

Abstract
Moorella thermoacetica ferments glucose to three acetic acids. In the oxidative part of the fermentation, the hexose is converted to 2 acetic acids and 2 CO2 molecules with the formation of 2 NADH and 2 reduced ferredoxin (Fdred2−) molecules. In the reductive part, 2 CO2 molecules are reduced to acetic acid, consuming the 8 reducing equivalents generated in the oxidative part. An open question is how the two parts are electronically connected, since two of the four oxidoreductases involved in acetogenesis from CO2 are NADP specific rather than NAD specific. We report here that the 2 NADPH molecules required for CO2 reduction to acetic acid are generated by the reduction of 2 NADP+ molecules with 1 NADH and 1 Fdred2− catalyzed by the electron-bifurcating NADH-dependent reduced ferredoxin:NADP+ oxidoreductase (NfnAB). The cytoplasmic iron-sulfur flavoprotein was heterologously produced in Escherichia coli, purified, and characterized. The purified enzyme was composed of 30-kDa (NfnA) and 50-kDa (NfnB) subunits in a 1-to-1 stoichiometry. NfnA harbors a [2Fe2S] cluster and flavin adenine dinucleotide (FAD), and NfnB harbors two [4Fe4S] clusters and FAD. M. thermoacetica contains a second electron-bifurcating enzyme. Cell extracts catalyzed the coupled reduction of NAD+ and Fd with 2 H2 molecules. The specific activity of this cytoplasmic enzyme was 3-fold higher in H2-CO2-grown cells than in glucose-grown cells. The function of this electron-bifurcating hydrogenase is not yet clear, since H2-CO2-grown cells additionally contain high specific activities of an NADP+-dependent hydrogenase that catalyzes the reduction of NADP+ with H2. This activity is hardly detectable in glucose-grown cells.
INTRODUCTION
Flavin-based electron bifurcation is a recently discovered mechanism of coupling endergonic to exergonic redox reactions in the cytoplasm of anaerobic bacteria and archaea. Via this novel mechanism, e.g., the endergonic reduction of ferredoxin (Fd) (two [4Fe4S] clusters, each with an Eo′ of <−400 mV) with NADH (Eo′ = −320 mV) is coupled to the exergonic reduction of crotonyl coenzyme A (CoA) to butyryl-CoA (Eo′ = −10 mV) with NADH (Eo′ = −320 mV) in butyric acid-forming clostridia (reaction 1). The coupled reaction is catalyzed by the cytoplasmic butyryl-CoA dehydrogenase/electron transfer flavoprotein complex (Bcd/EtfAB) containing only flavin adenine dinucleotides (FADs) as prosthetic groups (28, 37). The mechanism of coupling was proposed previously to proceed similarly to that of ubiquinone-based electron bifurcation in the cytochrome bc1 complex of the respiratory system (9, 29, 46). One of the main differences between flavin- and ubiquinone-based electron bifurcations appears to be that flavin-based electron bifurcation is associated with a cytoplasmic enzyme complex and operates at redox potentials around that of free flavins (−200 mV), whereas ubiquinone-based electron bifurcation is associated with a membrane enzyme complex and operates at around the redox potential of ubiquinone (+110 mV).
 |
Another example of flavin-based electron bifurcation is the coupling of ferredoxin reduction with H2 (Eo′ = −414 mV) to the reduction of the heterodisulfide CoM-S-S-CoB (Eo′ = −140 mV) with H2 in methanogenic archaea growing on H2 and CO2 (reaction 2) (ΔGo′ calculated by using an Eo′ of −400 mV for ferredoxin from Clostridium pasteurianum [65]). This coupled reaction is catalyzed by the cytoplasmic [NiFe]-hydrogenase/heterodisulfide reductase complex MvhADG/HdrABC, with the subunit HdrA harboring FAD (32, 69). Two other examples are the formation of 2 H2 molecules from NADH and Fdred2− catalyzed by the cytoplasmic [FeFe]-hydrogenase complex HydABC in Thermotoga maritima (reaction 3) (61) and the reduction of 2 NADP+ molecules with reduced ferredoxin and NADH catalyzed by the NADH-dependent reduced ferredoxin:NADP+ oxidoreductase complex NfnAB in Clostridium kluyveri (reaction 4) (72):
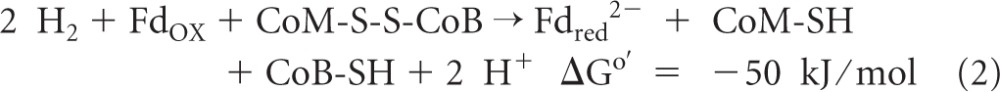 |
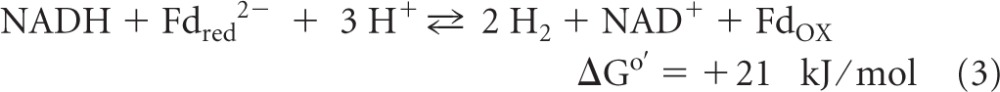 |
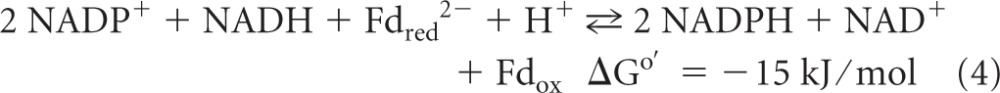 |
Reaction 3 was demonstrated previously by Schut and Adams only in the direction of H2 formation (61). In this direction, the enzyme catalyzing the reaction is actually confurcating rather than bifurcating. However, the flavin mononucleotide (FMN)-dependent enzyme should also catalyze the NAD+-dependent reduction of ferredoxin with H2, because it is this reaction that is exergonic under standard conditions. Indeed, it was recently shown that in Acetobacterium woodii, which can grow on H2 and CO2 and which contains only one hydrogenase of the T. maritima electron-bifurcating type, the hydrogenase catalyzes an NAD+-dependent reduction of ferredoxin with H2 (55). Also, NfnAB, catalyzing reaction 4, is confurcating in the forward direction and bifurcating in the backward direction.
For an understanding of the thermodynamics of reactions 1 to 4, it is important to know that in living cells, the redox potentials of the ox/red couples are quite different from those under standard conditions. Thus, the E′ of the H+/H2 couple in methanogenic environments is −320 mV (69), whereas the Eo′ is −414 mV; the E′ of the Fdox/Fdred2− couple in most anaerobes is −500 mV (69), whereas the Eo′ is −400 mV (redox potential of ferredoxin from C. pasteurianum) (65); the E′ of the NAD+/NADH couple in Escherichia coli is −280 mV, whereas the Eo′ is −320 mV; and the E′ of the NADP+/NADPH couple in E. coli is −370 mV, whereas the Eo′ is −320 mV. The reason for the almost 100-mV difference in the E′ of the NAD+/NADH couple and that of the NADP+/NADPH couple is that in living cells, the NAD+/NADH ratio is near 30/1, whereas the NADP+/NADPH ratio is near 1/50 (5). One indication for this is that in many aerobes and facultative organisms, transhydrogenation from NADH to NADP+ is driven by the proton motive force catalyzed via a membrane-associated transhydrogenase (59).
In this communication, evidence is presented to show that in Moorella thermoacetica (formerly Clostridium thermoaceticum), transhydrogenation from NADH to NADP+ proceeds via reaction 4 (Fig. 1). M. thermoacetica is a Gram-positive, endospore-forming, and cytochrome-containing anaerobic bacterium that can grow on glucose, forming three acetic acids (reactions 5 to 7) (22), or on H2 and CO2 (reactions 8 and 9) (12, 34) with a growth temperature optimum near 55°C (19):
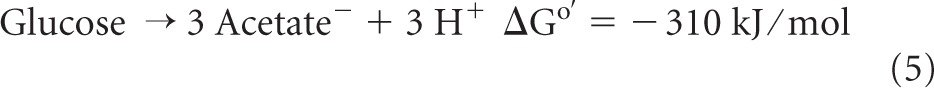 |
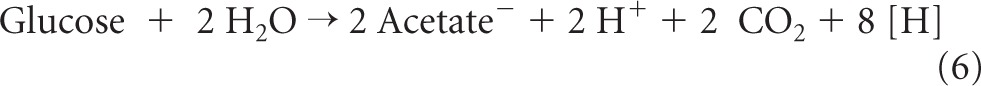 |
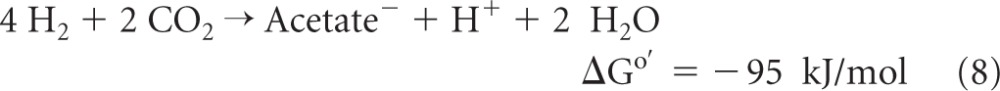 |
Fig 1.

Scheme of the metabolism of M. thermoacetica fermenting glucose to 3 acetic acids or 4 H2 plus 2 CO2 molecules to 1 acetic acid. H4F, tetrahydrofolate. The two cytoplasmic enzymes identified in this work as being electron bifurcating are highlighted by a blue disc and a green disc, respectively. Also described for the first time is the presence of an NADP+-reducing hydrogenase activity in H2-CO2-grown cells. All the enzymes involved in the oxidative and reductive part of the fermentation are cytoplasmic. The physiological electron donor of methylene-H4F reductase has not yet been identified. It appears not to be NADH, NADPH, or reduced ferredoxin. The genome of M. thermoacetica encodes an Ech-type [NiFe]-hydrogenase (see Table S1 in the supplemental material), an F1Fo-ATP synthase (14), and a sodium ion-dependent pyrophosphatase (43), which are membrane associated and which may be involved in energy conservation, especially when the organism grows on H2-CO2 as the energy source. Under this condition, net ATP is not formed via substrate-level phosphorylation.
In the oxidative part of the fermentation, glucose is oxidized to 2 acetic acids and 2 CO2 molecules, generating 8 reducing equivalents ([H]) (reaction 6), and in the reductive part, the two CO2 molecules are reduced to acetic acid, consuming the 8 reducing equivalents (reaction 7). Details of the two metabolic pathways (reactions 5 to 9) are shown in Fig. 1, which includes the findings made in this work.
Most of the enzymes involved in CO2 reduction to acetic acid (Wood-Ljungdahl pathway) (reaction 7) have been well studied (38, 39, 58). What we know of the mechanism of the total synthesis of acetate from CO2 is derived mostly from studies of the enzymes in M. thermoacetica, whose genome has been sequenced (54). However, a few important questions have remained open. One of these questions is how electron transport between reactions 6 (glycolysis) and 7 (Wood-Ljungdahl pathway) proceeds. The problem is that whereas the oxidoreductases involved in catalyzing glucose oxidation to acetic acid and CO2 are either NAD specific or ferredoxin specific, at least two of the oxidoreductases involved in catalyzing CO2 reduction to acetic acid are NADP specific (Fig. 1). Another question is what are the electron acceptors in reaction 9 catalyzed by the hydrogenases of M. thermoacetica, which have until now been assayed only with viologen dyes (12, 18, 33).
An answer to these questions is our finding that M. thermoacetica contains an electron-bifurcating enzyme catalyzing the coupled reduction of NADP+ with NADH and reduced ferredoxin. The specific activity of this enzyme in cell extracts was sufficient to account for its participation in energy metabolism. The nfnAB genes, putatively encoding the electron-bifurcating transhydrogenase, were expressed in E. coli, and the recombinant protein complex was found to catalyze the coupled reduction of 2 NADP+ molecules with 1 NADH and 1 reduced ferredoxin. Furthermore, cell extracts of H2-CO2-grown M. thermoacetica cells were shown to exhibit two different hydrogenase activities, namely, an electron-bifurcating hydrogenase catalyzing the NAD+-dependent reduction of ferredoxin with H2 and a second hydrogenase catalyzing the reduction of NADP+ with H2 (Fig. 1).
MATERIALS AND METHODS
Biochemicals.
Methyl-tetrahydrofolate (methyl-H4F) was purchased from Schircks Laboratories (Jona, Switzerland). Methylene-tetrahydrofolate (methylene-H4F) was prepared from tetrahydrofolate (H4F) and formaldehyde by a spontaneous reaction. H4F, glucose-6-phosphate, glucose-6-phosphate dehydrogenase from baker's yeast, glyceraldehyde-3-phosphate, benzyl viologen, and methyl viologen were obtained from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany). Ferredoxin (Fd) (60) and ferredoxin-dependent [FeFe]-hydrogenase (37) were purified from C. pasteurianum DSM 525, which was grown on glucose-ammonium medium (31).
Growth of M. thermoacetica DSM 521.
M. thermoacetica DSM 521 was grown at 55°C on glucose in 5-liter glass bottles containing 3 liters of medium with 100% CO2 as the gas phase at 1.3 × 105 Pa as described previously (53), with minor modifications. The medium contained 100 mmol glucose (component A); 40 mmol K2HPO4, 40 mmol KH2PO4, and 100 mmol NaHCO3 (components B); and 5 g tryptone, 5 g yeast extract, 7.6 mmol (NH4)2SO4, 0.5 mg resazurin, 4.4 mmol sodium thioglycolate, 1 mmol MgSO4, 0.3 μmol KAl(SO4)2, 0.1 mmol Fe(NH4)2(SO4)2, 0.1 mmol Co(NO3)2, 0.2 mmol CaCl2, 20 μmol NiSO4, 6.8 mmol NaCl, 5 μmol ZnCl2, 25 μmol MnCl2, 2.4 μmol H3BO3, 7 μmol Na2SeO3, 3 μmol Na2WO4, 25 μmol Na2MoO4, and 10 μmol EDTA · Na2 (components C) per liter. Components A, B, and C were dissolved separately in water (450 ml in a 1-liter bottle, 600 ml in a 1-liter bottle, and 1,740 ml in a 5-liter bottle, respectively, for 3 liters of medium), and the solutions were stirred in a type B vinyl anaerobic chamber (Coy, Grass Lake, MI) under 95% N2–5% H2 for 3 h. The bottles were then closed with rubber stoppers and autoclaved. After cooling, solutions A, B, and C were combined under N2 and subsequently sparged with 100% CO2 (the gases were passed through a filter for sterilization). Before inoculation, the 2,790-ml medium was supplemented with anaerobic filter-sterilized solutions D (6 ml), E (30 ml), and F (30 ml), followed by the injection via a sterile syringe of about 6 ml 25% HCl, resulting in a final pH of 6.8. Solution D contained 1 g each of d-biotin, cyanocobalamin, flavin mononucleotide, folic acid, nicotinic acid, pantothenic acid, p-aminobenzoic acid, and thiamine pyrophosphate per liter; solution E contained 36 g of Na2S · H2O per liter; and solution F contained 36 g of cysteine-HCl per liter. After inoculation with 300 ml of a preculture, the culture was allowed to grow for about 2 days to an optical density at 600 nm (OD660) of about 5 and then harvested by centrifugation under N2. The cells were stored at −80°C until use.
M. thermoacetica cells were grown at 55°C on H2-CO2 in 2-liter glass bottles containing 500 ml of medium and 80% H2–20% CO2 as the gas phase at 1.3 × 105 Pa. The medium was the same as that described above for growth on glucose, with exceptions that it did not contain glucose and that the concentrations of tryptone and yeast extract were only 2 g per liter. After inoculation with 50 ml of a glucose-grown preculture, the culture was allowed to grow for about 1 week until it no longer took up H2, as measured manometrically.
Under the cultivation conditions described above, M. thermoacetica grew on glucose with a minimum doubling time (td) of about 6 h (360 min) and a calculated growth yield (Y) of about 40 g cells (dry mass)/mol glucose. From these growth parameters, the specific rate of glucose fermentation (the specific rate of acetate formation from 2 CO2 molecules) in μmol per min per mg protein was calculated, assuming that the average amount of cells fermenting glucose during the doubling period (t2 − t1) is 1.5 times that at t1 and that the cells consist of 50% protein (factor 2) (62): specific rate = 2/(1.5 × td × Yglucose) = 2/(1.5 × 360 min × 40 g/mol) ≈ 0.1 μmol glucose per min per mg protein. For H2-CO2-grown cells (doubling time of about 24 h), the estimate for the specific rate of H2 oxidation was in the same order.
Preparation of cell extracts.
The glucose- or H2-CO2-grown cells (about 1 g of cell pellet) were suspended in 3 ml anaerobic 50 mM Tris-HCl (pH 7.5) containing 2 mM dithiothreitol and 10 μM FAD and were subsequently disrupted by the passage of the suspension through a French pressure cell 3 times at 120 MPa. Cell debris and membranes were removed by centrifugation at 115,000 × g at 4°C for 30 min. The supernatant was used for enzyme assays.
Enzyme activity assays.
Except where indicated, enzyme activity assays were performed at 45°C with 1.5-ml anaerobic cuvettes closed with rubber stoppers and filled with 0.8-ml reaction mixtures and 0.7 ml N2, H2, or CO at 1.2 × 105 Pa. After the start of the reactions with the enzyme, NAD(P) reduction or oxidation was monitored spectrophotometrically at 340 nm (ϵ = 6.2 mM−1 cm−1), ferredoxin reduction or oxidation was monitored at 430 nm (ϵΔox-red ≈ 13.1 mM−1 cm−1), benzyl viologen reduction or oxidation was monitored at 555 nm (ϵ = 12 mM−1 cm−1), and methyl viologen reduction was monitored at 600 nm (ϵ = 13 mM−1 cm−1). One unit was defined as the transfer of 2 μmol electrons per min. The ferredoxin used in the assays was from C. pasteurianum. The protein content was determined with a reagent from Bio-Rad, using bovine serum albumin as the standard.
Glyceraldehyde-3-phosphate dehydrogenase.
For glyceraldehyde-3-phosphate dehydrogenase, the reaction mixture contained 50 mM Tricine-NaOH (pH 8.5); 10 mM 2-mercaptoethanol; 10 mM sodium phosphate; 1 mM glyceraldehyde-3-phosphate; and 1 mM NAD+, 1 mM NADP+, or about 30 μM ferredoxin. The gas phase was 100% N2.
Pyruvate:ferredoxin oxidoreductase.
For pyruvate:ferredoxin oxidoreductase, the reaction mixture contained 100 mM Tris-HCl (pH 7.5); 2 mM dithiothreitol; 10 mM pyruvate; 1 mM thiamine pyrophosphate; 1 mM CoA; and about 30 μM ferredoxin, 1 mM NAD+, or 1 mM NADP+. The gas phase was 100% N2.
Formate dehydrogenase.
For formate dehydrogenase, the assay used was modified from a method described previously (40). The reaction mixture contained 100 mM Tris-HCl (pH 7.5); 2 mM dithiothreitol; 20 mM formate; and 1 mM NADP+, 1 mM NAD+, or about 30 μM ferredoxin as the electron acceptor. The gas phase was 100% N2.
Methylene-H4F dehydrogenase.
For methylene-H4F dehydrogenase, the assay used was modified from methods described previously (41, 50, 74). The reaction mixture contained 100 mM morpholinepropanesulfonic acid (MOPS)-KOH (pH 6.5); 50 mM 2-mercaptoethanol; 0.4 mM tetrahydrofolate; 10 mM formaldehyde; and 0.5 mM NADP+, 0.5 mM NAD+, or about 30 μM ferredoxin. The gas phase was 100% N2. After the start of the reaction with the enzyme, the reduction of NAD(P)+ and the formation of methenyl-H4F were monitored at 350 nm using an ϵ of 30.5 mM−1 cm−1 [5.6 mM−1 cm−1 for NAD(P)H plus 24.9 mM−1 cm−1 for methenyl-H4F]. The activity was measured at pH 6.5 rather than at pH 7.5 (as in the case of most of the other activity assays) because the dehydrogenation of methylene-H4F is thermodynamically favored at higher proton concentrations.
Methylene-H4F reductase.
For methylene-H4F reductase, the reaction mixture contained 100 mM Tris-HCl (pH 7.5), 20 mM ascorbate, and 10 μM FAD. For methylene-H4F reduction with reduced benzyl viologen, the reaction mixture was supplemented with 0.75 mM tetrahydrofolate, 10 mM formaldehyde, and 1 mM benzyl viologen. The gas phase was 100% N2. Before the start of the reaction with the enzyme, benzyl viologen was reduced to an ΔA555 of ∼1.3 with sodium dithionite. For benzyl viologen reduction with methyl-H4F, the assay was modified from a method described previously (11). The reaction mixture was supplemented with 20 mM benzyl viologen and 1 mM methyl-H4F. The gas phase was 100% N2. Before the start of the reaction with the enzyme, benzyl viologen was reduced to an ΔA555 of 0.3 with sodium dithionite. For methylene-H4F reduction with NAD(P)H or reduced ferredoxin, the reaction mixture was supplemented with 0.5 mM tetrahydrofolate; 10 mM formaldehyde; and 0.1 mM NADH, 0.1 mM NADPH, or 30 μM reduced ferredoxin (reduced to 80% with dithionite). The gas phase was 100% N2. NAD(P)H oxidation was monitored at 350 nm (ϵ = 5.6 mM−1 cm−1), and ferredoxin oxidation was monitored at 430 nm.
CO dehydrogenase.
For CO dehydrogenase, the reaction mixture contained 100 mM Tris-HCl (pH 7.5), 2 mM dithiothreitol, and about 30 μM ferredoxin, 1 mM NAD+, or 1 mM NADP+. The gas phase was 100% CO.
Hydrogenase.
For hydrogenase, the reaction mixture contained 100 mM potassium phosphate (pH 7.5) or Tris-HCl (pH 7.5), 10 μM FMN, and 2 mM dithiothreitol. The gas phase was 100% H2. For NAD+-dependent ferredoxin reduction with H2, the reaction mixture was supplemented with ferredoxin (about 30 μM) and 1 mM NAD+ (or as indicated). For methyl viologen, NAD+, or NADP+ reduction with H2, the reaction mixture was supplemented with 10 mM methyl viologen, 1 mM NADP+, or 1 mM NAD+.
NADH-dependent Fdred2−:NADP+ oxidoreductase.
For NADH-dependent Fdred2−:NADP+ oxidoreductase (NfnAB), all reaction mixtures contained 100 mM MOPS-KOH (pH 7.0), 10 mM 2-mercaptoethanol, and 10 μM FAD as basal ingredients. For NADH-dependent NADP+ reduction with reduced ferredoxin, the reaction mixture was supplemented with 2 mM NADP+, 0.5 mM NADH (or as indicated), about 30 μM ferredoxin, and 1 unit of hydrogenase from C. pasteurianum (Fdred2−-regenerating system). H2 was the gas phase. NADP+ reduction was monitored at 380 nm (ϵ = 1.2 mM−1 cm−1). For NAD+-dependent ferredoxin reduction with NADPH, the reaction mixture was supplemented with 0.5 mM NADP+, 40 mM glucose-6-phosphate, and 2 units of glucose-6-phosphate dehydrogenase (NADPH-regenerating system); 10 mM NAD+ (or as indicated); and about 30 μM ferredoxin. N2 was the gas phase. For Fdox-dependent NAD+ reduction with NADPH, the reaction mixture was supplemented with 0.5 mM NADP+, 40 mM glucose-6-phosphate, and 2 units glucose-6-phosphate dehydrogenase (NADPH-regenerating system); 10 mM NAD+; about 10 μM ferredoxin; and 1 unit of hydrogenase from C. pasteurianum (Fdox-regenerating system). N2 was the gas phase. NAD+ reduction was monitored at 380 nm (ϵ = 1.2 mM−1 cm−1).
Heterologous expression of nfnAB in E. coli.
The genes were cloned and expressed together with a tagged 3′ Strep cassette with a pET-51b(+) Ek/LIC vector kit (Merck, Darmstadt, Germany). M. thermoacetica genomic DNA was extracted and purified with a QIAamp DNA minikit (Qiagen, Hilden, Germany). The genes were amplified by PCR with KOD Hot Start DNA polymerase (Merck, Darmstadt, Germany) using M. thermoacetica genomic DNA as a template. The following primers were used: 5′-GACGACGACAAGATGTACCGCATTGTCCGCAAAGA-3′ (forward primer) (the inserted sequence specific for ligation-independent cloning is underlined) and 5′-GAGGAGAAGCCCGGTTATTTTTCCCGTAGATAGGCGTCAA-3′ (reverse primer) (the inserted sequence specific for ligation-independent cloning is underlined). After purification with a MinElute PCR purification kit (Qiagen, Hilden, Germany), the blunt PCR product was treated with T4 DNA polymerase in the presence of dATP to generate specific vector-compatible overhangs. It was then annealed into the linear pET-51b(+) Ek/LIC vector (Merck, Darmstadt, Germany) with single-stranded complementary overhangs, which was subsequently transformed into NovaBlue GigaSingles competent cells. After amplification, the construct was verified by DNA sequencing. It was then transformed into E. coli C41(DE3) cells for expression, which already harbored pCodonPlus and pRKISC (47). pRKISC contains the E. coli isc locus (66) and has been successfully used for the production of iron-sulfur proteins (27, 47).
For expression, the cells were aerobically grown in 2 liters of Terrific broth (TB) medium at 37°C with a high stirring speed (750 rpm). Before inoculation, the medium was supplemented with carbenicillin (50 mg liter−1), chloramphenicol (25 mg liter−1), and tetracycline (10 mg liter−1) to maintain the plasmids and with cysteine (0.12 g liter−1), ferrous sulfate (0.1 g liter−1), ferric citrate (0.1 g liter−1), and ferric ammonium citrate (0.1 g liter−1) for the enhancement of iron-sulfur cluster synthesis. When an OD600 of about 0.6 was reached, the stirring speed was lowered to 250 rpm. Concomitantly, the culture was supplemented with IPTG (isopropyl β-d-thiogalactoside) (0.5 mM) to induce gene expression. After 20 h at 37°C, stirring was stopped, and the culture was left for another 20 h at 4°C before harvesting by centrifugation. The recombinant E. coli cells were washed with anaerobic 100 mM Tris-HCl (pH 7.5) and stored at −80°C under N2 until use.
Purification of Strep-tagged proteins.
The steps for the purification of Strep-tagged proteins were performed at room temperature in a Coy type B vinyl anaerobic chamber filled with 95% N2–5% H2 and containing a palladium catalyst for O2 reduction with H2. The E. coli cells were resuspended in 100 mM Tris-HCl (pH 7.5) containing 2 mM dithiothreitol and 10 μM FAD and disrupted by sonication (10 times at 32 W for 2 min each time). Cell debris was removed by centrifugation at 115,000 × g at 4°C for 30 min. The supernatant was then heated at 55°C for 30 min. After the denatured protein was removed by centrifugation at 2,000 × g at 22°C for 10 min, the supernatant was applied onto a 5-ml Strep-Tactin Superflow column (IBA, Göttingen, Germany), which was equilibrated with 20 ml buffer W (100 mM Tris-HCl [pH 7.5], 150 mM NaCl, 2 mM dithiothreitol, and 10 μM FAD). The column was then washed with 35 ml buffer W. The recombinant protein was eluted with 15 ml buffer E (buffer W containing 2.5 mM desthiobiotin). The fractions containing the target protein were pooled and concentrated by ultrafiltration with an Amicon filter (50-kDa cutoff; Millipore). The purified protein was washed with 100 mM Tris-HCl (pH 7.5) containing 2 mM dithiothreitol and 10 μM FAD and then stored at −20°C under N2 until use.
FeS cluster reconstitution and measurement of iron content.
In order to improve the activity of the purified proteins, their FeS clusters were reconstituted in vitro (72). The reaction mixture contained 100 μmol Tris-HCl (pH 7.5), 8 μmol dithiothreitol, 10 nmol FAD, 2 mg enzyme, 2 μmol cysteine, and 1.5 μmol FeSO4 per ml. The reaction was done at room temperature for 1 h under strictly anoxic conditions. After centrifugation at 52,000 × g at 4°C for 30 min, the supernatant was ultrafiltrated by using Amicon filters (50-kDa cutoff; Millipore) and washed with 5 volumes of 50 mM Tris-HCl (pH 7.5) containing 2 mM dithiothreitol and 10 μM FAD.
The iron content of the enzyme was measured calorimetrically with 3-(2-pyridyl)-5,6-bis(5-sulfo-2-furyl)-1,2,4-triazinedisodium trihydrate (Ferene; Sigma) with Mohr's salt as a standard according to a method described previously (16).
Nucleotide sequence accession number.
The M. thermoacetica genome GenBank accession number is CP000232.1.
RESULTS
In cell extracts of M. thermoacetica, we found three new activities, which could be involved in the energy metabolism of this organism (Fig. 1): the coupled reduction of NADP+ with reduced ferredoxin and NADH, the coupled reduction of ferredoxin and NAD+ with H2, and the reduction of NADP+ with H2. To find out whether the three enzyme activities can have a function in energy metabolism, we compared their cell extract specific activities with those of known oxidoreductases (Table 1 and see Table 3) and with the specific rate of substrate consumption during the growth of the organisms on glucose and on H2-CO2, which was estimated to be about 0.1 μmol per min per mg protein (see Materials and Methods).
Table 1.
Oxidoreductase activities in cell extracts of glucose-grown M. thermoaceticad
| Oxidoreductase | Substrate | Sp act (U/mg) determined in: |
|
|---|---|---|---|
| This study | Literature (reference[s]) | ||
| Glyceraldehyde-3-phosphate dehydrogenase | GAP + NAD+ | 4.3 | Not reported; approximate Km for NAD+ reported to be 0.1 mM (67) |
| GAP + NADP+ | <0.01 | ||
| GAP + Fdox | <0.01 | ||
| Pyruvate:Fd oxidoreductase | Pyr + CoA + Fdox | 0.6 | Measured only with MV (21, 24, 71) |
| Pyr + CoA + NAD+ | <0.01 | ||
| Pyr + CoA + NADP+ | <0.01 | ||
| Formate dehydrogenase | Formate + NADP+ | 0.7 | 0.4–2.3 (40, 67, 75) |
| Formate + NAD+ | <0.01 | ||
| Formate + Fdox | <0.01 | ||
| Methylene-H4F dehydrogenase | Methylene-H4F + NADP+ | 1.4 | 1.7–2.2 (1, 41, 50) |
| Methylene-H4F + NAD+ | <0.01 | ||
| Methylene-H4F + Fdox | <0.01 | ||
| Methylene-H4F reductase | Methyl-H4F + BVox | 0.5 | 0.6 (51) |
| Methylene-H4F + BVred | 0.7 | ||
| Methylene-H4F + NADPH | <0.01 | ||
| Methylene-H4F + NADH | <0.01 | ||
| Methylene-H4F + Fdred | <0.01 | ||
| CO dehydrogenase | CO + Fdox | 0.9 | Measured only with MV (17, 20, 56) |
| CO + NAD+ | <0.01 | ||
| CO + NADP+ | <0.01 | ||
| NADH-dependent Fdred:NADP+ oxidoreductase | Fdreda + NADH + NADP+ | 0.8 | 0.15 (67) |
| Fdreda + NADP+ | 0.03 | ||
| NADH + NADP+ | <0.01 | ||
| NADPHb + NAD+ + Fdox | 0.4 | ||
| NADPHb + Fdox | <0.01 | ||
| NADPHb + Fdoxc + NAD+ | 0.3 | ||
| NADPHb + NAD+ | <0.01 | ||
| Hydrogenase | H2 + MVox | 0.4 | 1.2–8 (18, 33, 42) |
| H2 + NAD+ + Fdox | 0.1 | ||
| H2 + Fdox | <0.01 | ||
| H2 + NAD+ | <0.01 | ||
| H2 + NADP+ + Fdox | <0.01 | ||
| H2 + NADP+ | <0.01 | ||
Fdred-regenerating system (Fd, hydrogenase from C. pasteurianum, and 100% H2, keeping the ferredoxin about 50% reduced).
NADPH-regenerating system (NADP+, glucose-6-phosphate dehydrogenase, and glucose-6-phosphate).
Fdox-regenerating system (Fd, hydrogenase from C. pasteurianum, and 100% N2).
GAP, glyceraldehyde-3-phosphate; Fd, ferredoxin from C. pasteurianum; Pyr, pyruvate; H4F, tetrahydrofolate; BV, benzyl viologen; MV, methyl viologen. The assays were performed at 45°C. The specific activities were higher by a factor of 2 at 55°C, the growth temperature optimum of M. thermoacetica. For assay conditions, see Materials and Methods. The reduction or oxidation of the substrate listed last was monitored spectrophotometrically to measure the activity.
Table 3.
Activities of NADH-dependent reduced ferredoxin:NADP+ oxidoreductase (NfnAB) and hydrogenase in cell extracts of H2-CO2-grown M. thermoaceticad
| Oxidoreductase | Substrate | Sp act (U/mg) |
|---|---|---|
| NADH-dependent Fdred:NADP+ oxidoreductase | Fdreda + NADH + NADP+ | 2.4 |
| Fdreda + NADP+ | 1.7 | |
| NADH + NADP+ | <0.01 | |
| NADPHb + NAD+ + Fdox | 0.5 | |
| NADPHb + Fdox | <0.01 | |
| NADPHb + Fdoxc + NAD+ | 0.4 | |
| NADPHb + NAD+ | <0.01 | |
| Hydrogenase | H2 + MVox | 55.2 |
| H2 + NAD+ + Fdox | 0.3 | |
| H2 + Fdox | 0.02 | |
| H2 + NAD+ | <0.01 | |
| H2 + NADP+ + Fdox | 1.8 | |
| H2 + NADP+ | 1.7 |
Fdred-regenerating system (ferredoxin, hydrogenase from C. pasteurianum, and 100% H2, keeping the ferredoxin about 50% reduced).
NADPH-regenerating system (NADP+, glucose-6-phosphate dehydrogenase, and glucose-6-phosphate).
Fdox-regenerating system (Fd, hydrogenase from C. pasteurianum, and 100% N2).
For abbreviations and assay details, see Table 1. The reduction of the substrate listed last was monitored spectrophotometrically to measure the activity.
Specific activities of catabolic oxidoreductases in cell extracts of glucose-grown M. thermoacetica.
The specific activities of most of the oxidoreductases involved in the glucose fermentation of M. thermoacetica (Fig. 1) were described many years ago for cell extracts (Table 1). Surprisingly, however, data on the specific activity of glyceraldehyde-3-phosphate dehydrogenase are not found. For pyruvate:ferredoxin oxidoreductase and CO dehydrogenase, only specific activities with viologen dyes rather than with their physiological electron acceptor ferredoxin have been reported. We found that with ferredoxin, the specific activities of these two enzymes in cell extracts were significantly lower than those with viologen dyes, but they were still much higher than the estimated specific rate of glucose fermentation by growing M. thermoacetica cells (0.1 μmol per min per mg). This is also true for the specific activities of all the other oxidoreductases involved (Table 1).
The cell extracts catalyzed the reduction of NAD+ with glyceraldehyde-3-phosphate with a specific activity of 4.3 U/mg and the CoA-dependent reduction of ferredoxin with pyruvate with a specific activity of 0.6 U/mg (Table 1). NADP+ or ferredoxin was not reduced with glyceraldehyde-3-phosphate, and NAD+ or NADP+ was not reduced with pyruvate. The results confirm that in the oxidative part of the fermentation, two oxidoreductases are involved, namely, NAD-specific glyceraldehyde-3-phosphate dehydrogenase (67) and pyruvate:ferredoxin oxidoreductase (21, 24). Consistently, the genome of M. thermoacetica harbors a gene for glyceraldehyde-3-phosphate dehydrogenase and a gene for the monomeric pyruvate:ferredoxin oxidoreductase that has been purified (21, 24, 71) (see Table S1 in the supplemental material).
The cell extracts catalyzed the reduction of NADP+ with formate with a specific activity of 0.7 U/mg, the reduction of NADP+ with methylene-H4F (1.4 U/mg), the reduction of methylene-H4F with reduced benzyl viologen (0.7 U/mg), and the reduction of ferredoxin with CO (0.9 U/mg) (Table 1). Neither formate nor methylene-H4F reduced NAD+ or ferredoxin, and methylene-H4F was not reduced by NADH, NADPH, or reduced ferredoxin. These results confirm that in the reductive part of the fermentation, a NADP-specific formate dehydrogenase (75), a NADP-specific methylene-H4F dehydrogenase (41, 50), a methylene-H4F reductase with an as-yet-unknown physiological electron donor (51), and a ferredoxin-specific CO dehydrogenase (20, 36, 56) are involved. The genes encoding these four oxidoreductases are shown in Fig. 2 and in Table S2 and Fig. S1 in the supplemental material.
Fig 2.

The M. thermoacetica genomic region around the nfnAB genes encoding NADH-dependent reduced ferredoxin:NADP+ oxidoreductase (NfnAB). There is a 10-bp overlap between nfnA and nfnB. Downstream of the nfnAB gene, separated by a 22-bp intergenic region, is a gene (Moth_1516) encoding a bifunctional NADP+-dependent methylene-H4F dehydrogenase/methenyl-H4F cyclohydrolase.
For the connection of the oxidative part and the reductive part, we found an NADH-dependent reduced ferredoxin:NADP+ oxidoreductase activity. Cell extracts of M. thermoacetica catalyzed the reduction of NADP+ with reduced ferredoxin only in the presence of NADH and the reduction of ferredoxin with NADPH only in the presence of NAD+ (Table 1). This activity was first observed 40 years ago (67), but its dependence on NADH and NAD+, respectively, was wrongly interpreted to indicate that the ferredoxin:NADP+ oxidoreductase was allosterically regulated by the NAD+/NADH ratio (70). From recent work with C. kluyveri, it is much more likely that the activities are catalyzed by the electron-bifurcating NfnAB complex (72). Indeed, the nfnAB genes of M. thermoacetica show high sequence identity to NfnAB of C. kluyveri at the protein level, 54% in the case of nfnA and 62% in the case of nfnB. Above that, the nfnAB genes were found in the genome of M. thermoacetica adjacent to the gene for bifunctional NADP+-dependent methylene-H4F dehydrogenase and methenyl-H4F cyclohydrolase, consistent with a function of NfnAB in energy metabolism (Fig. 2).
NADH-dependent Fdred2−:NADP+ oxidoreductase (NfnAB).
In a previous study, it was shown that the nfnAB genes (both tagged with a His6 cassette) from C. kluyveri can be functionally expressed in E. coli BL21(DE3) cells. We therefore expressed the nfnAB genes from M. thermoacetica (Fig. 2) in E. coli under similar conditions but with some notable differences: the nfnA gene was tagged with a Strep cassette, and the nfnB gene was not tagged and was cloned together with the nfnA gene. For expression, a special host, E. coli C41(DE3) harboring pCodonPlus and pRKISC (47), was used, which has been successfully employed for the production of iron-sulfur proteins (27).
The recombinant NfnAB protein was found in the E. coli supernatant centrifuged at 115,000 × g, which catalyzed the NAD+-dependent reduction of ferredoxin with NADPH at a specific activity of only 0.02 U/mg, indicating that the expression level of the nfnAB genes was not high. After heat treatment at 55°C for 30 min, the purification of NfnAB was achieved by chromatography on a Strep tag affinity column eluted with 2.5 mM desthiobiotin. The elution of the activity was in a broad peak. The peak fractions contained NfnA and NfnB in an almost 1-to-1 ratio, as judged from a scan of the Coomassie brilliant blue-stained SDS-PAGE gels that showed only two bands, with apparent molecular masses of 30 and 50 kDa, respectively, in agreement with the calculated values.
The specific activity of purified NfnAB for the reduction of ferredoxin with NADPH in the presence of NAD+ was 6.8 U/mg. The activity increased up to 13.8 U/mg (Table 2) when, before its measurement, the purified enzyme complex was anaerobically incubated in the presence of Fe2+ (1.5 mM), cysteine (2 mM), dithiothreitol (8 mM), and FAD (10 μM) at pH 7.5 for 1 h at room temperature. After the removal of nonbound Fe2+ by ultrafiltration, the preparations contained up to 8.7 Fe per heterodimer. Based on the protein sequence derived from nfnAB, the presence of two [4Fe4S] clusters and one [2Fe2S] cluster is predicted (Fig. 2).
Table 2.
Reactions catalyzed by recombinant NADH-dependent reduced ferredoxin:NADP+ oxidoreductase (NfnAB) from M. thermoaceticad
| Substrate | Sp act (U/mg) |
|---|---|
| Fdreda + NADH + NADP+ | 22.4 |
| Fdreda + NADP+ | < 0.01 |
| NADH + NADP+ | < 0.01 |
| NADH + Fdoxb + NADP+ | < 0.01 |
| NADPHc + NAD+ + Fdox | 13.8 |
| NADPHc + Fdox | 0.2 |
| NADPHc + Fdoxb + NAD+ | 8.4 |
| NADPHc + Fdreda + NAD+ | 5.6 |
| NADPHc + NAD+ | < 0.01 |
Fdred-regenerating system (Fd, hydrogenase from C. pasteurianum, and 100% H2, keeping the ferredoxin about 50% reduced).
Fdox-regenerating system (Fd, hydrogenase from C. pasteurianum, and 100% N2).
NADPH-regenerating system (NADP+, glucose-6-phosphate dehydrogenase, and glucose-6-phosphate).
Fd, ferredoxin from C. pasteurianum. The activities were determined after in vitro iron-sulfur cluster reconstitution in NfnAB. For assay conditions, see Materials and Methods. The reduction of the substrate listed last was monitored spectrophotometrically to measure the activity.
The purified enzyme complex catalyzed the NADH-dependent reduction of NADP+ with reduced ferredoxin (22.4 U/mg), the NAD+-dependent reduction of ferredoxin with NADPH (13.8 U/mg), and the ferredoxin-dependent reduction of NAD+ with NADPH (8.4 U/mg) (Table 2). Per mol NAD+ added, 0.83 mol ferredoxin was reduced in the presence of NADPH (Fig. 3A), and per mol NADH added, 1.7 mol of NADP+ was reduced in the presence of reduced ferredoxin (Fig. 3B). The results indicate that the enzyme preparation catalyzed the reduction of NADP+ with reduced ferredoxin and NADH in a 2-to-1-to-1 stoichiometry, in agreement with reaction 4. The purified complex also catalyzed the reduction of viologen dyes with NADPH. This reaction was stimulated with but not dependent on NAD+ (72). With respect to its molecular properties (subunit molecular masses and N-terminal amino acid sequences), NfnAB is not identical to the NADPH-dependent artificial mediator acceptor pyridine nucleotide oxidoreductase (AMAPOR), isolated and characterized more than 10 years ago for C. thermoaceticum (26).
Fig 3.

Stoichiometry of the reaction catalyzed by recombinant NADH-dependent reduced ferredoxin:NADP+ oxidoreductase (NfnAB) from M. thermoacetica. (A) NAD+-dependent ferredoxin reduction with NADPH. (B) NADH-dependent NADP+ reduction with reduced ferredoxin. For assay conditions, see Materials and Methods. All the reactions were started by the addition of ∼0.2 units of NfnAB.
The energetic coupling of the endergonic reduction of ferredoxin from C. pasteurianum with NADPH to the exergonic reduction of NAD+ with NADPH was demonstrated by showing that the reduction of ferredoxin with NADPH proceeds beyond the equilibrium concentrations expected for an uncoupled reaction (Fig. 4). Ferredoxin was reduced to 70% (E′ = −410 mV) in the presence of NAD+ when the NADPH/NADP+ ratio was 10 to 1 (E′ = −350 mV). Without energetic coupling, a reduction of only less than 10% of the ferredoxin is thermodynamically possible. When, as a control, C. pasteurianum hydrogenase catalyzed the reduction of ferredoxin (Eo′ = −400 mV) with H2 at 105 Pa at pH 7 (Eo′ = −414 mV), the ferredoxin was found to be reduced to about 50%, in agreement with the thermodynamic prediction. The monomeric [FeFe]-hydrogenase from C. pasteurianum differs from the heterotrimeric [FeFe]-hydrogenase from T. maritima in not being electron bifurcating.
Fig 4.

Ferredoxin reduction with NADPH catalyzed by recombinant NfnAB from M. thermoacetica. As controls, ferredoxin (Fd) reduction with 100% H2 catalyzed by the monomeric ferredoxin-dependent hydrogenase from C. pasteurianum (see the text) was used, as was the spontaneous reduction of Fd with sodium dithionite. The reactions were performed with 1.5-ml anaerobic cuvettes filled with 0.8 ml 100 mM MOPS-KOH (pH 7.0) containing 20 μM Fd and, where indicated, hydrogenase (∼0.15 unit), NfnAB (∼0.1 unit), or 0.5 mM sodium dithionite. In the case of NAD+-dependent Fd reduction with NADPH, the gas phase was 100% N2 at 1.2 × 105 Pa. In the case of Fd reduction with H2, the gas phase was 100% H2 at 1.2 × 105 Pa. The temperature was 45°C. The reactions were started by the addition of enzyme or dithionite. With dithionite, 100% of Fd was reduced, and with H2 plus hydrogenase, about 50% of Fd was reduced.
Hydrogenases.
When M. thermoacetica grows on glucose, some hydrogen is formed in the case where the CO2 concentration is low, and therefore, the cells are expected to exhibit hydrogenase activity (33). Indeed, cell extracts catalyzed the reduction of methyl viologen with H2 at a specific activity of 0.4 μmol min−1 mg−1 (see also reference 18). The genome of M. thermoacetica harbors genes for a membrane-associated [NiFe]-hydrogenase of the Ech type and two gene clusters for cytoplasmic heteromeric [FeFe]-hydrogenase but not an isolated gene for a monomeric [FeFe]-hydrogenase of the C. pasteurianum type (see Table S3 in the supplemental material). One of the two gene clusters for the heteromeric [FeFe]-hydrogenases shows sequence similarity to the electron-bifurcating NAD+- and ferredoxin-dependent [FeFe]-hydrogenase (HydABC) from T. maritima (61), and the other one shows sequence similarity to the NADP+-reducing [FeFe]-hydrogenase (HndABCD) from Desulfovibrio fructosovorans (44). Both [FeFe]-hydrogenases also show sequence similarity to each other. We therefore looked for these activities and found that cell extracts of glucose-grown cells catalyzed the reduction of ferredoxin only in the presence of NAD+ (Table 1). Per mol NAD+ added, 0.83 mol of ferredoxin from C. pasteurianum was reduced (Fig. 5). The cell extracts did not catalyze the reduction of NADP+ with H2 at significant specific rates in either the absence or the presence of ferredoxin (Table 1).
Fig 5.

NAD+-dependent ferredoxin reduction with 100% H2 catalyzed by cell extracts of glucose-grown M. thermoacetica. (A) Time course of ferredoxin reduction in the absence and presence of NAD+ (1 mM). (B) Amount of ferredoxin reduced versus the amount of NAD+ added. NADP+ could not substitute for NAD+ in promoting ferredoxin reduction with H2. For assay conditions, see Materials and Methods. The reactions were started with ∼0.2 mg cell extracts with a specific activity of 0.1 unit per mg.
M. thermoacetica can grow on H2 and CO2 albeit with doubling times of only about 24 h and, in our hands, for only a few generations when starting from a glucose-grown culture. In cell extracts of such grown cells, the specific activity of NAD+- and ferredoxin-dependent hydrogenase was 3-fold higher than that in glucose-grown cells, indicating an induction of the enzyme upon the transfer of the organism from glucose to H2 and CO2 (Table 3).
Interestingly, cell extracts of H2-CO2-grown cells also catalyzed the reduction of NADP+ with H2, and the reduction was not dependent on ferredoxin (Table 3). The specific activity was 1.7 U/mg and, thus, more than 100 times higher than that in glucose-grown cells (Table 1).
DISCUSSION
We have shown that M. thermoacetica cells grown on glucose or on H2-CO2 contain an electron-bifurcating NfnAB complex catalyzing the coupled reduction of NADP+ with reduced ferredoxin and NADH (reaction 4) (Tables 1 to 3). The specific activities of NfnAB were almost the same in glucose- and H2-CO2-grown cells (Tables 1 and 3). Cell extracts of glucose-grown cells also catalyzed the coupled reduction of ferredoxin and NAD+ with H2 (reaction 3) (Table 1) albeit at only one-third of the specific activity of that of H2-CO2-grown cells. We have additionally found that during the growth of M. thermoacetica cells on H2-CO2, a non-electron-bifurcating NADP+-reducing hydrogenase is induced (Table 3). The possible function of these enzymes in the energy metabolism of M. thermoacetica is shown in Fig. 1. The scheme disregards that during the growth of M. thermoacetica on glucose, some H2 is formed (33).
During the growth of M. thermoacetica on glucose, two NAD+ and two ferredoxin molecules are reduced in the oxidative part of the energy metabolism (Fig. 1). One NADH and one reduced ferredoxin are used to regenerate, via the NfnAB complex, two NADPH molecules required for the reduction of CO2 to formate and of methenyl-H4F to methylene-H4F. This leaves one reduced ferredoxin for the reduction of CO2 to CO in the reductive part of the energy metabolism and one NADH. How this NADH is reoxidized and how methylene-H4F is reduced to methyl-H4F are not yet known.
Based on the genome sequence, M. thermoacetica contains a cytoplasmic methylene-H4F reductase that differs from that of E. coli by putatively being composed of 6 different subunits rather than of 1 (30, 64) (see Fig. S1 in the supplemental material). One of the six subunits is a flavoprotein with low sequence similarity to the NAD-specific methylene-H4F reductase of E. coli. The gene for the flavoprotein is neighbored by one for a conserved iron-sulfur zinc protein. The other four show sequence similarity to the subunits MvhD, HdrA, HdrB, and HdrC of the electron-bifurcating MvhADG/HdrABC complex from methanogenic archaea (see Fig. S1 in the supplemental material). The redox potential, Eo′, of the methylene-H4F/methyl-H4F couple has been determined to be −200 mV (73) and is thus much more positive than any of the other electron donors or acceptors involved in the energy metabolism of M. thermoacetica. These are indications that the methylene-H4F reductase of M. thermoacetica could be an electron-bifurcating enzyme.
During the growth of M. thermoacetica on H2 and CO2, the NADPH required for the reduction of CO2 to formate and for the reduction of methenyl-H4F to methylene-H4F is probably provided mainly via the NADP+-reducing hydrogenase (Fig. 1). The reduced ferredoxin for the reduction of CO2 to CO is probably regenerated via the electron-bifurcating hydrogenase. Again, how the NADH is reoxidized and how methylene-H4F is reduced are not yet known. The scheme also does not explain how net ATP is synthesized during growth on H2 and CO2. The ATP generated via substrate-level phosphorylation in the acetokinase reaction is required for the formyl-H4F synthetase reaction. A previously proposed additional energy-coupling site is the reduction of methylene-H4F to methenyl-H4F (68).
M. thermoacetica contains cytochromes and menaquinone (13, 25). Where could these electron carriers come into play? A hypothesis is that the cytochrome b found in the membrane of M. thermoacetica is involved in methylene-H4F reduction to methyl-H4F (3, 19, 23, 58). The involvement in this step appears, however, not to be obligatory, since membranes of cells grown on oxalate and nitrate apparently reducing CO2 to acetic acid under these conditions did not contain cytochrome b (63). In this respect, it is of interest that M. thermoacetica is aerotolerant and can rapidly reduce O2 when present at low levels (15). The electron transport chain to O2 has been shown to involve cytochrome b and other cytochromes (15), so it could well be that the cytochromes in M. thermoacetica are involved in O2 reduction rather than in CO2 reduction to acetic acid. The redox potential of menaquinone (−75 mV) is too positive to be involved in acetogenesis from CO2.
Besides acetogens that contain cytochromes and menaquinone (13, 25, 54), there are also acetogens that do not contain these membrane-associated electron carriers. There are many other important differences between these two groups of acetogens, as revealed by a comparison of M. thermoacetica with the non-cytochrome-containing A. woodii (55). Thus, A. woodii contains a membrane-associated and energy-conserving RnfABCDEFG complex that catalyzes the reduction of NAD+ with Fdred2− (6–8). The genes for this enzyme are not found in M. thermoacetica (54); vice versa, from the genome sequence, it is predicted that M. thermoacetica contains a membrane-associated energy-conserving Ech-type complex (54) that catalyzes the reduction of protons to H2 with Fdred2− (see Table S3 in the supplemental material). The genes for this enzyme are not found in A. woodii (55). The membrane-associated Ech-type [NiFe]-hydrogenase from M. thermoacetica is most similar to the hydrogenase 4 complex in E. coli (2, 4). Other differences are that A. woodii does not contain NfnAB, that the methylene-H4F dehydrogenase is NAD rather than NADP specific (57), and that methylene-H4F reductase is active with NADH (10), while the enzyme from M. thermoacetica is not. In this respect, it is of interest that in the genome of A. woodii, genes for homologues of MvhD, HdrA, HdrB, and HdrC, which are part of the M. thermoacetica methylene-H4F reductase gene cluster (see Fig. S1 in the supplemental material), are not found (55). In A. woodii, the methylene-H4F reductase gene cluster is composed of only three genes, predicted to encode a flavoprotein with sequence similarity to the methylene-H4 reductase of E. coli, the conserved iron-sulfur zinc protein also found in M. thermoacetica, and a second flavoprotein with sequence similarity to RnfC not found in M. thermoacetica. As for M. thermoacetica, the reduction of methylene-H4F to methyl-H4F in A. woodii and other non-cytochrome-containing acetogens was proposed previously to be the missing site of energy conservation (35, 55).
Acetogenesis from CO2 is the fourth type of energy metabolism dependent on the newly discovered mechanism of energy conservation via flavin-based electron bifurcation. The other three energy metabolisms employing this mechanism are methanogenesis from H2 and CO2 (32, 69), butyric acid-forming fermentations (37, 72), and glucose fermentation to 2 acetic acid, 2 CO2, and 4 H2 molecules (61), and this is most probably only the beginning of a longer list. Proposals for the involvement of electron-bifurcating enzymes in the energy metabolisms of other anaerobic microorganisms were reported recently (45, 48, 49, 52).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Max Planck Society and the Fonds der Chemischen Industrie.
We thank Yasuhiro Takahashi from Osaka University for providing E. coli C41(DE3) harboring pCodonPlus and pRKISC.
Footnotes
Published ahead of print 11 May 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Andreesen JR, Schaupp A, Neurauter C, Brown A, Ljungdahl LG. 1973. Fermentation of glucose, fructose, and xylose by Clostridium thermoaceticum: effect of metals on growth yield, enzymes, and the synthesis of acetate from CO2. J. Bacteriol. 114:743–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrews SC, et al. 1997. A 12-cistron Escherichia coli operon (hyf) encoding a putative proton-translocating formate hydrogenlyase system. Microbiology 143:3633–3647 [DOI] [PubMed] [Google Scholar]
- 3. Arendsen AF, Soliman MQ, Ragsdale SW. 1999. Nitrate-dependent regulation of acetate biosynthesis and nitrate respiration by Clostridium thermoaceticum. J. Bacteriol. 181:1489–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bagramyan K, Mnatsakanyan N, Poladian A, Vassilian A, Trchounian A. 2002. The roles of hydrogenases 3 and 4, and the F0F1-ATPase, in H2 production by Escherichia coli at alkaline and acidic pH. FEBS Lett. 516:172–178 [DOI] [PubMed] [Google Scholar]
- 5. Bennett BD, et al. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5:593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biegel E, Müller V. 2010. Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U. S. A. 107:18138–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Biegel E, Schmidt S, Gonzalez JM, Müller V. 2011. Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 68:613–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biegel E, Schmidt S, Müller V. 2009. Genetic, immunological and biochemical evidence for a Rnf complex in the acetogen Acetobacterium woodii. Environ. Microbiol. 11:1438–1443 [DOI] [PubMed] [Google Scholar]
- 9. Brandt U. 1996. Bifurcated ubihydroquinone oxidation in the cytochrome bc1 complex by proton-gated charge transfer. FEBS Lett. 387:1–6 [DOI] [PubMed] [Google Scholar]
- 10. Buchenau B. 2001. Are there tetrahydrofolate specific enzymes in methanogenic archaea and tetrahydromethanopterin specific enzymes in acetogenic bacteria? Diploma thesis. Philipps University, Marburg, Germany [Google Scholar]
- 11. Clark JE, Ljungdahl LG. 1984. Purification and properties of 5,10-methylenetetrahydrofolate reductase, an iron-sulfur flavoprotein from Clostridium formicoaceticum. J. Biol. Chem. 259:10845–10849 [PubMed] [Google Scholar]
- 12. Daniel SL, Hsu T, Dean SI, Drake HL. 1990. Characterization of the H2- and CO-dependent chemolithotrophic potentials of the acetogens Clostridium thermoaceticum and Acetogenium kivui. J. Bacteriol. 172:4464–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das A, Hugenholtz J, Van Halbeek H, Ljungdahl LG. 1989. Structure and function of a menaquinone involved in electron transport in membranes of Clostridium thermoautotrophicum and Clostridium thermoaceticum. J. Bacteriol. 171:5823–5829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Das A, Ljungdahl LG. 1997. Composition and primary structure of the F1F0 ATP synthase from the obligately anaerobic bacterium Clostridium thermoaceticum. J. Bacteriol. 179:3746–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Das A, Silaghi-Dumitrescu R, Ljungdahl LG, Kurtz DM., Jr 2005. Cytochrome bd oxidase, oxidative stress, and dioxygen tolerance of the strictly anaerobic bacterium Moorella thermoacetica. J. Bacteriol. 187:2020–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dickert S, Pierik AJ, Linder D, Buckel W. 2000. The involvement of coenzyme A esters in the dehydration of (R)-phenyllactate to (E)-cinnamate by Clostridium sporogenes. Eur. J. Biochem. 267:3874–3884 [DOI] [PubMed] [Google Scholar]
- 17. Diekert GB, Thauer RK. 1978. Carbon monoxide oxidation by Clostridium thermoaceticum and Clostridium formicoaceticum. J. Bacteriol. 136:597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Drake HL. 1982. Demonstration of hydrogenase in extracts of the homoacetate-fermenting bacterium Clostridium thermoaceticum. J. Bacteriol. 150:702–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drake HL, Daniel SL. 2004. Physiology of the thermophilic acetogen Moorella thermoacetica. Res. Microbiol. 155:869–883 [DOI] [PubMed] [Google Scholar]
- 20. Drake HL, Hu SI, Wood HG. 1980. Purification of carbon monoxide dehydrogenase, a nickel enzyme from Clostridium thermoaceticum. J. Biol. Chem. 255:7174–7180 [PubMed] [Google Scholar]
- 21. Drake HL, Hu SI, Wood HG. 1981. Purification of five components from Clostridium thermoaceticum which catalyze synthesis of acetate from pyruvate and methyltetrahydrofolate. Properties of phosphotransacetylase. J. Biol. Chem. 256:11137–11144 [PubMed] [Google Scholar]
- 22. Fontaine FE, Peterson WH, McCoy E, Johnson MJ, Ritter GJ. 1942. A new type of glucose fermentation by Clostridium thermoaceticum n. sp. J. Bacteriol. 43:701–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fröstl JM, Seifritz C, Drake HL. 1996. Effect of nitrate on the autotrophic metabolism of the acetogens Clostridium thermoautotrophicum and Clostridium thermoaceticum. J. Bacteriol. 178:4597–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furdui C, Ragsdale SW. 2002. The roles of coenzyme A in the pyruvate:ferredoxin oxidoreductase reaction mechanism: rate enhancement of electron transfer from a radical intermediate to an iron-sulfur cluster. Biochemistry 41:9921–9937 [DOI] [PubMed] [Google Scholar]
- 25. Gottwald M, Andreesen JR, LeGall J, Ljungdahl LG. 1975. Presence of cytochrome and menaquinone in Clostridium formicoaceticum and Clostridium thermoaceticum. J. Bacteriol. 122:325–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gunther H, Walter K, Kohler P, Simon H. 2000. On a new artificial mediator accepting NADP(H) oxidoreductase from Clostridium thermoaceticum. J. Biotechnol. 83:253–267 [DOI] [PubMed] [Google Scholar]
- 27. Hamann N, et al. 2007. A cysteine-rich CCG domain contains a novel [4Fe-4S] cluster binding motif as deduced from studies with subunit B of heterodisulfide reductase from Methanothermobacter marburgensis. Biochemistry 46:12875–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Herrmann G, Jayamani E, Mai G, Buckel W. 2008. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190:784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hunte C, Palsdottir H, Trumpower BL. 2003. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 545:39–46 [DOI] [PubMed] [Google Scholar]
- 30. Igari S, et al. 2011. Properties and crystal structure of methylenetetrahydrofolate reductase from Thermus thermophilus HB8. PLoS One 6:e23716 doi:10.1371/journal.pone.0023716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jungermann K, Thauer RK, Leimenstoll G, Decker K. 1973. Function of reduced pyridine nucleotide-ferredoxin oxidoreductases in saccharolytic clostridia. Biochim. Biophys. Acta 305:268–280 [DOI] [PubMed] [Google Scholar]
- 32. Kaster AK, Moll J, Parey K, Thauer RK. 2011. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. U. S. A. 108:2981–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kellum R, Drake HL. 1984. Effects of cultivation gas phase on hydrogenase of the acetogen Clostridium thermoaceticum. J. Bacteriol. 160:466–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kerby R, Zeikus JG. 1983. Growth of Clostridium thermoaceticum on H2/CO2 or CO as energy source. Curr. Microbiol. 8:27–30 [Google Scholar]
- 35. Köpke M, et al. 2010. Clostridium ljungdahlii represents a microbial production platform based on syngas. Proc. Natl. Acad. Sci. U. S. A. 107:13087–13092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kumar M, Lu WP, Liu LF, Ragsdale SW. 1993. Kinetic evidence that carbon-monoxide dehydrogenase catalyzes the oxidation of carbon-monoxide and the synthesis of acetyl-CoA at separate metal centers. J. Am. Chem. Soc. 115:11646–11647 [Google Scholar]
- 37. Li F, et al. 2008. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lindahl PA. 2012. Metal-metal bonds in biology. J. Inorg. Biochem. 106:172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ljungdahl LG. 1986. The autotrophic pathway of acetate synthesis in acetogenic bacteria. Annu. Rev. Microbiol. 40:415–450 [DOI] [PubMed] [Google Scholar]
- 40. Ljungdahl LG, Andreesen JR. 1978. Formate dehydrogenase, a selenium-tungsten enzyme from Clostridium thermoaceticum. Methods Enzymol. 53:360–372 [DOI] [PubMed] [Google Scholar]
- 41. Ljungdahl LG, O'Brien WE, Moore MR, Liu MT. 1980. Methylenetetrahydrofolate dehydrogenase from Clostridium formicoaceticum and methylenetetrahydrofolate dehydrogenase, methenyltetrahydrofolate cyclohydrolase (combined) from Clostridium thermoaceticum. Methods Enzymol. 66:599–609 [DOI] [PubMed] [Google Scholar]
- 42. Lundie LL, Jr, Drake HL. 1984. Development of a minimally defined medium for the acetogen Clostridium thermoaceticum. J. Bacteriol. 159:700–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Malinen AM, Belogurov GA, Baykov AA, Lahti R. 2007. Na+-pyrophosphatase: a novel primary sodium pump. Biochemistry 46:8872–8878 [DOI] [PubMed] [Google Scholar]
- 44. Malki S, et al. 1995. Characterization of an operon encoding an NADP-reducing hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 177:2628–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martin WF. 2012. Hydrogen, metals, bifurcating electrons, and proton gradients: the early evolution of biological energy conservation. FEBS Lett. 586:485–493 [DOI] [PubMed] [Google Scholar]
- 46. Mitchell P. 1976. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 62:327–367 [DOI] [PubMed] [Google Scholar]
- 47. Nakamura M, Saeki K, Takahashi Y. 1999. Hyperproduction of recombinant ferredoxins in Escherichia coli by coexpression of the ORF1-ORF2-iscS-iscU-iscA-hscB-hscA-fdx-ORF3 gene cluster. J. Biochem. 126:10–18 [DOI] [PubMed] [Google Scholar]
- 48. Nitschke W, Russell MJ. 2009. Hydrothermal focusing of chemical and chemiosmotic energy, supported by delivery of catalytic Fe, Ni, Mo/W, Co, S and Se, forced life to emerge. J. Mol. Evol. 69:481–496 [DOI] [PubMed] [Google Scholar]
- 49. Nitschke W, Russell MJ. 2012. Redox bifurcations: mechanisms and importance to life now, and at its origin. A widespread means of energy conversion in biology unfolds. Bioessays 34:106–109 [DOI] [PubMed] [Google Scholar]
- 50. O'Brien WE, Brewer JM, Ljungdahl LG. 1973. Purification and characterization of thermostable 5,10-methylenetetrahydrofolate dehydrogenase from Clostridium thermoaceticum. J. Biol. Chem. 248:403–408 [PubMed] [Google Scholar]
- 51. Park EY, Clark JE, DerVartanian DV, Ljungdahl LG. 1991. 5,10-Methylenetetrahydrofolate reductase:iron-sulfur-zinc flavoproteins of two acetogenic clostridia, p 389–400 In Muller F. (ed), Chemistry and biochemistry of flavoenzymes, vol 1 CRC Press, Boca Raton, FL [Google Scholar]
- 52. Pereira IA, et al. 2011. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pierce E, Becker DF, Ragsdale SW. 2010. Identification and characterization of oxalate oxidoreductase, a novel thiamine pyrophosphate-dependent 2-oxoacid oxidoreductase that enables anaerobic growth on oxalate. J. Biol. Chem. 285:40515–40524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pierce E, et al. 2008. The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ. Microbiol. 10:2550–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Poehlein A, et al. 2012. An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS One 7:e33439 doi:10.1371/journal.pone.0033439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ragsdale SW, Clark JE, Ljungdahl LG, Lundie LL, Drake HL. 1983. Properties of purified carbon monoxide dehydrogenase from Clostridium thermoaceticum, a nickel, iron-sulfur protein. J. Biol. Chem. 258:2364–2369 [PubMed] [Google Scholar]
- 57. Ragsdale SW, Ljungdahl LG. 1984. Purification and properties of NAD-dependent 5,10-methylenetetrahydrolate dehydrogenase from Acetobacterium woodii. J. Biol. Chem. 259:3499–3503 [PubMed] [Google Scholar]
- 58. Ragsdale SW, Pierce E. 2008. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784:1873–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E. 2004. The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J. Biol. Chem. 279:6613–6619 [DOI] [PubMed] [Google Scholar]
- 60. Schönheit P, Wascher C, Thauer RK. 1978. Rapid procedure for purification of ferredoxin from clostridia using polyethyleneimine. FEBS Lett. 89:219–222 [DOI] [PubMed] [Google Scholar]
- 61. Schut GJ, Adams MW. 2009. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191:4451–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schwörer B, Thauer RK. 1991. Activities of formylmethanofuran dehydrogenase, methylenetetrahydromethanopterin dehydrogenase, methylenetetrahydromethanopterin reductase, and heterodisulfide reductase in methanogenic bacteria. Arch. Microbiol. 155:459–465 [Google Scholar]
- 63. Seifritz C, Fröstl JM, Drake HL, Daniel SL. 2002. Influence of nitrate on oxalate- and glyoxylate-dependent growth and acetogenesis by Moorella thermoacetica. Arch. Microbiol. 178:457–464 [DOI] [PubMed] [Google Scholar]
- 64. Sheppard CA, Trimmer EE, Matthews RG. 1999. Purification and properties of NADH-dependent 5,10-methylenetetrahydrofolate reductase (MetF) from Escherichia coli. J. Bacteriol. 181:718–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Smith ET, Bennett DW, Feinberg BA. 1991. Redox properties of 2[4Fe-4S] ferredoxins. Anal. Chim. Acta 251:27–33 [Google Scholar]
- 66. Takahashi Y, Nakamura M. 1999. Functional assignment of the ORF2-iscS-iscU-iscA-hscB-hscA-fdx-ORF3 gene cluster involved in the assembly of Fe-S clusters in Escherichia coli. J. Biochem. 126:917–926 [DOI] [PubMed] [Google Scholar]
- 67. Thauer RK. 1972. CO2-reduction to formate by NADPH. The initial step in the total synthesis of acetate from CO2 in Clostridium thermoaceticum. FEBS Lett. 27:111–115 [DOI] [PubMed] [Google Scholar]
- 68. Thauer RK, Jungermann K, Decker K. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41:100–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6:579–591 [DOI] [PubMed] [Google Scholar]
- 70. Thauer RK, Rupprecht E, Ohrloff C, Jungermann K, Decker K. 1971. Regulation of the reduced nicotinamide adenine dinucleotide phosphate-ferredoxin reductase system in Clostridium kluyveri. J. Biol. Chem. 246:954–959 [PubMed] [Google Scholar]
- 71. Wahl RC, Orme-Johnson WH. 1987. Clostridial pyruvate oxidoreductase and the pyruvate-oxidizing enzyme specific to nitrogen fixation in Klebsiella pneumoniae are similar enzymes. J. Biol. Chem. 262:10489–10496 [PubMed] [Google Scholar]
- 72. Wang S, Huang H, Moll J, Thauer RK. 2010. NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 192:5115–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wohlfarth G, Diekert G. 1991. Thermodynamics of methylenetetrahydrofolate reduction to methyltetrahydrofolate and its implications for the energy metabolism of homoacetogenic bacteria. Arch. Microbiol. 155:378–381 [Google Scholar]
- 74. Wohlfarth G, Geerligs G, Diekert G. 1991. Purification and characterization of NADP+-dependent 5,10-methylenetetrahydrofolate dehydrogenase from Peptostreptococcus productus Marburg. J. Bacteriol. 173:1414–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yamamoto I, Saiki T, Liu SM, Ljungdahl LG. 1983. Purification and properties of NADP-dependent formate dehydrogenase from Clostridium thermoaceticum, a tungsten-selenium-iron protein. J. Biol. Chem. 258:1826–1832 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.