutant.
Broth-grown stationary-phase ΔenhC Legionella was defective in the macrophage acidification phenotype. Like with the ΔlpnE mutant, that defect was reversed by WS treatment (Fig. 2B). As expected, the ΔlpnE ΔenhC double mutant showed an acidification defect, and that defect was not reversed by WS treatment. These data showed that WS reversal of the acidification defect in one mutant required the other TPR-containing protein. Similar to above inferences from entry studies, these results suggest that LpnE and EnhC participate in the acidification phenotype under WS culture conditions, in functionally redundant pathways or interacting in a common pathway.
This defective acidification phenotype of broth stationary-phase JR32 ΔenhC was consistent with a previously reported defect in a ΔenhC mutant of dotA-negative Lp02, expressing plasmid dotA, assessed by LAMP-1 colocalization (15). However, broth stationary-phase cultures of an ΔenhC mutant of dotA+ Lp02 were not defective for this phenotype, unless the macrophages were pretreated with tumor necrosis factor alpha (TNF-α) (30). These phenotypic differences between JR32 and Lp02 strains and between the two ΔenhC mutants of Lp02 may be attributable to the absence of an Lvh T4SS in strain Lp02, use of macrophages derived from bone marrow of A/J mice in the Lp02 studies, and genetic differences between the two Lp02 strains.
Characterization of intracellular multiplication of ΔlpnE and ΔenhC mutants in macrophages and amoebae.
The JR32 ΔlpnE mutant showed no defect in intracellular multiplication in macrophages derived from human HL-60 cells or in the J774 or MH-S macrophage line, originating from mouse ascites or mouse lung, respectively (data not shown). Our results are consistent with those for the lpnE∷Km mutant of L. pneumophila strain 130b, which showed no defect in intracellular multiplication in THP-1 macrophages (42). With A. castellanii as the host, ΔlpnE JR32 showed no significant difference from strain JR32 over 3 days of infection at 28°C and 37°C with MOIs of 0.05 to 2.5. Titers of bacteria released into the infection medium from ΔlpnE JR32 were no more than 2-fold different from those for JR32 (data not shown). Our data differ from published results for intracellular multiplication of the lpnE∷Km mutant of Legionella strain 130b in A. castellanii at 37°C. In that instance, titers for the mutant at 24 and 48 h postinfection were decreased by factors of 10 and 4, respectively (42).
Like the JR32 ΔlpnE mutant, ΔenhC and ΔenhC ΔlpnE double mutants of JR32 showed no significant decrease in intracellular multiplication in J774 macrophages. Titers were never less than a factor of 2 to 3 below titers for strain JR32 at an MOI of 0.1 or 1.0 at 2 to 3 days postinfection (data not shown). Similarly, replication of JR32 ΔenhC and ΔenhC ΔlpnE mutants was not defective in A. castellanii (data not shown). These results contrast with findings for an ΔenhC mutant of Lp02 which was not defective for intracellular multiplication in the amoeba Dictyostelium discoideum but showed a pronounced delay in intracellular multiplication in macrophages derived from bone marrow of A/J mice at 1 day after infection (30). Differences in intracellular multiplication between ΔenhC mutants of strains Lp02 and JR32 and between lpnE mutants of strains 130b and JR32 may reflect differences in the bacterial genomes, in macrophage host cells, or both. Strain Lp02 does not contain the Lvh T4SS locus which is present in strain JR32. Strain 130b contains loci for two Lvh T4SSs and one Trb-1-like T4SS. All three strains contain a single locus for a Dot/Icm T4SS (47, 48).
Hydrogen peroxide and sodium ion resistance phenotypes of ΔlpnE and ΔenhC mutants.
Increased sensitivity to hydrogen peroxide was observed for the ΔenhC mutant of Lp02 (30). Likewise, our JR32 ΔenhC mutant showed increased hydrogen peroxide sensitivity, as did JR32 ΔlpnE and ΔlpnE ΔenhC mutants (Table 1). All three JR32 mutants with changes in TPR-containing proteins were less sensitive than a KatA and KatB catalase-peroxidase double mutant (1), which showed increased sensitivity at the lower concentration of hydrogen peroxide.
Table 1.
Resistance to hydrogen peroxide and sodium ion stressa
| Strain name or genotype | Resistance to hydrogen peroxideb |
Resistance to sodium (%)c | |
|---|---|---|---|
| 0.5 M | 1.0 M | ||
| JR32 | 27 ± 1 | 30 ± 1 | 14 ± 1 |
| ΔlpnE | 28 ± 1 | 36 ± 1 | 4.1 ± 0.7 |
| ΔenhC | 28 ± 1 | 37 ± 1 | 2.9 ± 0.8 |
| ΔlpnE ΔenhC | 26 ± 1 | 34 ± 1 | 5.5 ± 1.6 |
| katA katB | 34 ± 1 | 37 ± 1 | |
| katA | 52 ± 5 | ||
For hydrogen peroxide resistance at 1.0 M H2O2, P < 0.001 for JR32 versus all other strains. For resistance at 0.2 M H2O2, P = 0.02 for JR32 versus the ΔlpnE or ΔenhC mutant, P = 0.12 for JR32 versus the ΔlpnE ΔenhC mutant, and P < 0.001 for JR32 versus the katA katB strain. For sodium resistance, P < 0.001 for JR32 versus the ΔlpnE, ΔenhC, or the ΔlpnE ΔenhC mutant and P < 0.001 for the katA strain versus the ΔlpnE, ΔenhC, or ΔlpnE ΔenhC mutant.
Zone of inhibition (mm) from a 7-mm disk loaded with 10 μl of H2O2 of the indicated concentration.
Calculated as 100 × the titer of stationary-phase cultures on CYE with 100 mM NaCl/titer on CYE.
Some Legionella mutants with attenuated virulence traits exhibit increased resistance to sodium ion (10, 11, 46, 58). For dot/icm mutants, increased resistance to sodium ion has been attributed to an interaction between the Dot/Icm T4SS and systems maintaining Legionella Na+ homeostasis (46). In direct contrast, decreased resistance to sodium ion was reported for mutations in genes implicated in direct or indirect maintenance of cell envelope integrity and translocation: dotL, which is homologous to genes for T4SS coupling proteins, and enhC in Lp02 strain backgrounds (8, 15, 30). Our JR32 ΔenhC mutant showed decreased resistance to 100 mM sodium ion (Table 1), consistent with the Lp02 ΔenhC mutant (8, 15, 30). Decreased resistance, i.e., hypersensitivity, to sodium ion was also shown for our ΔlpnE single and ΔlpnE ΔenhC double mutants compared to parental strain JR32. A control, the JR32 katA mutant, showed increased resistance to sodium ion identical to that reported earlier (1).
Translocation of protein substrates of the Legionella T4SS is increased in ΔlpnE and ΔlpnE ΔenhC mutants.
To directly test lpnE and enhC involvement in T4SS function, implicated by hypersensitivity of mutants to sodium ion, translocation of five substrates of the Dot/Icm T4SS was quantified in ΔlpnE and ΔlpnE ΔenhC mutants using adenylate cyclase fusions. For each fusion, the cAMP produced in J774 macrophages after infection with mutant strains was compared to cAMP after infection with JR32 (Fig. 3).
Fig 3.
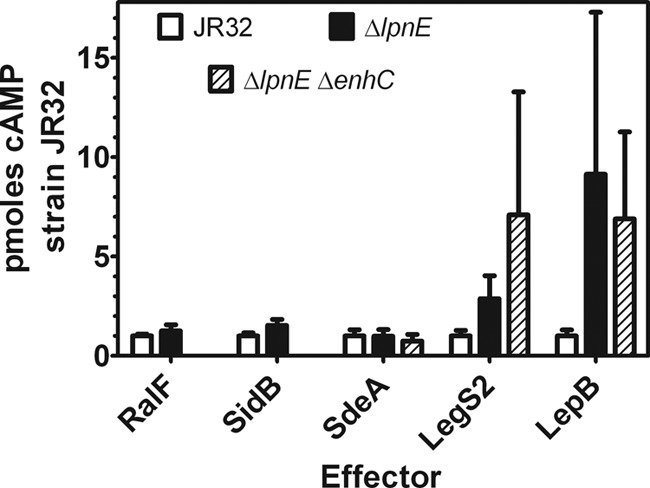
Translocation of Legionella effectors into macrophages by ΔlpnE and ΔlpnE ΔenhC mutants. J774 macrophages were infected with the ΔlpnE, ΔlpnE ΔenhC, or JR32 parental strain containing plasmid cyclase fusions to the N terminus of the listed Dot/Icm effector protein. Results are the ratio of cAMP for the listed strain to cAMP for strain JR32, from the mean of 12 to 30 determinations in 2 to 4 independent experiments. For the ΔlpnE mutant versus JR32, P < 0.001 for both LegS2/SplY and LepB. For the ΔlpnE ΔenhC mutant versus JR32, P < 0.02 and < 0.001 for LegS2/SplY.
Translocation was increased 3-fold and 7-fold, respectively, in ΔlpnE and ΔlpnE ΔenhC strains for the LegS2 (SplY) effector, a sphingosine 1-phosphate lyase that localizes in host cell mitochondria (17). Translocation was increased 9-fold and 7-fold in ΔlpnE and ΔlpnE ΔenhC strains for the LepB effector. LepB, a GTPase-activating protein, is required for nonlytic release of Legionella after replication in amoebae (12) and for binding to and inactivation of host Rab1 GTPase (27) after removal of posttranslationally incorporated AMP by SidD (39). Translocation was essentially unchanged in ΔlpnE and ΔlpnE ΔenhC mutants for cyclase fusions to RalF, a guanine nucleotide exchange factor (37), to SidB, a homolog of Vibrio cholerae Rtx toxin (32), and to SdeA, a member of the SidE family that binds to and requires IcmS for translocation (5).
WS reversal of defective entry and phagosome acidification phenotypes in the ΔlpnE single mutant was eliminated in the ΔlpnE ΔenhC double mutant. Therefore, translocation was compared in the ΔlpnE ΔenhC and ΔlpnE mutants to determine if the increased translocation was eliminated in the double mutant. However, the double mutant showed an increased or essentially unchanged translocation for the same T4SS effectors as did the ΔlpnE single mutant. These data establish that LpnE and/or EnhC selectively influences translocation of Legionella T4SS effector proteins. The significant increase in translocation for the ΔlpnE single and ΔlpnE ΔenhC double mutants suggest that LpnE and/or EnhC negatively affects translocation of LegS2 (SplY) and LepB. Possible roles for TPR-containing proteins in T4SS translocation are considered in the Discussion.
To test complementation, the ΔlpnE mutant containing the pcya∷lepB cyclase fusion in a derivative of pMMB207 was transformed with the empty pJN105-hygro vector and with pJN105-hygro containing lpnE. With the empty pJN105-hygro vector, cAMP from the lepB cyclase fusion was reduced by nearly an order of magnitude, precluding definitive complementation experiments. Effects of complementing vectors on L. pneumophila virulence phenotypes have been reported before, e.g., for mutants of strain 130b (41, 45) and of strain JR32 (50). In some instances, these interfering effects were attributed to mob genes in the vector and were eliminated by using mob-negative vector derivatives. However, pBBRMCS-1, from which pJN105-hygro is derived (3), is mob negative (19, 32), indicating a different interference mechanism. In addition, entry and acidification phenotypes of the ΔlpnE mutant containing plasmid GFP in pJN105-hygro were complemented using lpnE in a pMMB207 derivative (Fig. 1A and B and 2A). Thus, interference appears to depend on which vector has the complementing gene and/or the presence of the cyclase fusion construction.
DISCUSSION
TPR motifs have been implicated in the function of type III secretion systems (T3SSs) of bacterial pathogens. The TPR domains in the SycD chaperone of the Yersinia enterocolitica T3SS are involved in SycD dimerization and the secretion of YopB and YopD T3SS substrates (9). The TPR domains in the PcrH chaperone of the Pseudomonas aeruginosa T3SS are required for translocation of PopB and PopD substrates (7). In Xanthomonas axonopodis, the causative agent of citrus canker, the PthA T3SS substrate is a TPR-containing protein (35). The current work furthered understanding of the roles of L. pneumophila TPR-containing proteins LpnE and EnhC in T4SS-dependent virulence phenotypes in three areas.
(i) Virulence defects of ΔlpnE and ΔenhC mutants are reversed by mimicking the aquatic environment of L. pneumophila.
Mutants of strain JR32 with deletions of lpnE and enhC were defective in entry into amoeba and macrophage hosts and in a phagosome acidification phenotype in macrophages when broth-grown stationary-phase cultures of Legionella were used for infection. These results were consistent with published data for a mutation of lpnE in strain 130b (41, 42) and enhC mutations in strain AA100 and in dotA Lp02 expressing plasmid dotA (15). These defects were shown to be conditional, like entry and phagosome defects in dot/icm mutants (2, 3), and were reversed by exposing broth stationary-phase cultures to WS treatment, a mimic of the aquatic environment of L. pneumophila. Analysis of WS reversal may identify virulence factors that function preferentially in the environmental phase of the Legionella life cycle and are relevant to controlling the spread of Legionnaires' disease from environmental reservoirs to amoebae and humans.
Our ΔlpnE, ΔenhC, and ΔlpnE ΔenhC mutants of strain JR32 showed defective entry and phagosome acidification phenotypes but no significant defects in intracellular multiplication. Retention of parental strain intracellular multiplication can be understood by recognizing that the entry and acidification phenotypes are not totally defective. Some bacteria enter, alter phagosome acidification, and replicate exponentially in amoeba and macrophage hosts. Moreover, a linear change in entry or phagosome acidification phenotype, measured 1 h or less after infection, may not result in a detectable alteration in the intracellular multiplication phenotype, measured 1 to 3 days after infection and quantified exponentially.
(ii) TPR-containing proteins are implicated in WS reversal of Legionella virulence defects.
The lvh locus, encoding a T4SS highly homologous to Agrobacterium tumefaciens and Helicobacter pylori T4SSs (51), was required for WS reversal of virulence defects in JR32 dot/icm mutants (2, 3). Implication of the Lvh T4SS suggested that the Dot/Icm T4SS could be functionally replaced by the Lvh T4SS after WS treatment. These data support a model in which both Lvh and Dot/Icm T4SSs contribute to virulence phenotypes, with the Lvh T4SS playing a major role during the aquatic phase of the Legionella infectious cycle mimicked by WS treatment. Proteomic analysis identified a >10-fold induction of LpnE under conditions that reversed dot/icm virulence defects (3) and prompted the current study of LpnE and a second TPR-containing protein, EnhC, in WS reversal of L. pneumophila virulence defects.
WS treatment reversed defective entry and phagosome acidification phenotypes for broth-grown stationary-phase cultures of JR32 ΔlpnE and ΔenhC mutants, but the lvh locus was not required for WS reversal in the ΔlpnE mutant. Since the Lvh T4SS was required for WS reversal of virulence defects in mutants lacking a functional Dot/Icm T4SS, we hypothesized that WS reversal of defects in the ΔlpnE mutant required a second TPR-containing protein. Broth stationary-phase cultures of the ΔlpnE ΔenhC double mutants were defective in entry and vacuole acidification phenotypes, as expected. However, WS treatment produced no significant reversal of either defective phenotype in the double mutant. These results showed that EnhC was required for WS reversal of the ΔlpnE defects and, conversely, that LpnE was required for WS reversal of ΔenhC virulence defects. The data identify a second pathway for WS reversal and support a model in which these two TPR-containing proteins cooperate functionally in WS reversal of virulence defects. In sum, TPR-containing proteins are candidates for virulence factors acting in the aquatic, environmental phase of the L. pneumophila life cycle.
(iii) TPR-containing proteins influence the translocation of Dot/Icm T4SS-dependent effectors.
Many studies of L. pneumophila translocation focused on the role of the Dot/Icm T4SS. Whenever tested, translocation of effectors, which constitute ∼10% of the L. pneumophila genome (61), was shown to be dependent on a functional Dot/Icm T4SS by a substantial decrease in translocation in a dot/icm mutant, usually dotA or icmT (23, 28, 31, 36). Little is known about mutations outside the dot/icm loci that influence translocation (57) or about mutations that increase rather than decrease translocation.
The current studies are therefore significant in demonstrating increased translocation of certain Dot/Icm-dependent effectors by mutation of Legionella genes for TPR-containing proteins, genes which are not in the dot/icm loci. Translocation of cyclase fusions to the N terminus of SplY/LegS2 and LepB into J774 macrophages was increased 3- to 9-fold in lpnE and lpnE enhC double mutants. For fusions to RalF, SidB, and SdeA, translocation was changed less than 50%, demonstrating selectivity in which effectors are affected by mutations in TPR-containing proteins. To our knowledge, these are the first mutations associated with a significant increased translocation of L. pneumophila effectors. Increased translocation was proposed as an explanation for defective intracellular multiplication of a mutant with a change in the SdhA effector, but testing of SidC and LidA effectors showed no increase in translocation (29). If internalization of the ΔlpnE mutant is a prerequisite for translocation, then the observed increases in translocation for the SplY/LegS2 and LepB effectors are underestimates, because cAMP values were normalized to the number of macrophages present. Since entry of ΔlpnE and ΔenhC mutants into macrophages was 4- to 10-fold less than for strain JR32, translocation per infected macrophage would be 4- to 10-fold larger than the values observed. However, translocation of SplY/LegS2 and LepB might occur from the medium without internalization, as shown for a RalF cyclase fusion in opsonized L. pneumophila interacting with CHO cells expressing the Fc receptor (38). In that case, translocation increases would not be influenced by attenuated entry of ΔlpnE and ΔenhC mutants.
One model for explaining increased translocation of LegS2 and LepB is competitive inhibition. Since LpnE is translocated by the Dot/Icm T4SS (59), LpnE may compete with LegS2 and LepB effectors for substrate binding to and translocation by the Dot/Icm T4SS machinery. According to this model, when LpnE is absent, LegS2 and LepB have increased access to the secretion machinery and show increased translocation. EnhC is associated with the bacterial envelope in strain Lp02 (30) but is not thus far known to be a translocation substrate (61). Chaperone function is a second model for increased translocation in the ΔlpnE mutant. Analogous to a model proposed for the DotB ATPase of the Dot/Icm T4SS, LpnE may function in selection of substrates destined for translocation (52). LpnE might selectively bind to and chaperone or sequester Legionella T4SS substrates and potentially influence the timing and sequence of effector translocation. In either model, defective entry and acidification of the ΔlpnE mutant could be attributable directly to the absence of LpnE and/or indirectly to the imbalance in translocation of T4SS effector proteins. As cited above, there is precedent for TPR domains in T3SS translocation substrates and for TPR-containing proteins functioning as chaperones in translocation of T3SS effectors.
Other studies in L. pneumophila implicated TPR-containing proteins in the function or assembly of T4SS machinery. EnhC and the TPR-containing protein LidL were identified in a screen for mutations that increase the viability of an Lp02 dotA mutant containing a plasmid overexpressing dotA (15). It was proposed that EnhC is involved in Dot/Icm function and/or biogenesis of structures on the cell envelope (30). Second, mutations in proteins involved in maintenance of the L. pneumophila envelope have been associated with an increased sensitivity to sodium ion (8, 30). Both JR32 lpnE and enhC mutants are hypersensitive to sodium ion, suggestive of a role in maintaining the bacterial envelope and secretion machinery dependent on envelope integrity.
In sum, this study demonstrated that virulence defects associated with deletion of lpnE and enhC are reversible by WS treatment in strain JR32. In contrast to virulence defects in dot/icm mutants, this WS reversal does not require the Lvh T4SS but utilizes an alternate reversal pathway or alternative pathways involving a second TPR-containing protein. Our studies showing increased translocation in ΔlpnE JR32 and on hypersensitivity to sodium of ΔlpnE and ΔenhC strains suggest that these TPR-containing proteins are involved in the function of the T4SS machinery and/or other cell envelope structures. Our results lay a foundation for further studies of LpnE, EnhC, and other TPR-containing proteins as T4SS substrates and virulence factors outside the L. pneumophila dot/icm loci. Our studies have implicated TPR-containing proteins with both Dot/Icm and Lvh T4SSs. Since some L. pneumophila laboratory strains contain Lvh or Lvh-like T4SSs and others do not (47, 48), future studies of TPR-containing proteins may yield insights into the relative contributions of those two secretion systems to virulence phenotypes.
ACKNOWLEDGMENTS
We thank Craig Roy, Yale University School of Medicine, for the CyaA-RalF fusion plasmid, Patrick Hearing, SUNY Stony Brook, for monoclonal antibody to M45 epitope, the reviewers for suggesting additional roles for LpnE in translocation, and our colleagues at Albert Einstein College of Medicine: Anne Bresnick, Dianne Cox, and the Albert Einstein Analytical Imaging Facility for use of epifluorescence microscopes and Anne Etgen for use of the plate reader and data analysis program.
This work was supported by Public Health Service grant AI 072487 to H.M.S. from the National Institutes of Health.
Footnotes
Published ahead of print 4 May 2012
REFERENCES
- 1. Bandyopadhyay P, Steinman HM. 2000. Catalase-peroxidases of Legionella pneumophila: cloning of the katA gene and studies of KatA function. J. Bacteriol. 182:6679–6686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bandyopadhyay P, Liu S, Gabbai CB, Venitelli Z, Steinman HM. 2007. Environmental mimics and the Lvh type IVA secretion system contribute to virulence-related phenotypes of Legionella pneumophila. Infect. Immun. 75:723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bandyopadhyay P, Xiao H, Coleman HA, Price-Whelan A, Steinman HM. 2004. Icm/dot-independent entry of Legionella pneumophila into amoeba and macrophage hosts. Infect. Immun. 72:4541–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bandyopadhyay P, Byrne B, Chan Y, Swanson MS, Steinman HM. 2003. Legionella pneumophila catalase-peroxidases are required for proper trafficking and growth in primary macrophages. Infect. Immun. 71:4526–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bardill JP, Miller JL, Vogel JP. 2005. IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56:90–103 [DOI] [PubMed] [Google Scholar]
- 6. Blatch GL, Lassle M. 1999. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays 21:932–939 [DOI] [PubMed] [Google Scholar]
- 7. Bröms JE, Edqvist PJ, Forsberg A, Francis MS. 2006. Tetratricopeptide repeats are essential for PcrH chaperone function in Pseudomonas aeruginosa type III secretion. FEMS Microbiol. Lett. 256:57–66 [DOI] [PubMed] [Google Scholar]
- 8. Buscher BA, et al. 2005. The DotL protein, a member of the TraG-coupling protein family, is essential for viability of Legionella pneumophila strain Lp02. J. Bacteriol. 187:2927–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Büttner CR, Sorg I, Cornelis GR, Heinz DW, Niemann HH. 2008. Structure of the Yersinia enterocolitica type III secretion translocator chaperone SycD. J. Mol. Biol. 375:997–1012 [DOI] [PubMed] [Google Scholar]
- 10. Byrne B, Swanson MS. 1998. Expression of Legionella pneumophila virulence traits in response to growth conditions. Infect. Immun. 66:3029–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Catrenich CE, Johnson W. 1989. Characterization of the selective inhibition of growth of virulent Legionella pneumophila by supplemented Mueller-Hinton medium. Infect. Immun. 57:1862–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J, et al. 2004. Legionella effectors that promote nonlytic release from protozoa. Science 303:1358–1361 [DOI] [PubMed] [Google Scholar]
- 13. Chien M, et al. 2004. The genomic sequence of the accidental pathogen Legionella pneumophila. Science 305:1966–1968 [DOI] [PubMed] [Google Scholar]
- 14. Cirillo SL, Lum J, Cirillo JD. 2000. Identification of novel loci involved in entry by Legionella pneumophila. Microbiology 146:1345–1359 [DOI] [PubMed] [Google Scholar]
- 15. Conover GM, Derre I, Vogel JP, Isberg RR. 2003. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48:305–321 [DOI] [PubMed] [Google Scholar]
- 16. D'Andrea LD, Regan L. 2003. TPR proteins: the versatile helix. Trends Biochem. Sci. 28:655–662 [DOI] [PubMed] [Google Scholar]
- 17. Degtyar E, Zusman T, Ehrlich M, Segal G. 2009. A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell. Microbiol. 11:1219–1235 [DOI] [PubMed] [Google Scholar]
- 18. Delahay RM, et al. 2008. The highly repetitive region of the Helicobacter pylori CagY protein comprises tandem arrays of an alpha-helical repeat module. J. Mol. Biol. 377:956–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elzer PH, et al. 1995. In vivo and in vitro stability of the broad-host-range cloning vector pBBR1MCS in six Brucella species. Plasmid 33:51–57 [DOI] [PubMed] [Google Scholar]
- 20. Ensminger AW, Isberg RR. 2009. Legionella pneumophila Dot/Icm translocated substrates: a sum of parts. Curr. Opin. Microbiol. 12:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feeley JC, et al. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10:437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fields BS, Benson RF, Besser RE. 2002. Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15:506–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franco IS, Shuman HA, Charpentier X. 2009. The perplexing functions and surprising origins of Legionella pneumophila type IV secretion effectors. Cell. Microbiol. 11:1435–1443 [DOI] [PubMed] [Google Scholar]
- 24. Gatsos X, et al. 2008. Protein secretion and outer membrane assembly in Alphaproteobacteria. FEMS Microbiol. Rev. 32:995–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hilbi H, Segal G, Shuman HA. 2001. Icm/dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol. Microbiol. 42:603–617 [DOI] [PubMed] [Google Scholar]
- 26. Horwitz MA, Silverstein SC. 1980. Legionnaires' disease bacterium (Legionella pneumophila) multiplies intracellularly in human monocytes. J. Clin. Invest. 66:441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingmundson A, Delprato A, Lambright DG, Roy CR. 2007. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450:365–369 [DOI] [PubMed] [Google Scholar]
- 28. Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U. S. A. 103:18745–18750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu M, Conover GM, Isberg RR. 2008. Legionella pneumophila EnhC is required for efficient replication in tumour necrosis factor alpha-stimulated macrophages. Cell. Microbiol. 10:1906–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luo ZQ. 2011. Targeting one of its own: expanding roles of substrates of the Legionella pneumophila Dot/Icm type IV secretion system. Front. Microbiol. 2:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luo ZQ, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U. S. A. 101:841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Molofsky AB, Swanson MS. 2004. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53:29–40 [DOI] [PubMed] [Google Scholar]
- 34. Molofsky AB, Swanson MS. 2003. Legionella pneumophila CsrA is a pivotal repressor of transmission traits and activator of replication. Mol. Microbiol. 50:445–461 [DOI] [PubMed] [Google Scholar]
- 35. Murakami MT, et al. 2010. The repeat domain of the type III effector protein PthA shows a TPR-like structure and undergoes conformational changes upon DNA interaction. Proteins 78:3386–3395 [DOI] [PubMed] [Google Scholar]
- 36. Nagai H, Kubori T. 2011. Type IVB secretion systems of Legionella and other Gram-negative bacteria. Front. Microbiol. 2:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679–682 [DOI] [PubMed] [Google Scholar]
- 38. Nagai H, et al. 2005. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc. Natl. Acad. Sci. U. S. A. 102:826–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neunuebel MR, et al. 2011. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science 333:453–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Newton HJ, Ang DK, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23:274–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Newton HJ, Sansom FM, Bennett-Wood V, Hartland EL. 2006. Identification of Legionella pneumophila-specific genes by genomic subtractive hybridization with Legionella micdadei and identification of lpnE, a gene required for efficient host cell entry. Infect. Immun. 74:1683–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Newton HJ, et al. 2007. Sel1 repeat protein LpnE is a Legionella pneumophila virulence determinant that influences vacuolar trafficking. Infect. Immun. 75:5575–5585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ninio S, Roy CR. 2007. Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol. 15:372–380 [DOI] [PubMed] [Google Scholar]
- 44. Reisenauer A, Mohr CD, Shapiro L. 1996. Regulation of a heat shock sigma32 homolog in Caulobacter crescentus. J. Bacteriol. 178:1919–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossier O, Starkenburg SR, Cianciotto NP. 2004. Legionella pneumophila type II protein secretion promotes virulence in the A/J mouse model of Legionnaires' disease pneumonia. Infect. Immun. 72:310–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sadosky AB, Wiater LA, Shuman HA. 1993. Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect. Immun. 61:5361–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samrakandi MM, Cirillo SL, Ridenour DA, Bermudez LE, Cirillo JD. 2002. Genetic and phenotypic differences between Legionella pneumophila strains. J. Clin. Microbiol. 40:1352–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schroeder GN, et al. 2010. Legionella pneumophila strain 130b possesses a unique combination of type IV secretion systems and novel Dot/Icm secretion system effector proteins. J. Bacteriol. 192:6001–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Segal G, Shuman HA. 1999. Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67:2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Segal G, Shuman HA. 1998. Intracellular multiplication and human macrophage killing by Legionella pneumophila are inhibited by conjugal components of IncQ plasmid RSF1010. Mol. Microbiol. 30:197–208 [DOI] [PubMed] [Google Scholar]
- 51. Segal G, Russo JJ, Shuman HA. 1999. Relationships between a new type IV secretion system and the icm/dot virulence system of Legionella pneumophila. Mol. Microbiol. 34:799–809 [DOI] [PubMed] [Google Scholar]
- 52. Sexton JA, Yeo HJ, Vogel JP. 2005. Genetic analysis of the Legionella pneumophila DotB ATPase reveals a role in type IV secretion system protein export. Mol. Microbiol. 57:70–84 [DOI] [PubMed] [Google Scholar]
- 53. Shin S, Roy CR. 2008. Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell. Microbiol. 10:1209–1220 [DOI] [PubMed] [Google Scholar]
- 54. Steinert M, Hentschel U, Hacker J. 2002. Legionella pneumophila: an aquatic microbe goes astray. FEMS Microbiol. Rev. 26:149–162 [DOI] [PubMed] [Google Scholar]
- 55. Stone BJ, Kwaik YA. 1999. Natural competence for DNA transformation by Legionella pneumophila and its association with expression of type IV pili. J. Bacteriol. 181:1395–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sturgill-Koszycki S, Swanson MS. 2000. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 192:1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vincent CD, et al. 2006. Identification of non-dot/icm suppressors of the Legionella pneumophila DeltadotL lethality phenotype. J. Bacteriol. 188:8231–8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vogel JP, Roy C, Isberg RR. 1996. Use of salt to isolate Legionella pneumophila mutants unable to replicate in macrophages. Ann. N. Y. Acad. Sci. 797:271–272 [DOI] [PubMed] [Google Scholar]
- 59. Weber SS, Ragaz C, Hilbi H. 2009. The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell. Microbiol. 11:442–460 [DOI] [PubMed] [Google Scholar]
- 60. Wiater LA, Sadosky AB, Shuman HA. 1994. Mutagenesis of Legionella pneumophila using Tn903 dlllacZ: identification of a growth-phase-regulated pigmentation gene. Mol. Microbiol. 11:641–653 [DOI] [PubMed] [Google Scholar]
- 61. Zhu W, et al. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638 doi:10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]