


Abstract
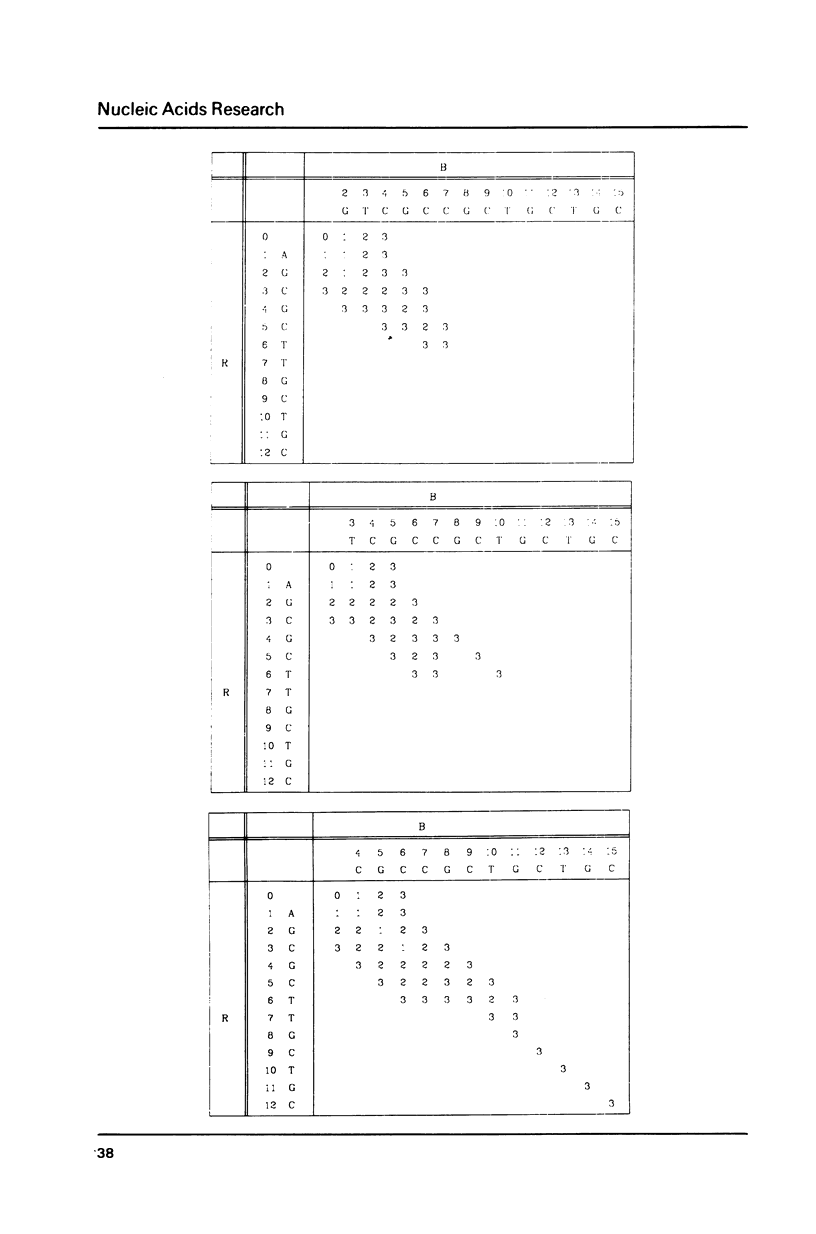
There are a few algorithms designed to solve the problem of the optimal alignment of one sequence, the pattern, of length m, with another, longer sequence the text, of length n. These algorithms allow mismatches, deletions and insertions. Algorithms to date run in O(mn) time. Let us define an integer, k, which is the maximal number of differences allowed. We present a simple algorithm showing that sequences can be optimally aligned in O(k2n) time. For long sequences the gain factor over the currently used algorithms is very large.
Full text
PDF















Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Dumas J. P., Ninio J. Efficient algorithms for folding and comparing nucleic acid sequences. Nucleic Acids Res. 1982 Jan 11;10(1):197–206. doi: 10.1093/nar/10.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickett J. W. Fast optimal alignment. Nucleic Acids Res. 1984 Jan 11;12(1 Pt 1):175–179. doi: 10.1093/nar/12.1part1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goad W. B., Kanehisa M. I. Pattern recognition in nucleic acid sequences. I. A general method for finding local homologies and symmetries. Nucleic Acids Res. 1982 Jan 11;10(1):247–263. doi: 10.1093/nar/10.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn L. J., Queen C. L., Wegman M. N. Computer analysis of nucleic acid regulatory sequences. Proc Natl Acad Sci U S A. 1977 Oct;74(10):4401–4405. doi: 10.1073/pnas.74.10.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizel J. V., Jr, Lenk R. P. Enhanced graphic matrix analysis of nucleic acid and protein sequences. Proc Natl Acad Sci U S A. 1981 Dec;78(12):7665–7669. doi: 10.1073/pnas.78.12.7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman S. B., Wunsch C. D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970 Mar;48(3):443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Nussinov R. An efficient code searching for sequence homology and DNA duplication. J Theor Biol. 1983 Jan 21;100(2):319–328. doi: 10.1016/0022-5193(83)90355-7. [DOI] [PubMed] [Google Scholar]
- Nussinov R., Jacobson A. B. Fast algorithm for predicting the secondary structure of single-stranded RNA. Proc Natl Acad Sci U S A. 1980 Nov;77(11):6309–6313. doi: 10.1073/pnas.77.11.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Matching sequences under deletion-insertion constraints. Proc Natl Acad Sci U S A. 1972 Jan;69(1):4–6. doi: 10.1073/pnas.69.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers P. H. Pattern recognition in genetic sequences. Proc Natl Acad Sci U S A. 1979 Jul;76(7):3041–3041. doi: 10.1073/pnas.76.7.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco I., Jr, Uhlenbeck O. C., Levine M. D. Estimation of secondary structure in ribonucleic acids. Nature. 1971 Apr 9;230(5293):362–367. doi: 10.1038/230362a0. [DOI] [PubMed] [Google Scholar]
- Wilbur W. J., Lipman D. J. Rapid similarity searches of nucleic acid and protein data banks. Proc Natl Acad Sci U S A. 1983 Feb;80(3):726–730. doi: 10.1073/pnas.80.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]