

Abstract
The dnaN159 allele encodes a temperature-sensitive mutant form of the β sliding clamp (β159). SOS-induced levels of DNA polymerase IV (Pol IV) confer UV sensitivity upon the dnaN159 strain, while levels of Pol IV ∼4-fold higher than those induced by the SOS response severely impede its growth. Here, we used mutations in Pol IV that disrupted specific interactions with the β clamp to test our hypothesis that these phenotypes were the result of Pol IV gaining inappropriate access to the replication fork via a Pol III*-Pol IV switch relying on both the rim and cleft of the clamp. Our results clearly demonstrate that Pol IV relied on both the clamp rim and cleft interactions for these phenotypes. In contrast to the case for Pol IV, elevated levels of the other Pols, including Pol II, which was expressed at levels ∼8-fold higher than the normal SOS-induced levels, failed to impede growth of the dnaN159 strain. These findings suggest that the mechanism used by Pol IV to switch with Pol III* is distinct from those used by the other Pols. Results of experiments utilizing purified components to reconstitute the Pol III*-Pol II switch in vitro indicated that Pol II switched equally well with both a stalled and an actively replicating Pol III* in a manner that was independent of the rim contact required by Pol IV. These results provide compelling support for the Pol III*-Pol IV two-step switch model and demonstrate important mechanistic differences in how Pol IV and Pol II switch with Pol III*.
INTRODUCTION
Cellular DNA is subject to frequent damage by endogenous and exogenous agents. If left unchecked, this damage leads to mutations and genome instability and, in extreme cases, serves to block transcription and replication, leading to cellular death (reviewed in reference 16). To cope with damage, organisms have evolved a variety of accurate repair and damage tolerance functions. Faithful duplication of cellular DNA relies on the proper coordination of these functions with the replication machinery. Generally speaking, DNA repair functions either excise damaged bases to enable accurate resynthesis of the damaged region, or act to directly reverse the damage (reviewed in reference 16). In contrast, damage tolerance functions do not repair damaged DNA but rather catalyze replication over lesions that persist in the DNA via a process termed translesion DNA synthesis (TLS) (38, 47). Due to their remarkably high fidelity, replicative DNA polymerases (Pols) are typically unable to catalyze TLS. Thus, most organisms possess one or more specialized Pols capable of replicating imperfect DNA templates, often termed TLS Pols (36). Because most TLS Pols lack intrinsic proofreading activity and display low fidelity compared to that of well-studied replicative Pols, even when replicating undamaged DNA, the actions of these Pols must be tightly controlled to limit unwanted mutations (reviewed in reference 41).
Although multiple mechanisms contribute to the coordinate regulation of replicative and TLS Pols, roles played by the DnaN family of sliding-clamp proteins have gained considerable attention (35). The Escherichia coli β (DnaN) sliding-clamp protein was initially discovered based on its ability to confer processivity upon the bacterial Pol III replicase (8). However, it has since been determined that the β clamp interacts with all five E. coli Pols (25, 26, 29, 46). Pol III catalyzes most replication in E. coli and is comprised of three subassemblies: core (αεθ), DnaX (τ2γδδ′ψχ), and β clamp (reviewed in references 32 and 33). The α subunit of Pol III core possesses intrinsic Pol activity. The β clamp is “loaded” onto the 3′ end of a primed or nicked double-stranded DNA (dsDNA) by DnaX in a reaction that requires ATP (reviewed in reference 5). Once loaded, DnaX chaperones α onto β, tethering core to DNA (12). The α subunit also interacts with the τ subunit of DnaX. Since DnaX has two τ subunits, it tethers two Pol III core complexes for simultaneous leading- and lagging-strand replication. The complex comprised of two cores, DnaX, and two β clamps is termed Pol III holoenzyme (Pol III HE), whereas the form lacking a β clamp is termed Pol III* (32, 33).
In addition to Pol III, E. coli has at least four other Pols, named Pol I, Pol II, Pol IV, and Pol V. Pol I, like Pol III, contributes to DNA replication, particularly on the lagging strand during Okazaki fragment maturation (reviewed in reference 16). In contrast, Pol II, Pol IV, and Pol V are thought to play minimal roles in replication and instead participate in TLS (41). Consistent with this view, Pol II, Pol IV, and Pol V are regulated as part of the global SOS response to DNA damage (reviewed in reference 16). As a result, their steady-state levels increase dramatically following replication-blocking DNA damage via a mechanism involving RecA-mediated autodigestion of the LexA transcriptional repressor (9). Although Pol I interacts physically with the β clamp (26, 29), structural features of this interaction have not yet been described. In contrast, the other four Pols are each known to possess a clamp-binding motif (CBM) that interacts specifically with a hydrophobic cleft located near the C tail of each clamp protomer (10). Pols II to V require interaction with this cleft for processive replication and biological activity (4, 11, 25, 28, 29). In addition to the cleft, Pols also contact noncleft surfaces (7, 19, 20, 28, 42–44). These surfaces, together with a single cleft on the clamp, are sufficient for supporting viability of E. coli and managing the actions of its different Pols during UV-induced mutagenesis (45).
E. coli Pol II and Pol IV are each able to replace Pol III* on a β clamp assembled on a primed DNA template in vitro (17, 19, 22). When added at high levels to in vitro reaction mixtures containing a preformed replisome comprised of a purified circular DNA template, β clamp, Pol III*, DnaB helicase, DnaG primase, and single-stranded DNA binding protein (SSB), these Pols slowed fork progression to ∼1 bp/s (22). Likewise, overproduction of either of these Pols using plasmids containing their respective genes under the control of the araBAD promoter inhibited chromosomal replication in vivo (22, 48). Overproduction of mutant Pols lacking functional CBMs failed to do so, indicating that these Pols must bind the clamp to impede replication in vivo. Taken together, these findings indicate that both Pol II and Pol IV are able to dynamically switch with Pol III* in vivo.
Although structural information regarding the β clamp-Pol II complex is not yet available, Bunting and colleagues described the structure of the complex comprising the β clamp and the Pol IV little finger (Pol IVLF) domain (7). In the crystal, Pol IVLF contacted two discrete clamp surfaces: residues 303VWP305 of Pol IV contacted E93 and L98 on the rim of the β clamp, while the C-terminal six residues of Pol IV containing the CBM associated with the clamp cleft. Based on characterization of mutant forms of these proteins, we previously determined that the rim contact was dispensable for processive Pol IV replication in vitro, while the cleft contact was absolutely required (19). We additionally determined that Pol IV switched with a stalled but not with an actively replicating Pol III* in vitro, consistent with the view that Pol IV gains access to the fork only if Pol III* is unable to extend the 3′ end of the primer (19). Importantly, a single cleft on the clamp was sufficient to support this switch, and both the rim and cleft contacts were required (19). Taken together, these findings suggest that Pol IV must first contact the rim of the β clamp immediately adjacent to the cleft that is bound by Pol III* prior to gaining access to the same cleft previously associated with the stalled Pol III* (19, 45) (Fig. 1).
Fig 1.

Pol IV utilizes a two-step switch to replace a stalled Pol III*. Pol III* requires a single cleft in the β clamp for processive replication (a). Pol IV replaces a stalled Pol III* at the replication fork via a two-step switch. In this switch, Pol IV first binds the rim of the clamp (b). Next, it binds the same cleft that was previously bound by the stalled Pol III* (c). Pol III* may stay associated with the clamp by contacting surfaces other than the cleft (c) or may be displaced from the clamp once Pol IV acquires the cleft (d). See the text for further details.
The model that Pols utilize unique contacts with the β clamp (such as the rim) as part of a mechanism to compete with one another for binding the clamp cleft and subsequently gaining access to the replication fork was based, in large part, on phenotypes of the E. coli dnaN159 strain (42–44). The dnaN159 allele encodes a temperature-sensitive mutant form of the β sliding clamp (β159) bearing G66E (glycine-66 to glutamic acid) and G174A substitutions, and strains bearing this allele display DNA replication defects due to an inability to properly manage the actions of the five E. coli Pols, particularly Pol IV (28, 30, 42–44). Residue G66 is located on the rim of the β clamp, near where Pol IV binds. The G66E substitution impairs interactions of the β clamp with Pol IV and the δ subunit of the DnaX clamp loader (28). In contrast, G174 is located within the hydrophobic cleft of the clamp. As a result, the G174A substitution affects interactions of the clamp with the CBMs present in δ, as well as the different Pols (28, 42). Despite the fact that the G66E and G174A substitutions in β159 affect Pol IV function in vitro (28), Pol IV is nevertheless required for two striking phenotypes of the dnaN159 strain. First, dnaN159 strains display sensitivity to UV. Importantly, UV sensitivity was fully suppressed by (not epistatic with) inactivation of the dinB gene, which encodes Pol IV (42, 43). Second, growth of the dnaN159 strain was severely impaired by a low-copy-number plasmid expressing slightly higher-than-physiological levels of Pol IV (28, 30). We interpret these findings to indicate that β159 supports Pol switching in vivo but is impaired for regulating the timing and/or order of the switches, due to the differential effects of the G66E and/or G174A substitutions on interactions of β159 with the different Pols. Consistent with this view, the dnaN159 strain displays a modest increase in spontaneous mutation frequency that is dependent on Pol V (29), as well as an increased frequency of UV-induced mutagenesis that is dependent upon both Pol IV and loss of nucleotide excision repair (43). Here, we used amino acids substitutions in Pol IV that disrupted specific interactions with the β clamp to test our hypothesis that these phenotypes were the result of Pol IV gaining inappropriate access to the replication fork via a two-step Pol III*-Pol IV switch relying on both the rim and cleft of the clamp (Fig. 1). Our results support this model and provide insight into why the other Pols fail to exert similar phenotypes in the dnaN159 strain.
MATERIALS AND METHODS
E. coli strains, plasmid DNAs, and bacteriological techniques.
The E. coli strains and plasmid DNAs used in this study are described in Table 1. Strains were constructed using P1vir-mediated generalized transduction (34) and were routinely grown in Luria-Bertani medium (LB) (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl), unless otherwise indicated. When necessary, the following antibiotics were used at the indicated concentrations: ampicillin (Amp), 150 μg/ml; tetracycline (Tet), 12 μg/ml; kanamycin (Kan), 60 μg/ml; and chloramphenicol (Cam), 20 μg/ml.
Table 1.
E. coli strains and plasmid DNAs used in this study
| E. coli strain or plasmid | Relevant genotype or characteristics | Source, reference, or construction |
|---|---|---|
| E. coli strains | ||
| JW0733 | ΔnadA721∷kan | CGSC (3) |
| SMR5923 | Δ(attλ)∷sulApΩgfp-mut2 | S. Sandler (31) |
| JJC213 | Δrep∷kan | B. Michel (6) |
| RW118 | thr-1 araD139 Δ(gpt-proA)62 lacY1 tsx-33 supE44 galK2 hisG4(Oc) rpsL31 xyl-5 mtl-1 argE3(Oc) thi-1 sulA211 dnaN+ | R. Woodgate (21) |
| MS100 | RW118 dnaN+ tnaA300∷Tn10 | 42 |
| MS101 | MS100 dnaN159 tnaA300∷Tn10 | 42 |
| MS102 | MS100 dnaN+ tnaA300∷Tn10 lexA3(Ind−) | 42 |
| MS103 | MS100 dnaN159 tnaA300∷Tn10 lexA3(Ind−) | 42 |
| MS104 | MS100 dnaN+ tnaA300∷Tn10 lexA51(Def) | 42 |
| MS105 | MS100 dnaN159 tnaA300∷Tn10 lexA51(Def) | 42 |
| MS116 | MS100 dnaN159 tnaA300∷Tn10 ΔuvrB∷cat Δ(dinB-yafN)∷kan | 42 |
| MS125 | MS100 dnaN159 tnaA300∷Tn10 Δ(dinB-yafN)∷kan | 43 |
| JH103 | MS100 dnaN+ tnaA300∷Tn10 ΔnadA721∷kan | P1(JW0733) × (MS100) |
| JH104 | MS100 dnaN159 tnaA300∷Tn10 ΔnadA721∷kan | P1(JW0733) × (MS101) |
| JH105 | MS100 dnaN+ tnaA300∷Tn10 lexA3(Ind−) ΔnadA721∷kan | P1(JW0733) × (MS102) |
| JH106 | MS100 dnaN+ tnaA300∷Tn10 lexA51(Def) ΔnadA721∷kan | P1(JW0733) × (MS104) |
| JH107 | MS100 dnaN+ tnaA300∷Tn10 Δ(attλ)∷sulApΩgfp-mut2 | P1(SMR5923) × (JH103) |
| JH108 | MS100 dnaN159 tnaA300∷Tn10 Δ(attλ)∷sulApΩgfp-mut2 | P1(SMR5923) × (JH104) |
| JH109 | MS100 dnaN+ tnaA300∷Tn10 lexA3(Ind−) Δ(attλ)∷sulApΩgfp-mut2 | P1(SMR5923) × (JH105) |
| JH110 | MS100: dnaN+ tnaA300∷Tn10 lexA51(Def) Δ(attλ)∷sulApΩgfp-mut2 | P1(SMR5923) × (JH106) |
| RW542 | RW118 lexA51(Def) | R. Woodgate (14) |
| JH111 | MS100 tnaA+ lexA51(Def) Δrep∷kan | P1(JJC213) × (RW542) |
| Plasmids | ||
| pRM200 | Apr; pET11a directing expression of Pol II-D547N(exo−) | This work |
| pWSK29 | Apr; pSC101 origin | 51 |
| pRM100 | pWSK29 bearing polA+ (Pol I) | 30 |
| pRM101 | pWSK29 bearing polB+ (Pol II) | 30 |
| pRM103 | pWSK29 bearing umuD′ and umuC+ (Pol V) | 30 |
| pRM102 | pWSK29 bearing dinB+ (Pol IV) | 30 |
| pJH101 | pWSK29 bearing dinB(V303A-W304G-P305A) (Pol IVR) | 19 |
| pJH102 | pWSK29 bearing dinB(Δ346-351) (Pol IVC) | 19 |
| pJH100 | pWSK29 bearing dinB(D103N) (Pol IV-D103N) | 19 |
| pJH103 | pWSK29 bearing dinB(V303A-W304G-P305A-D103N) (Pol IVR-D103N) | 19 |
| pJH104 | pWSK29 bearing dinB(Δ346-351-D103N) (Pol IVC-D103N) | 19 |
| pJH105 | pWSK29 bearing dinB(D8A) (Pol IV-D8A) | This work |
| pJH107 | pWSK29 bearing dinB(D103A) (Pol IV-D103A) | This work |
The Δattλ∷sulApΩgfp-mut2 allele was introduced into strains MS100, MS101, MS102, and MS104 using a two-step approach that relied on the close linkage between nadA and attλ. We first used P1vir to transduce ΔnadA721∷kan into all four strains by selecting for kanamycin resistance. The presence of the ΔnadA721∷kan allele was verified by plating transductants on supplemented M9 agar plates with or without 1 μM nicotinic acid. The ΔnadA721∷kan allele was then replaced with Δattλ∷sulApΩgfp-mut2 using P1vir by selecting for growth on M9 agar lacking nicotinic acid and screening for Kan sensitivity. The structure of the attλ region was analyzed in representative clones using colony PCR. Primers att lambda For (5′-GTC ACG CCA AAA GCC AAT GC-3′) and att lambda Rev (5′-GTT AAT CAC TCT GCC AGA TG-3′) flank the E. coli attλ locus and yield an ∼250-bp fragment for the strain lacking Δattλ∷sulApΩgfp-mut2 and an ∼1,100-bp fragment for strains bearing Δattλ∷sulApΩgfp-mut2.
D8A and D103A substitutions were introduced into pRM102 using the QuikChange procedure (Stratagene) and primers (Integrated DNA Technologies) dinB-D8A-Top (5′-C ATT CAT GTG GCC ATG GAC TGC-3′), dinB-D8A-orig (5′-GCA GTC CAT GGC CAC ATG AAT G-3′), dinB-D103A-Top (5′-CCG TTG TCA CTG GCT GAG GCC TAT CTC GAT GTC-3′), and dinB-D103A-orig (5′-GAC ATC GAG ATA GGC CTC AGC CAG TGA CAA CGG-3′). Following DpnI restriction, reaction products were transformed into chemically competent DH5α E. coli, and the complete nucleotide sequences of representative dinB(D8A) and dinB(D103A) mutants were verified (Roswell Park Cancer Institute Biopolymer Facility, Buffalo, NY), as described previously (42).
The Pol II-D547N(exo−)-overproducing plasmid (pRM200) was derived from the wild-type Pol II overproducer (pRM107 [28]) using the QuikChange procedure as described above and primers (Sigma) PolB-D547N top (5′-C TAC GGC GAT ACG AAT TCA ACG TTT GTC TGG C-3′), PolB-D547N bottom (5′-G CCA GAC AAA CGT TGA ATT CGT ATC GCC GTA G-3′), PolB[exo−] top (5′-G GTT TCT ATA GCT ATT GCA ACC ACC CGC CAT GGT GAG CTG-3′), and PolB[exo−] bottom (5′-CAG CTC ACC ATG GCG GGT GGT TGC AAT AGC TAT AGA AAC C-3′).
UV sensitivity.
Sensitivity to 254-nm UV light was measured using a germicidal lamp (General Electric) as described previously (42). Briefly, cultures were grown in LB at 30°C to an optical density at 595 nm of ∼0.5. The cultures were then washed once with sterile 0.8% saline, resuspended in saline, transferred to sterile 60-mm petri dishes, and either mock irradiated or irradiated with 1, 2, or 3 J/m2 UV. Appropriate 10-fold serial dilutions of each sample were spotted onto LB plates supplemented with Amp and photographed following overnight incubation at 30°C.
Quantitative transformation assay.
Quantitative transformation assays were performed as described previously using chemically competent cells and 50 ng of the indicated plasmid DNA per reaction (28, 30). Selection of transformants was performed using LB agar supplemented with appropriate antibiotics. Plates were incubated overnight at the indicated temperature.
Quantitative Western blot analysis.
Quantitative Western blotting was performed with whole-cell lysates as described previously (43, 44) using polyclonal antibodies specific to Pol I, Pol II, or Pol IV and chemiluminescence detection (Pierce). The abundance of each Pol was calculated relative to a standard curve of the respective purified Pol using the formula P = (M × A)/(N × W), where P is the number of Pol molecules/cell (Pol I, Pol II, or Pol IV), M is grams of the respective Pol present in the whole-cell lysate, N is the number of cells examined (based on plating experiments), A is Avogadro's number (6.023 × 1023 molecules/mol), and W is the molecular mass of the Pol (Pol I, 103.1 kDa; Pol II, 90 kDa; Pol IV, 39.5 kDa). Antibodies specific to Pol I or Pol II were generated against recombinant forms of these Pols using New Zealand White rabbits (Sigma), while those specific to Pol IV were a generous gift from Takehiko Nohmi (23). The steady-state levels of each Pol reported in Table 3 represent the averages of three determinations using at least two independent lysates, each run in a separate SDS-PAGE with a standard curve of the appropriate purified Pol.
Table 3.
Steady-state levels of E. coli Pols expressed from the chromosome or plasmids
| Polyclonal antibody specificity | Molecules/cell of respective DNA Pol as a function of SOS statusa |
|||||
|---|---|---|---|---|---|---|
| SOS repressed |
SOS induced |
|||||
| Chromosome | Chromosome plus plasmid | Plasmid aloneb | Chromosome | Chromosome plus plasmid | Plasmid alone | |
| Pol I | 588 ± 60 | 3,265 ± 959 | ≈2,677 | 495 ± 107 | 4,124 ± 1,700 | ≈3,629 |
| Pol II | 75 ± 44 | 1,819 ± 786 | ≈1,744 | 1,073 ± 486 | 8,648 ± 328 | ≈7,575 |
| Pol IV | ≥156 | 1,233 ± 224 | ≈1,077 | 1,161 ± 155 | 4,944 ± 1,153 | ≈3,783 |
| Pol Vc | NDd | ND | ND | 60 | 300 | ≈240 |
Steady-state levels of each Pol present under SOS-repressed (lexA+) and SOS-induced [lexA51(Def)] states are indicated. Results shown represent the averages of triplicates ± standard deviations.
The approximate level of each Pol expressed from the respective plasmid was calculated by subtracting the level observed for the strain bearing the Pol-expressing plasmid from the level observed for the same strain bearing the control plasmid.
Values shown for Pol V (UmuD′2C) are from reference 40.
ND, none detected.
Quantitation of SOS induction by flow cytometry.
Flow cytometry was performed using a Becton Dickinson FACSCalibur 4-color flow cytometer. Overnight cultures of each strain were subcultured at 1:25 in M9 medium (supplemented with 1 mM CaCl2, 1 mM MgSO4, 5 μg/ml thiamine, 0.2% glucose, and 0.2% Casamino Acids) and grown at the indicated temperature (30°C, 34°C, or 37°C) to an optical density at 595 nm of ∼0.5, at which point 100,000 cells of each strain were immediately analyzed using the linear mode and 1.00-A gain for forward scatter, side scatter, and fluorescent scatter parameters. Results were analyzed using CellQuest (BD). Strains JH109 [dnaN+ lexA3(Ind−) Δattλ∷sulApΩgfp-mut2] and JH110 [dnaN+ lexA51(Def) Δattλ∷sulApΩgfp-mut2] were used to assign gating for “SOS-off” (intrinsic cell fluorescence) and “SOS-on” (sulAp-gfp-expressing cells) states. Gating parameters were assigned based on averaged results from 3 independent experiments for each strain. Cells of strain JH107 (dnaN+ lexA+ Δattλ∷sulApΩgfp-mut2) or JH108 (dnaN159 lexA+ Δattλ∷sulApΩgfp-mut2) were concluded to be induced for SOS (e.g., expressing sulAp-gfp) if their level of fluorescence was greater than that observed for strain JH109 [dnaN+ lexA3(Ind−) Δattλ∷sulApΩgfp-mut2]. The results shown in Fig. 3 represent the averages from 6 independent determinations.
Fig 3.
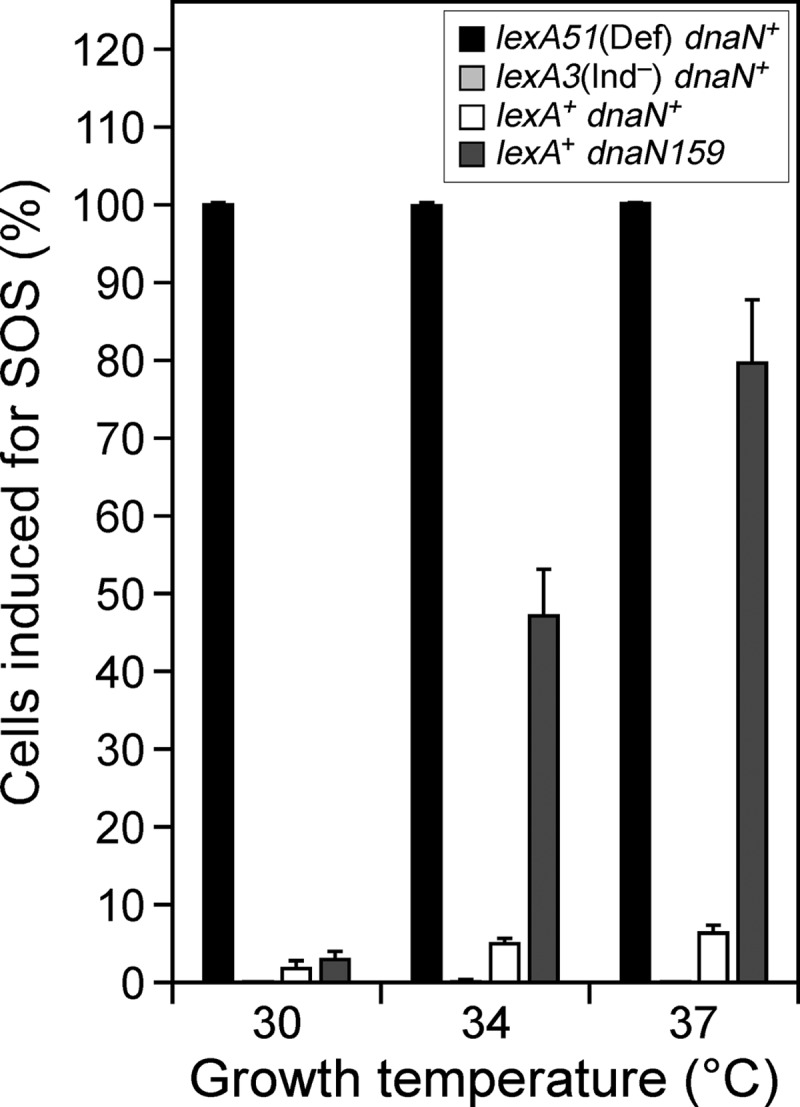
A subpopulation of the dnaN159 cells is induced for the SOS response. Strains JH107 (dnaN+ lexA+), JH108 (dnaN159 lexA+), JH109 [dnaN+ lexA3(Ind−)], and JH110 [dnaN+ lexA51(Def)] bearing the sulApΩgfp-mut2 SOS reporter were analyzed using flow cytometry as described in Materials and Methods. The results shown are averages from 3 to 6 independent experiments. Error bars represent standard deviations.
Purification of recombinant proteins.
Wild-type β and βR clamps (13, 19), Pol II and Pol II-D547N(exo−) (28), Pol III* [consisting of (αεθ)2(τ2γδδ′ψχ) (19, 28, 37)], the γ3δδ′ form of the DnaX clamp loader (39), the δ subunit of DnaX (39), and single-strand DNA binding protein (SSB) (27) were purified as described in the noted references.
Primer extension assay.
β clamp-dependent replication activity of Pol II and Pol II-D547N(exo−) was measured in vitro using an M13mp18 single-stranded DNA (ssDNA) viral template primed with PAGE-purified SP20 (5′-ACG CCT GTA GCA TTC CAC AG-3′; Sigma) as described previously (19, 28). Reaction mixtures contained replication assay buffer (20 mM Tris-HCl [pH 7.5], 8.0 mM MgCl2, 0.1 mM EDTA, 5 mM dithiothreitol, 1 mM ATP, 5% glycerol, 0.8 μg/ml bovine serum albumin), a 0.133 mM concentration of the four deoxynucleoside triphosphates (dNTPs), 2 μM SSB, a 10 nM concentration of the γ3δδ′ form of DnaX, 40 nM β or βR clamp (as indicated), 5 nM SP20-primed M13mp18 ssDNA, and 10 nM Pol II or Pol II-D547N(exo−), as indicated. Reaction mixtures were incubated for 5 min at 37°C. For competition experiments, Pol II and Pol II-D547N(exo−) were mixed prior to addition to the reaction mixture. Nucleotide incorporation by Pol II (2.6 pmol) was set equal to 100%.
Pol III* switch assay.
The ability of Pol II-D547N(exo−) or the δ subunit of DnaX to switch with a stalled or an actively replicating Pol III* was measured in vitro using an assay described previously (19). Reaction conditions were similar to those described above for the primer extension assay and are summarized schematically in Fig. 5A. Nucleotide incorporation following a 15-s incubation at 37°C by β-Pol III* in the absence of Pol II (4.4 pmol) or δ (12.3 pmol) or by βR-Pol III* in the absence of Pol II (3.5 pmol) was set equal to 100%. Replication activity in the presence of the challenging protein is expressed relative to this value as percent replication.
Fig 5.
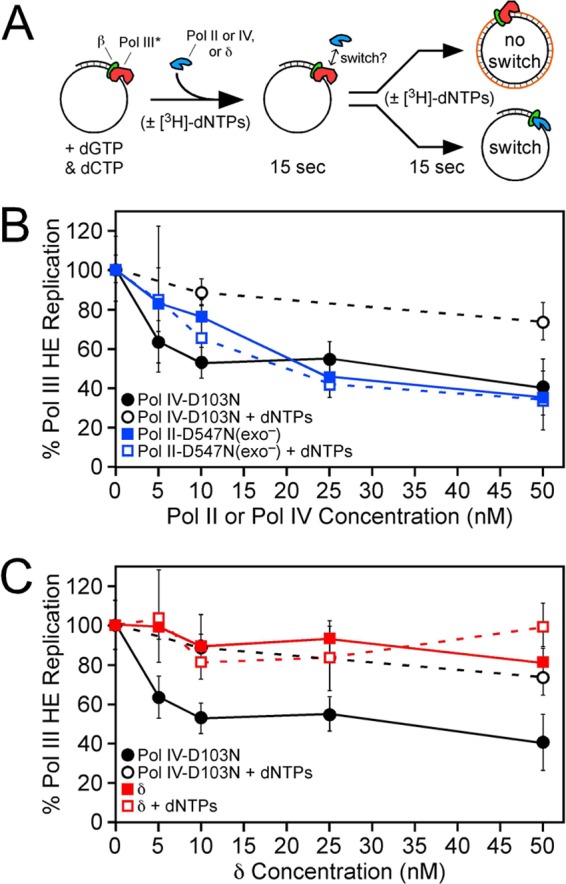
Switching between Pol III* and Pol II or δ. (A) Cartoon depiction of the assay (reprinted from Proceedings of the National Academy of Sciences of the United States of America [19]). (B and C) Results for Pol III*-Pol II-D547N(exo−) (B) and Pol III*-δ (C) switching. All reaction mixtures contained 5 nM Pol III* and the indicated concentration (5 to 50 nM) of Pol II-D547N(exo−) or δ. Replication activity is expressed relative to that of the Pol III* (5 nM) control, which was set equal to 100%. Reactions in which dNTPs were added at the same time as the competing protein (Pol II, Pol IV, or δ) are designated by “+dNTPs.” Values shown for Pol III*-Pol IV-D103N switching were determined at the same time as those for Pol II-D547N(exo−) and δ but were published previously (19). They are included here for comparison to Pol II-D547N(exo−) and δ. The results shown are the averages of at least 4 independent determinations. Error bars represent standard deviations.
RESULTS
The ability of Pol IV to confer UV sensitivity and to interfere with growth of the dnaN159 strain requires both the rim and cleft of the β clamp.
We recently described a model for the Pol III*-Pol IV switch, in which Pol IV binds the rim of the clamp prior to binding the cleft that was previously occupied by a stalled Pol III* (19). In light of this model, we hypothesized that the ability of Pol IV to confer UV sensitivity upon the dnaN159 strain (42) and to impede growth when expressed at an elevated level (28, 30) was due to the inability of the mutant β159 clamp to properly regulate the Pol III*-Pol IV switch. As a test of this hypothesis, we asked whether Pol IV mutants impaired for interaction with the rim (Pol IVR) or the cleft (Pol IVC) of the β clamp retained an ability to confer these dnaN159 phenotypes. Pol IVR contains 303VWP305-to-303AGA305 substitutions that abrogate its interaction with the rim of the clamp, while Pol IVC lacks its C-terminal six residues (Δ346QLVLGL351) encompassing the CBM that interacts with the clamp cleft (19). Of relevance to this experiment, both Pol IVR and Pol IVC retain Pol activity in vitro (19, 49), and each is expressed at a steady-state level similar to that observed for wild-type Pol IV in vivo (19).
The importance of the rim and cleft of the clamp to UV sensitivity was examined first. Since UV sensitivity was enhanced in strains deficient in nucleotide excision repair (42), we measured this phenotype in a dnaN159 ΔuvrB ΔdinB strain (MS116) expressing either wild-type Pol IV, Pol IVR, or Pol IVC from a low-copy-number plasmid. As summarized in Fig. 2, the strain expressing wild-type Pol IV was significantly more UV sensitive than that carrying the pWSK29 control. In contrast, strains expressing either Pol IVR or Pol IVC were indistinguishable from that carrying the pWSK29 control. Taken together, these results indicate that Pol IV contacts both the rim and cleft of the clamp to interfere with Pol III* function following UV irradiation in the dnaN159 strain.
Fig 2.

Pol IV requires both the rim and cleft of the β clamp to confer UV sensitivity upon the dnaN159 strain. The UV sensitivity of strain MS116 bearing plasmid pWSK29 (control), pRM102 (Pol IV), pJH101 (Pol IVR), or pJH102 (Pol IVC) was measured as described in Materials and Methods. This experiment was performed twice; representative results are shown.
We next asked whether the rim and cleft of the clamp were required for elevated levels of Pol IV to impede growth of the dnaN159 strain. For this experiment, we used a dnaN159 lexA51(Def) strain. The lexA51(Def) allele is defective for repression of SOS-regulated genes, including the dinB-encoded Pol IV. Using an established quantitative transformation assay (28, 30), we confirmed that elevated levels of Pol IV expressed from the native, SOS-regulated dinB promoter cloned in pWSK29 were toxic to the dnaN159 lexA51(Def) strain (MS105) but not to the isogenic dnaN+ partner (MS104) when plated at 30°C (28, 30) (Table 2). In contrast, neither Pol IVR nor Pol IVC was able to impede growth of these strains. As a control, we examined the same pWSK29 plasmid directing expression of Pol I, Pol II, or Pol V. Consistent with earlier findings (30), elevated levels of these Pols failed to significantly affect growth of either the dnaN+ or the dnaN159 strain (data not shown).
Table 2.
Effects of plasmids expressing mutant Pol IV proteins on viability of the dnaN159 lexA51(Def) strain
| Plasmid | Transformation efficiency with straina: |
|
|---|---|---|
| MS104 [dnaN+ lexA51(Def)] | MS105 [dnaN159 lexA51(Def)] | |
| pWSK29 (control) | (1.09 ± 0.59) × 104 (≡1.0) | (3.35 ± 1.84) × 104 (≡1.0) |
| pRM102 (Pol IV) | (1.24 ± 0.04) × 104 (1.1) | (2.78 ± 2.78) × 102 (8.3 × 10−3) |
| pJH101 (Pol IVR) | (1.10 ± 1.95) × 104 (1.0) | (1.99 ± 0.88) × 104 (0.6) |
| pJH102 (Pol IVC) | (1.65 ± 0.71) × 104 (1.5) | (2.94 ± 1.98) × 104 (0.9) |
| pJH105 (Pol IV-D8A) | (7.18 ± 2.42) × 103 (0.7) | (7.01 ± 3.03) × 103 (0.2) |
| pJH107 (Pol IV-D103A) | (9.61 ± 5.50) × 103 (0.9) | (<2.78 ± 0.00) × 101 (<8.3 × 10−4) |
| pJH100 (Pol IV-D103N) | (4.40 ± 2.31) × 103 (0.4) | (<2.78 ± 0.00) × 101 (<8.3 × 10−4) |
| pJH103 (Pol IVR-D103N) | (1.50 ± 0.08) × 104 (1.4) | (<2.78 ± 0.00) × 101 (<8.3 × 10−4) |
| pJH104 (Pol IVC-D103N) | (1.16 ± 1.32) × 104 (1.1) | (2.45 ± 0.55) × 104 (0.7) |
Transformants were selected at 30°C. Values shown are the averages from three independent experiments ± standard deviations. Values in parentheses represent transformation efficiencies relative to the frequency observed for the pWSK29 control, which was set equal to 1.0.
We questioned whether the ability of Pol IV to impede growth of the dnaN159 strain was simply the result of it being expressed at a higher level than the other Pols examined. Based on quantitative Western blot analysis (Table 3), Pol IV was expressed at a level intermediate to those of the other Pols. Moreover, if steady-state levels of each Pol are expressed in relation to their endogenous level, Pol IV was expressed at levels ∼4-fold higher than the normal SOS-induced levels, which represent the smallest fold increase among the four Pols examined. By comparison, Pol V was present at ∼5-fold-higher levels (40) (Table 3), while levels of Pol I and Pol II were each ∼8-fold elevated. These findings, taken together with the results discussed above, suggest that Pol IV is unique among the E. coli Pols in terms of its ability to impede growth of the dnaN159 strain.
The ability of Pol IV to impede growth of the dnaN159 strain is independent of its catalytic activity.
Since Pol IV catalytic activity was dispensable for switching with a stalled Pol III* in vitro (17, 19), we hypothesized that catalytic activity would likewise be unnecessary for elevated levels of Pol IV to impede growth of the dnaN159 strain. As a test of this hypothesis, we examined the Pol IV-D8A and Pol IV-D103N mutants, which lack detectable catalytic activity but are still proficient for switching with Pol III* in vitro (17, 19). As summarized in Table 2, Pol IV-D103N impaired growth of the dnaN159 strain, while Pol IV-D8A did not. We therefore constructed a Pol IV-D103A mutant and analyzed its phenotype to distinguish whether phenotypes of the D8A and D103N mutations differed due to the nature of their positions or their substitutions. As summarized in Table 2, Pol IV-D103A was indistinguishable from Pol IV-D103N with respect to its ability to impede growth of the dnaN159 lexA51(Def) strain. Taken together, these findings indicate that while catalytic activity of Pol IV is dispensable for its ability to impede growth of the dnaN159 strain, D8 is nevertheless required (see Discussion).
We next asked whether the ability of the Pol IV-D103 mutants to impede growth of the dnaN159 strain relied on interaction of the mutant with the rim and/or cleft of the clamp. Since the D103N and D103A mutants behaved similarly, we focused on Pol IV-D103N. As summarized in Table 2, Pol IVC-D103N (bearing both D103N and Δ346QLVLGL351 mutations) was unable to impede growth. In contrast, Pol IVR-D103N was indistinguishable from Pol IV-D103N. We previously demonstrated that each of these mutants was expressed at a level similar to that of the wild-type Pol IV (19). These results, indicating that Pol IV-D103N required the clamp cleft contact but not the rim to impede growth of the dnaN159 strain, are reminiscent of our previous observation that Pol IV may gain access to the DNA by at least two distinct mechanisms: one involving a two-step switch with a stalled Pol III*, which relies on both the rim and cleft of the clamp, and a second in which a naked clamp directly recruits Pol IV to an ssDNA nick or gap via a mechanism that is dependent on the clamp cleft but independent of the rim (19).
Physiological levels of Pol IV-D103N impede growth of the dnaN159 strain via a mechanism that requires both the rim and the cleft of the β clamp.
Transformation frequencies for plasmids expressing Pol IV-D103N or Pol IV-D103A were consistently lower than those for the same plasmid expressing wild-type Pol IV (Table 2), suggesting that these mutants were more toxic to the dnaN159 strain than wild-type Pol IV. In light of their phenotypes, we hypothesized that these mutant Pol IV proteins switch normally with Pol III* in the dnaN159 strain but, due to their catalytic defect, interfere with replication and/or ssDNA gap repair after gaining access to the DNA to confer the more severe growth defect. We further hypothesized that if Pol IV gains access to the DNA when expressed at physiological levels, these mutants might impede replication when expressed in the lexA+ strain, leading to SOS induction. Since dinB is SOS regulated, we postulated that SOS induction in the dnaN159 lexA+ strain would essentially mimic the dnaN159 lexA51(Def) phenotype. As a test of these hypotheses, we used our quantitative transformation assay. As summarized in Table 4, the dnaN159 lexA+ strain was transformed with a plasmid expressing the Pol IV-D103A or Pol IV-D103N mutant as efficiently as with the pWSK29 control plasmid or pWSK29 expressing wild-type Pol IV. However, colonies expressing Pol IV-D103N were significantly smaller than those expressing wild-type Pol IV or Pol IV-D103A and grew more poorly when cultured in liquid broth at 30°C under selective conditions (Table 4). In order to determine whether this poor-growth phenotype was the result of the Pol IV-D103N mutant inducing the SOS response in the dnaN159 lexA+ strain, we measured transformation efficiencies for these same plasmids using the isogenic dnaN159 lexA3(Ind−) strain (MS103); in contrast to the lexA+ allele, lexA3(Ind−) is refractory to RecA-mediated autodigestion (24), and as a result, strains bearing the lexA3(Ind−) allele are unable to induce the SOS response. Although transformation frequencies were similar to those for the dnaN159 lexA+ strain (Table 4), the health of the dnaN159 lexA3(Ind−) strain expressing Pol IV-D103N was significantly improved, indicating a role for SOS induction in the poor-growth phenotype of the Pol IV-D103N strain.
Table 4.
Effects of elevated levels of Pol IV on viabilities of different dnaN159 strains at 30° and 34°C
| Plasmid | Transformation efficiency with straina: |
|||||
|---|---|---|---|---|---|---|
| MS101 (dnaN159 dinB+
lexA+) |
MS116 [dnaN159 Δ(dinB-yafN)∷kan lexA+] |
MS103 [dnaN159 dinB+
lexA3(Ind−)] |
||||
| 30°C | 34°C | 30°C | 34°C | 30°C | 34°C | |
| pWSK29 (control) | (1.09 ± 0.36) × 105 (≡1.0) | (1.12 ± 0.09) × 105 (≡1.0) | (1.16 ± 0.20) × 105 (≡1.0) | (2.50 ± 0.18) × 105 (≡1.0) | (3.76 ± 0.61) × 104 (≡1.0) | (7.89 ± 2.16) × 103 (≡1.0) |
| pRM102 (Pol IV) | (8.17 ± 1.87) × 104 (0.8) | (7.95 ± 0.11) × 104 (0.7) | (7.14 ± 1.81) × 104 (0.6) | (1.70 ± 0.24) × 105 (0.7) | (3.10 ± 0.67) × 104 (0.8) | (9.51 ± 2.22) × 103 (1.2) |
| pJH101 (Pol IVR) | (8.66 ± 2.74) × 104(0.8) | (1.20 ± 0.11) × 105 (1.0) | NDc | ND | ND | ND |
| pJH102 (Pol IVC) | (9.67 ± 2.40) × 104 (0.9) | (9.56 ± 0.47) × 104 (0.9) | ND | ND | ND | ND |
| pJH107 (Pol IV-D103A) | (7.34 ± 2.28) × 104 (0.7) | (7.90 ± 0.20) × 104 (0.7) | (1.01 ± 0.06) × 105 (0.9) | (1.28 ± 0.32) × 105 (0.5) | (2.33 ± 0.44) × 104 (0.6) | (1.20 ± 0.10) × 104 (1.5) |
| pJH100 (Pol IV-D103N) | (5.57 ± 0.84) × 104 (0.5)b | (1.05 ± 0.35) × 102 (9.4 × 10−4) | (3.95 ± 0.64) × 104 (0.3) | (8.60 ± 1.10) × 101 (3.4 × 10−4) | (1.35 ± 0.39) × 104 (0.4) | (6.96 ± 1.03) × 103 (0.9) |
| pJH103 (Pol IVR-D103N) | (1.30 ± 0.35) × 105 (1.2) | (7.92 ± 3.43) × 104 (0.7) | ND | ND | ND | ND |
| pJH104 (Pol IVC-D103N) | (8.30 ± 2.65) × 104 (0.8) | (9.09 ± 0.11) × 104 (0.8) | ND | ND | ND | ND |
Transformants were selected at 30 or 34°C, as indicated. Values shown are the averages from three independent experiments ± standard deviations. Values in parentheses represent transformation efficiencies relative to the frequency observed for the pWSK29 control, which was set equal to 1.0.
Colonies were tiny.
ND, not determined.
To support results from the transformation experiments, we used a sulAp-gfp reporter developed by McCool et al. (31) to analyze the level of SOS induction in the dnaN+ and dnaN159 strains. As summarized in Fig. 3, we confirmed that SOS response was modestly induced in a subpopulation of dnaN159 cells grown at 30°C (42). Importantly, the size of this population increased proportionally with increasing temperature (Fig. 3), as expected since β159 is temperature sensitive and is therefore less proficient for replication at elevated temperatures. Given that ∼50% of the dnaN159 population was induced for SOS at 34°C, we examined transformation efficiencies at this temperature to see whether we could distinguish the Pol IV-D103N phenotype from that of either wild-type Pol IV or Pol IV-D103A. As summarized in Table 4, only Pol IV-D103N impaired growth of the dnaN159 lexA+ strain at 34°C. In contrast, plasmids expressing Pol IV-D103N mutants impaired for interaction with the rim (Pol IVR-D103N) or the cleft (Pol IVC-D103N) of the β clamp were well tolerated (Table 4). Furthermore, transformation efficiencies of the plasmids expressing wild-type Pol IV or Pol IV-D103A were indistinguishable from that of the pWSK29 control. Finally, deletion of the chromosomal dinB gene, which is responsible for ∼20 to 25% of Pol IV expressed in these strains, irrespective of SOS induction (Table 3), failed to improve transformation efficiency for the dnaN159 lexA+ strain with the Pol IV-D103N plasmid (Table 4). Taken together, these findings indicate that expression of Pol IV-D103N at a level somewhere in between chromosomal (∼1,200 molecules/cell) and plasmid-expressed (∼3,800 molecules/cell) levels serves to interfere with Pol III* function in the dnaN159 lexA+ strain via a mechanism involving the rim and cleft of the clamp. Furthermore, our finding that Pol IV-D8A, Pol IV-D103A, and Pol IV-D103N each displayed a distinct phenotype suggests that Pol IV catalysis either contributes in some way to the ability of Pol IV to switch with Pol III* in vivo or influences the kinetics of the Pol switch to impair cell growth (see Discussion).
Elevated levels of Pol IV impede growth of an E. coli rep strain in a manner that relies on the rim and cleft of the clamp.
Since β159 is impaired for conferring processive replication upon Pol III* (8, 28), we hypothesized that Pol IV may abrogate growth of the dnaN159 strain by targeting a less processive, or possibly stalled, Pol III*. In this case, elevated levels of Pol IV should impede growth of strains bearing mutations in genes other than dnaN that, when mutated, result in an increased frequency of Pol III* stalling. As a test of this hypothesis, we compared the ability of elevated levels of Pol IV to impede growth of an E. coli strain lacking rep function. The rep gene encodes the Rep protein, an important yet nonessential helicase that acts as a second motor at the replication fork. In addition to displacing proteins from the DNA (1, 6, 18), Rep also interacts physically with the main replicative helicase, DnaB, to increase the rate at which the replisome moves (18). Thus, strains lacking rep function display slower replication kinetics and increased frequencies of fork stalling due to recurrent collisions between the replisome and protein-DNA blocks (6).
As summarized in Table 5, elevated levels of Pol IV impaired growth of the rep strain, while elevated levels of the other Pols had only a minimal effect. Neither Pol IVR nor Pol IVC impeded growth of the rep strain (Table 5). We next examined the ability of Pol IV-D103N to impede growth of the rep strain. As summarized in Table 5, Pol IV-D103N was ∼10-fold more effective than wild-type Pol IV at impeding growth of the rep strain. Importantly, this phenotype relied on the ability of Pol IV to interact with both the rim and the cleft of the clamp, as neither Pol IVR-D103N nor Pol IVC-D103N was able to impede growth. These results, taken together with those discussed above, indicate that elevated levels of Pol IV impede growth of E. coli strains bearing either the dnaN159 or Δrep mutation, suggesting that this phenotype results from a switch involving Pol IV and a stalled Pol III*.
Table 5.
Effects of elevated levels of various Pols on viability of the Δrep lexA51(Def) strain
| Plasmid | Transformation efficiency with straina: |
|
|---|---|---|
| MS104 [rep+ lexA51(Def)] | JH111 [Δrep∷kan lexA51(Def)] | |
| pWSK29 (control) | (1.11 ± 1.38) × 104 (≡1.0) | (3.60 ± 2.30) × 103 (≡1.0) |
| pRM100 (Pol I) | (3.32 ± 1.20) × 103 (0.3) | (3.98 ± 0.24) × 102 (0.1) |
| pRM101 (Pol II) | (7.38 ± 4.12) × 103 (0.7) | (1.13 ± 0.32) × 103 (0.3) |
| pRM103 (Pol V) | (9.60 ± 3.20) × 103 (0.9) | (1.29 ± 1.13) × 103 (0.4) |
| pRM102 (Pol IV) | (4.68 ± 2.24) × 103 (0.4) | (7.23 ± 7.23) × 101 (2.0 × 10−2) |
| pJH101 (Pol IVR) | (4.72 ± 0.72) × 103 (0.4) | (3.16 ± 2.67) × 103 (0.9) |
| pJH102 (Pol IVC) | (1.18 ± 0.14) × 104 (1.1) | (2.66 ± 2.02) × 103 (0.7) |
| pJH100 (Pol IV-D103N) | (4.72 ± 0.96) × 103 (0.4) | (<1.15 ± 0.00) × 101 (<3.2 × 10−3) |
| pJH103 (Pol IVR-D103N) | (5.34 ± 0.20) × 103 (0.5) | (4.20 ± 6.84) × 102 (0.1) |
| pJH104 (Pol IVC-D103N) | (4.32 ± 1.08) × 103 (0.4) | (2.14 ± 0.37) × 103 (0.6) |
Transformants were selected at 37°C. Values shown are the averages from three independent experiments ± standard deviations. Values in parentheses represent transformation efficiencies relative to the frequency observed for the pWSK29 control, which was set equal to 1.0.
Pol II switches equally well with both a stalled and an actively replicating Pol III* in vitro.
The results discussed above suggest that Pol IV is unique in its ability to switch with a stalled Pol III*. Since Pol II was previously demonstrated to switch with Pol III* (22), we used an assay developed in our lab to determine whether the Pol III*-Pol II switch relied on a stalled Pol III HE in vitro and to compare its requirements with those of the Pol III*-Pol IV switch. Since our assay utilizes inhibition of Pol III HE replication to monitor Pol switching (19), we cloned and purified a mutant form of Pol II bearing D156A, E158A, and D547N substitutions, referred to as Pol II-D547N(exo−). Residues D156 and E158 are part of the 3′-to-5′ proofreading exonuclease (exo) domain of Pol II, and their replacement with alanine abolishes exo activity (50). Residue D156 coordinates Mg2+ and is therefore essential for Pol activity (50). As summarized in Fig. 4, Pol II-D547N(exo−) lacked detectable polymerase activity in an in vitro primer extension assay reconstituted with purified components. Furthermore, when mixed 1:1 with purified wild-type Pol II prior to its addition to the replication reaction mix, Pol II-D547N(exo−) inhibited replication by roughly 50% (Fig. 4). Since this assay measures β-dependent Pol II replication (28), these results indicate that Pol II-D547N(exo−) retains normal affinity for both the β clamp and the DNA template.
Fig 4.

Replication activity of Pol II-D547N(exo−). (A) Cartoon depiction of the assay. (B) The ability of Pol II+ or Pol II-D547N(exo−) together with β+ or βR (as noted) to catalyze replication in vitro was measured as described in Materials and Methods. The results shown are averages of at least 4 independent determinations. Error bars represent standard deviations.
To measure Pol III*-Pol II switching, we incubated Pol III* with β clamp, SP20-primed M13mp18 ssDNA, ATP, and dGTP/dCTP. Under these conditions, The Pol III HE complex assembles on the DNA template but remains in a “stuttering” state since it is unable to extend the primer more than a few nucleotides, due to the absence of dATP/dTTP (19). Importantly, we have previously demonstrated that this stalled Pol III HE is stable on the DNA template, with a half-life of >2 min under these conditions, and that it is efficiently rescued by addition of dATP and [3H]dTTP (here referred to as the [3H]dNTP cocktail) (19). By adding increasing concentrations (5 to 50 nM) of Pol II-D547N(exo−) to this reaction mixture, we were able to probe the switch between a stalled Pol III* and Pol II. Fifteen seconds after adding Pol II-D547N(exo−), the [3H]dTTP cocktail was added, and nascent strand synthesis by Pol III HE was measured using liquid scintillation spectroscopy (19) (Fig. 5A). Alternatively, simultaneous addition of Pol II-D547N(exo−) and the [3H]dNTP cocktail was used to measure the ability of Pol II to switch with an actively replicating Pol III* (Fig. 5A). In contrast to Pol IV, which switched exclusively with a stalled Pol III*, Pol II-D547N(exo−) switched equally well with both a stalled and an actively replicating Pol III* in vitro (Fig. 5B). Addition of Pol II-D547N(exo−) at concentrations of from 5 to 25 nM inhibited replication by Pol III* in a concentration-dependent manner. Interestingly, Pol II was slightly less efficient at switching with Pol III* than was Pol IV: whereas an ∼2-fold molar excess (10 nM) of Pol IV-D103N inhibited Pol III* activity by ∼50%, an ∼5-fold molar excess (25 nM) of Pol II-D547N(exo−) over Pol III* was required to inhibit Pol III* to a similar extent (Fig. 5B).
As a control for these experiments, we asked whether the δ subunit of the DnaX clamp loader complex was capable of switching with Pol III*. Since δ (KD [equilibrium dissociation constant] = ∼18 nM) and Pol II (KD = ∼30 nM) display similar affinities for the clamp (20, 39), we reasoned that use of δ in place of Pol II would distinguish between the ability of Pol II to utilize a concerted mechanism to switch with Pol III* and a simple in vitro mass action effect. As summarized in Fig. 5C, addition of δ at concentrations (5 to 50 nM) up to a 10-fold molar excess over Pol III* (5 nM) failed to impede replication, regardless of whether it was added prior to or together with the [3H]dNTP cocktail. These results not only indicate that δ cannot “switch” with Pol III*, supporting the conclusion that Pol II and Pol IV utilize distinct mechanisms to undergo a switch with Pol III*, but also confirm that Pol III* remains bound to the β clamp under the conditions of our switch assay, as δ would have otherwise unloaded clamp from the DNA template, thereby preventing processive Pol III* replication.
Unlike Pol IV, Pol II does not require the rim of the β clamp to switch with Pol III*.
In addition to requiring a stalled Pol III*, Pol IV also relied on physical contact with the rim (E93 and L98) of the β clamp to undergo a switch with Pol III* in vitro (19). βR contains E93K and L98K substitutions that abrogate interaction of Pol IV with the rim of the clamp (19). Importantly, βR was proficient for Pol III* replication, and the stability of the stalled Pol III HE complex assembled with βR is similar to that of the complex formed with β+ (19). As part of an ongoing effort to determine whether the clamp rim (e.g., residues E93 and L98) represented a unique contact for Pol IV or defined a surface used by multiple Pols, we asked whether βR was able to support Pol II function in vitro. As summarized in Fig. 6, βR was indistinguishable from β+ with respect to stimulation of Pol II replication activity. This result indicates that Pol II does not require contact with E93 or L98 of the β clamp for processive replication. We next asked whether βR was able to support Pol III*-Pol II switching. Since 25 nM Pol II-D547N(exo−) was required to inhibit 5 nM Pol III* by ∼50% in our switch assay (Fig. 5B), we chose this concentration to analyze the ability of βR to support the Pol III*-Pol II switch. As summarized in Fig. 6, βR was indistinguishable from β+ with respect to its ability to support the Pol III*-Pol II switch, regardless of whether Pol II-D547N(exo−) was added prior to or together with the [3H]dNTP cocktail. Thus, in summary, these results indicate that the clamp rim is dispensable for both Pol II replication and Pol III*-Pol II switching.
Fig 6.

Contact with the clamp rim is dispensable for the Pol III*-Pol II switch. Pol III* (5 nM) was assembled on SP20-primed M13mp18 ssDNA in the presence of either β+ or βR, as indicated, prior to challenging with a 5-fold molar excess (25 nM) of Pol II-D547N(exo−) as for Fig. 5. The results shown are the averages of at least 4 independent determinations. Error bars represent standard deviations.
DISCUSSION
The results discussed in this report demonstrate that Pol IV requires the ability to interact with both the rim and cleft of the β clamp to confer UV sensitivity upon the dnaN159 strain and to impede its growth when expressed at levels ∼4-fold higher than the normal SOS-induced levels. Elevated levels of Pol IV also impaired growth of an E. coli rep strain in a manner that relied on the rim and cleft of the clamp (Table 5). Inasmuch as Pol III* is prone to more frequent stalling in dnaN159 or rep strains, due to reduced levels of processivity (28) or difficulties in replicating protein-bound DNA (1, 2, 6, 18), respectively, these findings support the model that Pol IV switches with a stalled Pol III* in vivo. The ability of Pol IV to impede growth upon switching may result from the ability of frequent switching to impair replication or from the recently described ability of overproduced levels of Pol IV to incorporate oxidized deoxyguanosine into nascent DNA (15). In contrast to these results with Pol IV, similar levels of Pol I, II, or V failed to impair growth of these strains (Tables 2 and 5), suggesting that these Pols do not require that Pol III* be stalled in order to undergo a switch. Alternatively, these Pols may switch less efficiently with Pol III*. In this case, these Pols may be able to impede growth of the dnaN159 strain if expressed at levels higher than those examined here. The results of an in vitro assay using purified components were consistent with this conclusion: not only did Pol II switch equally well with both a stalled and an actively replicating Pol III*, but it also switched with Pol III* less efficiently than did Pol IV (Fig. 5). Taken together, these results support the model that Pol IV replaces a stalled Pol III* at the 3′-OH of a primer-template junction via a two-step switch that relies on both the rim and the cleft of the β clamp (Fig. 1). In contrast, the other Pols, exemplified by Pol II, appear either to be less efficient at switching with Pol III* or to utilize one or more distinct mechanisms to switch with Pol III*.
Based on results of quantitative Western blotting (Table 3), Pol IV is normally expressed at ∼150 molecules/cell (∼250 nM) in the absence of SOS induction and can reach levels as high as ∼1,161 molecules/cell (∼1.9 μM) under chronically SOS-induced conditions. These values are similar to those reported previously by Kim et al. (23). Although the affinity of Pol IV for the sliding clamp in solution is ∼450 nM (this value represents the sum of the rim and cleft contacts), its affinity for the clamp rim is ∼1.3 μM (19). Thus, SOS-induced levels of Pol IV appear to be sufficient to drive formation of the Pol IV-clamp complex in vivo. However, our finding that 10 nM Pol IV switches effectively with 5 nM stalled Pol III* in vitro (19) (Fig. 5B) suggests that Pol IV associates with a stalled Pol III HE complex with greater affinity than it does with the free clamp in solution. The affinity of Pol IV for the clamp rim may be greater when the clamp is on DNA. Alternatively, Pol IV may interact with one or more components of Pol III* in addition to the clamp. In either case, SOS-repressed levels of Pol IV may be sufficient for switching with a stalled Pol III* in vivo. Thus, Pol IV might associate with the replisome irrespective of SOS induction to confer upon the replisome a capacity to cope with DNA lesions that block Pol IIIα progression. Viewed in this way, elevated levels of Pol IV may impede growth of strains bearing dnaN159 or rep mutations by switching excessively with the repeatedly stalled Pol III*. This type of repeated switching would almost certainly impede Pol III HE function, resulting in the growth defect observed for the dnaN159 or rep strains expressing elevated levels of Pol IV.
Despite the fact that Pol IV-D8A, Pol IV-D103A, and Pol IV-D103N are similar in that they lack catalytic activity, each nevertheless displayed a distinct phenotype when expressed in the dnaN159 strain: whereas D8A was unable to impede growth (Table 2), both D103A and D103N did, with D103N displaying the more severe phenotype (Table 4). Importantly, Pol IV-D103N relied on both the rim and the cleft of the β clamp to impede growth of the dnaN159 lexA+ strain (Table 4). In contrast, Pol IVR-D103N impeded growth of the dnaN159 lexA51(Def) strain (Table 2). We suggest that this difference is the result of Pol IV levels being higher in the lexA51(Def) strain than in the lexA+ strain, even when grown at 34°C (Fig. 3). This conclusion is supported by our finding that the level of green fluorescent protein (GFP) expressed from the sulAp-gfp reporter in the dnaN159 lexA+ strain grown at 34° or 37°C was lower than that observed in the isogenic dnaN159 lexA51(Def) strain grown at 30°C, 34°, or 37°C, based on fluorescence measurements (data not shown), presumably due to the ability of LexA to dampen the extent of SOS induction. Thus, in contrast to the dnaN159 lexA+ strain, where Pol IVR-D103N impaired Pol III* function, the higher levels of Pol IVR-D103N present in the dnaN159 lexA51(Def) strain likely impeded both Pol III* function and ssDNA gap repair. Since Pol IV is recruited to ssDNA gaps independently of the clamp rim (19), disruption of this contact would not be expected to relieve the growth defect under these conditions. In contrast to the case for the dnaN159 strain, Pol IV-D103N neither impaired growth of the dnaN+ strain (Table 4) nor induced the SOS response (data not shown). Thus, its ability to impede growth of the dnaN159 strain appears to be specific to one or more features of the β159 clamp and/or to strains prone to replisome stalling.
The results of a biochemical assay using purified components to reconstitute Pol switching provided strong support for our model that Pol II and Pol IV utilize distinct mechanisms to switch with Pol III* (28, 42). In contrast to Pol IV, which switched specifically with a stalled Pol III* via a mechanism that relies on the rim and cleft of the clamp, Pol II switched equally well with both an actively replicating and a stalled Pol III* (Fig. 5) in a manner that was independent of the clamp rim (Fig. 6). Despite our finding that Pol II (20) (KD = ∼30 nM) has considerably higher affinity for the clamp in solution than does Pol IV (19, 20) (KD = ∼460 nM), Pol II nevertheless switched less efficiently with Pol III* in vitro than did Pol IV in vitro: whereas a 2-fold molar excess of Pol IV inhibited Pol III HE replication by ∼50% in vitro (19), an ∼5-fold molar excess of Pol II was required for a similar effect (Fig. 5). These findings suggest that Pol IV has a higher affinity for a stalled Pol III HE than does Pol II. Consistent with this conclusion, Indiani et al. (22) also noted that Pol II switched less efficiently with Pol III* than did Pol IV.
In contrast to the case for Pol II and Pol IV, the δ subunit of the DnaX clamp loader was unable to “switch” with Pol III* in vitro (Fig. 5). This finding demonstrates that an ability to switch with Pol III* in our in vitro assay relies on more than a competing partner simply binding to the β clamp. It is possible that Pol III* sterically occludes access of δ to the free cleft on the β clamp that is not bound by Pol III* (39). Since Pol IV binds the clamp rim adjacent to the cleft that is bound by Pol IV, it does not require access to the free cleft on the clamp to undergo a switch (19). Likewise, Pol II may also associate with one or more surfaces of the clamp that are accessible in the presence of Pol III* and/or components of Pol III* to effect the switch. Consistent with this conclusion, Pol II (and Pol IV) interact physically with residues H148 to R152 of the clamp, and replacement of these residues with alanines severely impeded the ability of clamp to confer processivity upon Pol II (and Pol IV) in vitro (20). Structural characterization of the clamp-Pol II complex will help to answer this question.
The inability of modestly elevated levels of Pol II (∼8-fold elevated) or Pol IV (∼4-fold elevated) to impede growth of E. coli strains proficient for processive replication (e.g., the dnaN+ strain) (Table 2) contrasts with the finding that high-level overexpression of these same Pols served to block replication in an otherwise wild-type E. coli strain (22, 48). Although Pol II levels were not measured in the cited work, the ability of Pol IV to block replication relied on levels that were ∼72-fold higher than the normal SOS-induced levels. It was suggested that elevated levels of Pol II and Pol IV have the ability to act like a break on replication (22, 48). Since these Pols are regulated as part of the SOS response, they were suggested to replace Pol III* at the fork following DNA damage to slow replication, thereby allowing additional time for accurate DNA repair mechanisms, similar to a eukaryotic intra-S-phase checkpoint control (22, 48). In the case of Pol IV, this checkpoint-like function was suggested by one group to involve physical displacement of Pol III* from the clamp (48) (Fig. 1). However, it is unclear whether Pol IV displaces Pol III* from the clamp under physiological conditions in vivo. It is possible that the ability of Pol IV to impede growth of the dnaN159 strain results from its checkpoint-like function. However, in this case, it seems likely that elevated levels of Pol II should have a similar effect, particularly since β159 is proficient for stimulating Pol II replication in vitro (28). Since elevated levels of Pol II failed to exert a phenotype in the dnaN159 or rep strain (Tables 2 and 5), the ability of these Pols to impede replication in a wild-type E. coli strain when overproduced to a higher level may result from their ability to switch excessively with Pol III*, effectively disrupting replication. Regardless of the mechanism underlying these phenotypes, the results discussed in this report provide compelling support for the model that Pol IV utilizes a two-step switch to replace a stalled Pol III* (Fig. 1), while Pol II (and the other E. coli Pols) utilizes a different mechanism that is independent of both the clamp rim and a stalled Pol III*. Further work is required to determine whether Pol II and Pol IV displace Pol III* from the clamp in vivo, as well as the relationship between Pol switching and the proposed checkpoint functions of these Pols. Continued use of the approaches described in this study to probe Pol III*-Pol II and Pol III*-Pol IV switching will help to address these and related questions.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant GM066094 (M.D.S.) from the National Institute of General Medical Sciences.
We thank Steven Sandler (University of Massachusetts at Amherst) and Bénédicte Michel (CNRS, Centre de Génétique Moléculaire, France) for E. coli strains, Takehiko Nohmi (National Institute of Health Sciences, Japan) for anti-Pol IV antibodies, Jill Duzen (University at Buffalo) for excellent technical assistance, Ray Kelleher (University at Buffalo) and The University at Buffalo School of Medicine and Biomedical Sciences Confocal Microscopy and Flow Cytometry Facility for assistance with flow cytometry experiments, and the members of our lab for helpful discussions.
Footnotes
Published ahead of print 27 April 2012
REFERENCES
- 1. Atkinson J, Gupta MK, McGlynn P. 2011. Interaction of Rep and DnaB on DNA. Nucleic Acids Res. 39:1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atkinson J, et al. 2011. Localization of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Res. 39:949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baba T, et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008 doi:10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Becherel OJ, Fuchs RP, Wagner J. 2002. Pivotal role of the beta-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair (Amst.) 1:703–708 [DOI] [PubMed] [Google Scholar]
- 5. Bloom LB. 2009. Loading clamps for DNA replication and repair. DNA Repair (Amst.) 8:570–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boubakri H, de Septenville AL, Viguera E, Michel B. 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 29:145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bunting KA, Roe SM, Pearl LH. 2003. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J. 22:5883–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burgers PM, Kornberg A, Sakakibara Y. 1981. The dnaN gene codes for the beta subunit of DNA polymerase III holoenzyme of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 78:5391–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. 2001. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. U. S. A. 98:11627–11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dohrmann PR, McHenry CS. 2005. A bipartite polymerase-processivity factor interaction: only the internal beta binding site of the alpha subunit is required for processive replication by the DNA polymerase III holoenzyme. J. Mol. Biol. 350:228–239 [DOI] [PubMed] [Google Scholar]
- 12. Downey CD, McHenry CS. 2010. Chaperoning of a replicative polymerase onto a newly assembled DNA-bound sliding clamp by the clamp loader. Mol. Cell 37:481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duzen JM, Walker GC, Sutton MD. 2004. Identification of specific amino acid residues in the E. coli beta processivity clamp involved in interactions with DNA polymerase III, UmuD and UmuD′. DNA Repair (Amst.) 3:301–312 [DOI] [PubMed] [Google Scholar]
- 14. Fernandez De Henestrosa AR, et al. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 35:1560–1572 [DOI] [PubMed] [Google Scholar]
- 15. Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Friedberg EC, et al. 2006. DNA repair and mutagenesis, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 17. Furukohri A, Goodman MF, Maki H. 2008. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J. Biol. Chem. 283:11260–11269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guy CP, et al. 2009. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol. Cell 36:654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heltzel JM, Maul RW, Scouten Ponticelli SK, Sutton MD. 2009. A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc. Natl. Acad. Sci. U. S. A. 106:12664–12669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heltzel JMH, et al. 2009. Sliding clamp-DNA interactions are required for viability and contribute to DNA polymerase management in Escherichia coli. J. Mol. Biol. 387:74–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho C, Kulaeva OI, Levine AS, Woodgate R. 1993. A rapid method for cloning mutagenic DNA repair genes: isolation of umu-complementing genes from multidrug resistance plasmids R391, R446b, and R471a. J. Bacteriol. 175:5411–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Indiani C, Langston LD, Yurieva O, Goodman MF, O'Donnell M. 2009. Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc. Natl. Acad. Sci. U. S. A. 106:6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. 2001. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol. Genet. Genomics 266:207–215 [DOI] [PubMed] [Google Scholar]
- 24. Little JW, Edmiston SH, Pacelli LZ, Mount DW. 1980. Cleavage of the Escherichia coli lexA protein by the recA protease. Proc. Natl. Acad. Sci. U. S. A. 77:3225–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lopez de Saro FJ, Georgescu RE, Goodman MF, O'Donnell M. 2003. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 22:6408–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopez de Saro FJ, O'Donnell M. 2001. Interaction of the beta sliding clamp with MutS, ligase, and DNA polymerase I. Proc. Natl. Acad. Sci. U. S. A. 98:8376–8380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lusetti SL, et al. 2006. The RecF protein antagonizes RecX function via direct interaction. Mol. Cell 21:41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maul RW, Ponticelli SK, Duzen JM, Sutton MD. 2007. Differential binding of Escherichia coli DNA polymerases to the beta-sliding clamp. Mol. Microbiol. 65:811–827 [DOI] [PubMed] [Google Scholar]
- 29. Maul RW, Sanders LH, Lim JB, Benitez R, Sutton MD. 2007. Role of Escherichia coli DNA polymerase I in conferring viability upon the dnaN159 mutant strain. J. Bacteriol. 189:4688–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maul RW, Sutton MD. 2005. Roles of the Escherichia coli RecA protein and the global SOS response in effecting DNA polymerase selection in vivo. J. Bacteriol. 187:7607–7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCool JD, et al. 2004. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol. Microbiol. 53:1343–1357 [DOI] [PubMed] [Google Scholar]
- 32. McHenry CS. 2003. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 49:1157–1165 [DOI] [PubMed] [Google Scholar]
- 33. McHenry CS. 2011. DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 80:403–436 [DOI] [PubMed] [Google Scholar]
- 34. Miller JH. 1999. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Moldovan GL, Pfander B, Jentsch S. 2007. PCNA, the maestro of the replication fork. Cell 129:665–679 [DOI] [PubMed] [Google Scholar]
- 36. Ohmori H, et al. 2001. The Y-family of DNA polymerases. Mol. Cell 8:7–8 [DOI] [PubMed] [Google Scholar]
- 37. Pritchard AE, Dallmann HG, Glover BP, McHenry CS. 2000. A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of delta-delta′ with DnaX(4) forms DnaX(3)-delta-delta′. EMBO J. 19:6536–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reuven NB, Arad G, Maor-Shoshani A, Livneh Z. 1999. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 274:31763–31766 [DOI] [PubMed] [Google Scholar]
- 39. Scouten Ponticelli SK, Duzen JM, Sutton MD. 2009. Contributions of the individual hydrophobic clefts of the Escherichia coli beta sliding clamp to clamp loading, DNA replication and clamp recycling. Nucleic Acids Res. 37:2796–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sommer S, Boudsocq F, Devoret R, Bailone A. 1998. Specific RecA amino acid changes affect RecA-UmuD′C interaction. Mol. Microbiol. 28:281–291 [DOI] [PubMed] [Google Scholar]
- 41. Sutton MD. 2010. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim. Biophys. Acta 1804:1167–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sutton MD. 2004. The Escherichia coli dnaN159 mutant displays altered DNA polymerase usage and chronic SOS induction. J. Bacteriol. 186:6738–6748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sutton MD, Duzen JM. 2006. Specific amino acid residues in the beta sliding clamp establish a DNA polymerase usage hierarchy in Escherichia coli. DNA Repair (Amst.) 5:312–323 [DOI] [PubMed] [Google Scholar]
- 44. Sutton MD, Duzen JM, Maul RW. 2005. Mutant forms of the Escherichia coli beta sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol. Microbiol. 55:1751–1766 [DOI] [PubMed] [Google Scholar]
- 45. Sutton MD, Duzen JM, Scouten Ponticelli SK. 2010. A single hydrophobic cleft in the Escherichia coli processivity clamp is sufficient to support cell viability and DNA damage-induced mutagenesis in vivo. BMC Mol. Biol. 11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sutton MD, Opperman T, Walker GC. 1999. The Escherichia coli SOS mutagenesis proteins UmuD and UmuD′ interact physically with the replicative DNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 96:12373–12378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tang M, et al. 1998. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc. Natl. Acad. Sci. U. S. A. 95:9755–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uchida K, et al. 2008. Overproduction of Escherichia coli DNA polymerase DinB (Pol IV) inhibits replication fork progression and is lethal. Mol. Microbiol. 70:608–622 [DOI] [PubMed] [Google Scholar]
- 49. Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP. 2000. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 1:484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang F, Yang W. 2009. Structural insight into translesion synthesis by DNA Pol II. Cell 139:1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199 [PubMed] [Google Scholar]