

Abstract
T cell dysfunction in the presence of ongoing antigen exposure is a cardinal feature of chronic viral infections with persistent high viremia, including HIV-1. Although interleukin-10 (IL-10) has been implicated as an important mediator of this T cell dysfunction, the regulation of IL-10 production in chronic HIV-1 infection remains poorly understood. We demonstrated that IL-10 is elevated in the plasma of individuals with chronic HIV-1 infection and that blockade of IL-10 signaling results in a restoration of HIV-1-specific CD4 T cell proliferation, gamma interferon (IFN-γ) secretion, and, to a lesser extent, IL-2 production. Whereas IL-10 blockade leads to restoration of IFN-γ secretion by HIV-1-specific CD4 T cells in all categories of subjects investigated, significant enhancement of IL-2 production and improved proliferation of CD4 T helper cells are restricted to viremic individuals. In peripheral blood mononuclear cells (PBMCs), this IL-10 is produced primarily by CD14+ monocytes, but its production is tightly controlled by regulatory T cells (Tregs), which produce little IL-10 directly. When Tregs are depleted from PBMCs of viremic individuals, the effect of the IL-10 signaling blockade is abolished and IL-10 production by monocytes decreases, while the production of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), increases. The regulation of IL-10 by Tregs appears to be mediated primarily by contact or paracrine-dependent mechanisms which involve IL-27. This work describes a novel mechanism by which regulatory T cells control IL-10 production and contribute to dysfunctional HIV-1-specific CD4 T cell help in chronic HIV-1 infection and provides a unique mechanistic insight into the role of regulatory T cells in immune exhaustion.
INTRODUCTION
T cell responses are critical to the containment of many viral infections. However, some infections, such as HIV-1, hepatitis C virus (HCV), and hepatitis B virus (HBV), can evade these responses and develop long-lasting persistence. A cardinal feature of these chronic viral infections is the development of immune dysfunction, or “exhaustion,” in the presence of ongoing antigen exposure (4, 38). This exhaustion is gradual and results in the hierarchical impairment of T cell effector functions, with an initial loss of proliferative capacity and interleukin-2 (IL-2) production and the late loss of the ability to secrete proinflammatory cytokines, such as gamma interferon (IFN-γ) (46).
A number of inhibitory pathways have been shown to mediate immune exhaustion in murine models of viral infection (reviewed in references 38 and 44). Several of these immunoregulatory networks, including programmed death 1 (PD-1), cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), T cell Ig and mucin-3 (TIM-3), and IL-10, have been shown to be upregulated in the setting of chronic HIV-1 infection in vivo and to mediate a reversible virus-specific immune dysfunction in vitro (reviewed in references 14, 25, 28, and 38). Although these different pathways have been suggested to be important in HIV-1 pathogenesis, only IL-10 has been shown to have an impact on disease progression and long-term outcome in HIV-1 infection (7, 18, 39).
IL-10 was initially identified as an important mediator of T cell exhaustion and a crucial determinant of viral persistence in the lymphocytic choriomeningitis virus (LCMV) mouse model (6, 13). In this system, chronic infection of animals with LCMV can be cleared if the mice are treated with an IL-10 receptor α (IL-10Rα)-blocking antibody or by animals deficient in the IL-10 gene. IL-10 blockade or deficiency can also augment clearance of several other pathogens, including Listeria monocytogenes, Mycobacterium avium, and Trypanosoma cruzi (10, 11, 33). The role of IL-10 in immune regulation, however, is complex, and it appears that this pleiotropic cytokine controls the delicate balance between the robust immune responses necessary for pathogen clearance and excessive inflammation that leads to collateral immune damage.
Studies of humans with naturally occurring polymorphisms in the promoter region of IL-10 show that changes associated with higher levels of IL-10 production are linked with asymptomatic chronic HBV infection (29), whereas changes leading to lower IL-10 levels are found in those with more-rapid progression to hepatic fibrosis (27). Similarly, IL-10 appears to critically regulate multiple aspects of HIV-1 pathogenesis. In chronic, untreated HIV-1 infection, IL-10 is increased in the plasma, with levels that closely correlate with HIV-1 viral load (5, 42). Additionally, our work previously showed that blockade of IL-10 signaling with an IL-10Rα antibody can restore both CD4 and CD8 T cell proliferation and cytokine secretion, suggesting that IL-10 may play a critical role in HIV-1-mediated immune exhaustion. IL-10 also directly modulates HIV-1 replication in peripheral blood mononuclear cell (PBMC) subsets with somewhat contrasting results depending on the model used (1, 40). However, genetic studies suggest that the anti-inflammatory effects of IL-10 may be protective in the setting of HIV-1-driven chronic immune activation. Individuals with genetic polymorphisms in the IL-10 gene that are associated with higher levels of IL-10 production show attenuated progression to AIDS, slower CD4 decline, and longer survival (7, 18, 39). In addition, a recent publication demonstrated that higher levels of IL-10 expression were associated with significantly higher plasma viral loads in acute infection but not in chronic infection (30). Taken together, these studies suggest that IL-10 can play significant and contrasting roles in HIV-1-associated immune dysfunction and disease progression.
Despite the key role of IL-10 in the regulation of inflammation, many of the fundamental details regarding the control of IL-10 production in HIV-1 infection remain poorly understood. Among these is the cellular source of IL-10, which is unclear. IL-10 has been reported to be produced by a variety of cell types, including B cells, T cells, NK cells, and monocytes, although reports of the source in humans have been conflicting, likely in part due to different assessment methods (5, 20, 43). Regulatory T cell (Treg) subsets have been reported to mediate their immune inhibitory function via IL-10, but via an uncertain mechanism (35).
In this study, we examined the role of Tregs in IL-10-mediated inhibition of virus-specific CD4 T cell responses in chronic HIV-1 infection. We show that monocytes are the primary producer of IL-10 in ex vivo PBMCs from individuals with untreated HIV-1 infection and that this production is tightly regulated by Tregs, which themselves produce little IL-10 ex vivo. We thus demonstrate a novel mechanism by which regulatory T cells inhibit HIV-1-specific immune responses by modulating the cytokine secretion pattern of a major cell subset involved in both innate and adaptive immunity, shifting it toward an inhibitory profile with high levels of IL-10 and low levels of TNF-α (IL-10hi/TNF-αlo).
MATERIALS AND METHODS
Ethics statement.
The study was performed according to local ethical regulations following approval by the institutional review board (Partners Human Research Committee) of the Massachusetts General Hospital, Boston, MA, USA. Written informed consent was obtained from all study participants prior to enrollment in the study.
Study subjects.
HIV-1-negative or chronically infected individuals were enrolled at Massachusetts General Hospital in accordance with an institutional review board-approved protocol. Blood was collected from individuals from the following clinical groups: (i) HIV-1 uninfected, (ii) HIV-1 controllers (having HIV-1 viral loads of <2,000 RNA copies/ml in the absence of antiretroviral therapy [ART]), (iii) chronically infected untreated (having HIV-1 viral loads of >2,000 RNA copies/ml; not on ART), and (iv) ART treated (having viral loads of <50 RNA copies/ml on fully suppressive ART).
Measurement of plasma IL-10 levels.
Plasma IL-10 levels were measured by a multiplex immunoassay based on a Luminex platform (Milliplex ultrasensitive assay; Millipore) according to the manufacturer's instructions using a Bio-Plex 200 array system instrument (Bio-Rad Laboratories).
Measurement of ex vivo cytokine secretion.
Freshly isolated PBMCs were cultured in R10 medium (RPMI 1640 medium [Invitrogen] supplemented with 10% human AB serum [Gemini Bio-Products]). In experiments where CD8 cells were depleted, RosetteSep CD8 depletion reagent (StemCell Technologies) was used prior to Ficoll purification of PBMCs. Cells (1 × 106) were cultured in 1 ml of R10 in the presence of an anti-IL-10Rα-blocking antibody (clone 37607; R&D Systems) or an IgG1 isotype control antibody at 10 μg/ml, and cells were left unstimulated or were stimulated with recombinant HIV-1 p24 protein (Protein Sciences) at 10 μg/ml and then cultured for 3 days. Supernatants were collected, and IFN-γ and IL-2 levels were measured using a Luminex immunoassay as described above.
Magnetic bead cell depletion.
Magnetic beads conjugated to CD25, CD14, or CD19 antibodies (Invitrogen) were used to deplete specific cellular subsets from PBMCs. Beads were incubated with PBMCs per the manufacturer's recommendations. Cells were then placed in a magnetic field, and the remaining cells were collected and resuspended in R10. Small portions of undepleted and depleted cells were analyzed by flow cytometry (LSR II flow cytometer; BD Biosciences) to verify that depletion of the targeted cell lineage was >85% efficient and that there was <20% depletion of nontargeted cell types.
Proliferation assay.
Antigen-specific CD4 T cell proliferation was measured using a 5- (and 6-)carboxyfluoresceindiacetate succinimidyl ester (CFSE) assay as previously described (24). Briefly, RosetteSep CD8-depleted PBMCs were labeled with 1.25 μM CFSE dye (Molecular Probes) and then washed. Cells were then depleted of CD25+ cells or mock depleted as described above and then stimulated with phytohemagglutinin (PHA; 5 μg/ml) or recombinant HIV-1 p24 (10 μg/ml; Protein Sciences) or left unstimulated in the presence of an anti-IL-10Rα-blocking antibody (clone 37607; R&D Systems) or an IgG1 isotype control antibody at 10 μg/ml. Cells were incubated for 7 days and then stained with CD3 phycoerythrin (PE)-Cy7, CD8 Alexa Fluor 700, and CD4 allophycocyanin (APC) antibodies (all from BD Biosciences) before acquisition on an LSR II flow cytometer.
Intracellular cytokine staining.
PBMCs were depleted of CD25 cells or mock depleted and then left unstimulated or stimulated with an HIV-1 Gag peptide pool (0.5 μg/ml/peptide) or staphylococcal enterotoxin B (SEB; 0.25 μg/ml; Sigma) and lipopolysaccharide (LPS; 0.25 μg/ml; Sigma). GolgiStop (0.9 μl/ml; BD Biosciences) was added, followed by an additional 14 h of incubation at 37°C/5% CO2. Cells were then stained with viability dye (Molecular Probes) and the following surface antibodies: CD3 Q605, CD4 Q655 (both from Invitrogen), CD8 PE-Cy5, CD25 PE-Cy7, CD19 APC-H7, CD14 Pacific blue, and CD127 APC (all from BD Biosciences). Cells were fixed and permeabilized (fixation medium A/permeabilization medium B; Invitrogen) and then stained with the following intracellular antibodies: IL-10 PE, tumor necrosis factor alpha (TNF-α) Alexa Fluor 700, and IFN-γ fluorescein isothiocyanate (FITC) (BD Biosciences). For staining of dendritic cell (DC) subsets, surface stains CD3/CD14/CD19 Pacific blue, CD11c APC, CD123 PE-Cy5, HLA-DR APC-Cy7, and CD103b FITC were used, followed by intracellular staining with IL-10 PE and TNF-αAlexa Fluor 700 (all from BD Biosciences). Both an isotype control antibody conjugate to PE and a fluorescence minus one (FMO) control were used to determine the negative cutoff for IL-10 staining. Fluorescence information was acquired by flow cytometry, and data were analyzed using FlowJo (Tree Star).
Transwell assay.
PBMCs were stained with CD14 APC-H7, CD11c APC, CD25 PE-Cy7, CD127-PE, CD8/CD19/CD56 Pacific blue (BD Biosciences), and violet viability dye (Invitrogen). CD14+ monocytes and CD25hi CD127lo Tregs were all sorted using a FACSAria fluorescence-activated cell sorter (FACS; BD Biosciences). Cells were then plated alone or in combination in a Transwell plate (Corning) at a ratio of 1:2 (T cell to monocytes). The supernatants were then collected after 3 days of incubation, and IL-10 levels were measured by Luminex as described above.
Measurement of IL-27 and IL-27Rα mRNA.
Cells were lysed in RNA lysis solution (Qiagen), and total RNA was extracted using an RNeasy minikit (Qiagen). cDNA was generated using an oligo(dT) primer and MultiScribe reverse transcriptase enzyme and reagents (Applied Biosystems). IL-27 and IL-27Rα mRNA cDNA was analyzed by quantitative PCR using Brilliant II SYBR green enzyme mix (Stratagene) on an Mx3005P real-time PCR instrument (Stratagene). Results were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) copies for each sample.
RESULTS
Elevated plasma IL-10 levels in chronic, untreated HIV-1 infection and restoration of CD4 T cell function with IL-10 signaling blockade.
We began our studies by characterizing the plasma levels of IL-10 in HIV-1-infected individuals with various clinical outcomes (Fig. 1A). Quantitation of IL-10 protein demonstrated that levels were significantly higher in those with chronic, untreated infection (CU; having a mean HIV-1 viral load of 34,028 RNA copies/ml) than in HIV-1-uninfected controls (P < 0.05). However, subjects who were able to spontaneously maintain low levels of viremia in the absence of treatment, so-called “HIV-1 controllers” (Cont; having <2,000 copies of HIV-1 RNA/ml), had IL-10 levels that were indistinguishable from those of uninfected controls. Treatment with fully suppressive antiretroviral therapy led to various degrees of plasma IL-10 that appeared to be intermediate to those found in HIV-1-uninfected and chronically infected, untreated patients. The results in these cohorts are thus consistent with our previous results showing that IL-10 protein and mRNA levels in the blood correlate with HIV-1 viral load in untreated subjects (5).
Fig 1.

Elevated IL-10 plasma levels in chronic HIV-1 infection and the effects of IL-10Rα blockade. (A) Plasma IL-10 levels were measured in HIV-1-seronegative individuals (HIV-1−; n = 10), in individuals with chronic HIV-1 infection and spontaneous control of viremia (Cont; n = 13), in patients receiving treatment with antiretroviral therapy (ART; n = 10), and in patients with chronic, untreated infection (CU; n = 21). Intergroup comparisons were performed by Kruskal-Wallis test (*, P < 0.05). Solid lines indicate medians; n.s., nonsignificant (P > 0.05). (B) CD8-depleted PBMCs were stimulated with recombinant HIV-1 p24 protein in the presence of an isotype control antibody or an IL-10Rα-blocking antibody. Proliferation was then measured in a 7-day CFSE assay, and net proliferation was calculated by subtracting the background in the absence of antigen. The fold increase in proliferation was calculated by taking the ratio of the net percentage of CD4+ CFSElo cells treated with blocking antibody versus the percentage of those treated with an isotype control antibody (***, P < 0.001). The dashed line indicates no effect (fold increase = 1). (C and D) CD8-depleted PBMCs were stimulated with HIV-1 p24 in the presence of an isotype control or IL-10Rα-blocking antibody, and IFN-γ (C) and IL-2 (D) were measured in supernatants 3 days after stimulation. The increase in IFN-γ production was greater than that of IL-2 following IL-10Rα blockade for all subjects (E), including HIV-1 controllers (F), subjects on ART (G), and those with chronic, untreated infection (H).
We next measured the ability of CD4 T cells to proliferate and to secrete cytokines when stimulated with recombinant HIV-1 p24 protein in the presence of an IL-10Rα-blocking antibody or an isotype control. Blockade of IL-10 signaling resulted in increased HIV-1-specific CD4 T cell proliferation in individuals with chronic, untreated disease, in whom IL-10 levels are the most elevated; in contrast, it had no effect on HIV-1 controllers or those treated with ART (Fig. 1B). Inhibition of the IL-10 pathway led to increased IFN-γ secretion by HIV-1-specific CD4 T cells in all categories of subjects examined (Fig. 1C), with the strongest effect observed in individuals with uncontrolled viremia. The impact on IL-2 levels, however, was more modest (Fig. 1D) and noted only in subjects with ongoing viral replication (P = 0.0165), therefore presenting the same pattern as the proliferation assays (Fig. 1B), consistent with the known role of IL-2 in T cell proliferation. A significant variability in the effect on IL-2 secretion was noted, and differences among groups did not reach statistical significance. Thus, in individuals with uncontrolled HIV-1 infection, IL-10 expression is upregulated and IL-10Rα blockade restores HIV-1-specific CD4 T cell proliferation as well as IFN-γ and IL-2 secretion. We next directly compared the relative impact of IL-10 blockade on IFN-γ and IL-2 secretion in the same individuals (Fig. 1E, F, G, and H). Analyses performed on the entire cohort showed that IL-10 blockade enhanced IFN-γ production significantly more than IL-2 production (Fig. 1E; P < 0.0001). We then looked at each of the three groups of subjects separately and found that this difference was present in each cohort examined (Fig. 1F, G, and H). These data show that blockade of the IL-10 pathway differentially affects HIV-1-specific CD4 T cells, depending on both the disease stage and the function examined.
Depletion of CD25+ regulatory CD4 T cells diminishes the effect of IL-10Rα blockade.
IL-10 has been suggested to play an important role in the mechanism of regulatory T cell immune modulation, although it remains unclear if Tregs secrete IL-10 directly or mediate their effects through interactions with other cell types (4, 35). In order to determine the role of “natural” CD25+ Tregs in IL-10-mediated exhaustion in chronic HIV-1 infection, we used magnetic beads coated with anti-CD25 antibodies to deplete CD25hi regulatory T cells from PBMCs drawn from individuals with chronic untreated disease (HIV-1 viral load of >10,000 RNA copies/ml). Depletion with these beads removed CD25hi FoxP3hi regulatory T cells with high efficiency (Fig. 2A; average of 89% depletion; data not shown). Other T cell subsets were minimally depleted (see Fig. S1 in the supplemental material). When CD25-depleted PBMCs were subsequently stimulated with recombinant HIV-1 p24 protein, CD4 T cell proliferation increased relative to that in undepleted cells (Fig. 2B and C, middle panel; P = 0.0085, Wilcoxon test), consistent with the negative regulatory function of these cells. Once again, blockade with an IL-10Rα antibody in the same subjects also produced an increase in proliferation (Fig. 2C, left panel; P = 0.0001). Interestingly, however, following CD25 depletion, treatment with IL-10Rα antibody had only a minimal effect on HIV-1-specific CD4 T cell proliferation (Fig. 2C, right panel; P = 0.4631, Wilcoxon test), and the median fold increase in proliferation with IL-10 blockade decreased from 1.8 without CD25+ cell depletion to 1.1 with CD25+ cell depletion (Fig. 2D; P = 0.0020, Wilcoxon test). In contrast to the effects observed in subjects with uncontrolled viremia, no significant impact of CD25 depletion and IL-10Rα blockade on HIV-1-specific CD4 T cell-proliferative responses was observed in four individuals with undetectable viral loads or in individuals with low, spontaneous IL-10 production (data not shown). Thus, depletion of CD25hi FoxP3hi regulatory T cells abolished the effect of IL-10Rα blockade on proliferation, suggesting that these cells play an important role in IL-10-mediated T cell exhaustion.
Fig 2.

CD25+ cell depletion abrogates the effects of IL-10Rα blockade. PBMCs from 12 chronically infected, untreated individuals were depleted of CD8 cells and then further depleted with anti-CD25 antibody-coated beads or were mock depleted. (A) A representative FACS plot of the depletion efficiency of CD25+ FoxP3+ cells is shown. (B) CD25-depleted or mock-depleted cells were stimulated with recombinant HIV-1 p24 protein in the presence of IL-10Rα-blocking antibody or an isotype control. CD4 T cell proliferation was then measured using a CFSE assay. An increase in CD4 T cell proliferation was seen with IL-10Rα blockade (C, left panel), and CD25 depletion (C, middle panel), but IL-10Rα blockade had minimal impact when the assays were performed with CD25-depleted PBMCs (C, right panel). (D) The fold increase in CD4 T cell proliferation in response to HIV-1 p24 stimulation with IL-10Rα blockade was decreased following depletion of CD25+ cells (median,1.8× increase without depletion and 1.1× with depletion).
CD14+ monocytes are the major producers of IL-10 in uncontrolled HIV-1 infection.
In order to determine the cell type primarily responsible for spontaneous IL-10 secretion, we performed IL-10 intracellular cytokine staining (ICS) on fresh PBMCs isolated from chronically infected subjects with uncontrolled viremia. The gating strategy is illustrated in Fig. S2 in the supplemental material. The data show that CD14+ monocytes were primarily responsible for spontaneous IL-10 production in unstimulated PBMCs (median percentage of IL-10+ monocytes, 0.87%; range, 0.01 to 9.06%) (Fig. 3A and B; P < 0.0001, Kruskal-Wallis test). Myeloid and plasmacytoid dendritic cell subsets only occasionally demonstrated spontaneous IL-10 secretion. B cell and T cell subsets, including CD4, CD8, and regulatory T cells, produced negligible IL-10 protein in the absence of stimulation. When PBMCs were stimulated with a Gag peptide pool, we identified only infrequent populations of HIV-1-specific IL-10-secreting CD4, CD8, and CD3+ CD4+ CD25hi CD127lo Treg cells (see Fig S3 in the supplemental material). T cells were able to secrete IL-10, however, if given strong nonspecific stimulation with CD3/CD28 antibodies, consistent with previously reported results (45; also data not shown). Nonetheless, CD14+ monocytes were the primary subset to spontaneously produce significant IL-10 in our cohort of chronically infected subjects in the absence of exogenous stimulation.
Fig 3.

IL-10 production by cellular subsets. (A) CD4+ T cells, CD8+ T cells, CD25+ FoxP3+ Tregs, CD14+ monocytes, CD19+ B cells, CD11c+ mDCs, and CD123+ pDCs from chronically HIV-1-infected, untreated subjects were analyzed for spontneous production of IL-10 by ICS. (B) Analysis of IL-10 secretion in unstimulated PBMC subsets in 15 untreated, chronically HIV-1-infected individuals. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Tregs induce production of IL-10 by CD14+ monocytes.
Since regulatory T cells appeared to have significant effects on IL-10-mediated T cell impairment yet did not directly produce IL-10, we next investigated whether Tregs could modulate production of IL-10 by monocytes. We used magnetic beads to deplete CD14+ or CD25+ cells from unstimulated PBMCs taken from subjects with chronic, untreated HIV-1 infection. For a control, we depleted CD19+ B cells. This method allowed for the efficient depletion of targeted populations from PBMCs (Fig. 4A). Flow cytometry was performed on all samples to ensure that only the targeted cell population was depleted without nonspecific depletion of other cell types. The same numbers of depleted or mock-depleted cells were then cultured for 3 days, and supernatants were collected and measured for IL-10 levels by Luminex assay. The amount of IL-10 produced following depletion was normalized to IL-10 levels in mock-depleted PBMCs (Fig. 4B). This demonstrated that depletion of both CD14+ monocytes and CD25+ T cells significantly reduced the amount of IL-10 secreted, whereas CD19 depletion did not (P < 0.0001, Kruskal-Wallis test).
Fig 4.

Treg depletion decreases IL-10 production by monocytes. Magnetic beads coated with antibodies against CD14, CD19, or CD25 were used to deplete specific cellular subsets from PBMCs from 15 chronically HIV-1-infected, untreated subjects. (A) Representative FACS plots are shown. (B) IL-10 in tissue culture supernatants was measured after 3 days of culture. IL-10 levels were normalized to those of mock-depleted PBMCs for each condition. *, P < 0.05; ***, P < 0.001. (C and D) Depletion of CD25+ cells resulted in a decrease in IL-10 production by CD14+ monocytes and a simultaneous increase in TNF-α production as measured by ICS. Representative FACS plots are shown (C) as well as group statistics for a cohort of 8 chronically infected, untreated individuals (D).
The effect of Tregs on monocyte production of IL-10 was also observed using IL-10 ICS. PBMCs from chronically infected individuals were bead depleted of CD25+ cells and then stimulated with SEB to activate regulatory T cells and LPS to augment the production of IL-10 by monocytes. Depletion of Tregs resulted in a significant decrease in IL-10 production by monocytes (Fig. 4C and D; P = 0.0188, Wilcoxon test), with a simultaneous reciprocal increase in TNF-α production (P = 0.0148, Wilcoxon test). These data suggest that regulatory T cells increase the production of inhibitory cytokines like IL-10 while simultaneously suppressing the production of proinflammatory cytokines, such as TNF-α. CD25 depletion did not affect IL-10 production by other cellular subsets.
Tregs regulate IL-10 production via contact/paracrine-mediated mechanisms.
To confirm the interplay between Tregs and monocytes in a reduced system and to determine if the regulation of IL-10 production by monocytes was mediated through soluble factors or cell contact, we live-sorted purified populations of CD14+ monocytes and CD25hi CD127lo regulatory T cells (see Fig. S4 in the supplemental material). We then plated monocytes alone or cocultured monocytes with Tregs, either in direct contact or physically separated by a Transwell membrane. Cells were stimulated with LPS and SEB, and the supernatants were collected after 3 days of culture. IL-10 production was measured by Luminex. When cultured alone, purified monocytes produced significantly greater amounts of IL-10 than regulatory T cells (Fig. 5A; P = 0011, Mann Whitney test). Coculture of monocytes with Tregs, however, resulted in a substantial increase in IL-10 production (Fig. 5B; median fold increase, 1.57; P = 0.0156, Wilcoxon test). Interestingly, the augmentation of IL-10 production by Tregs was abrogated by separation of the monocytes and Tregs by a Transwell membrane (Fig. 5C and D). This suggests that the mechanism of induction of increased IL-10 production is mediated primarily by direct contact between monocytes and Tregs, or, alternatively, requires close spatial proximity and paracrine activity. Incubation of monocytes and Tregs with anti-PD-1 ligand 1 (anti-PDL1) or anti-CTLA-4 antibodies did not affect the induction of IL-10 production (see Fig. S5 in the supplemental material).
Fig 5.
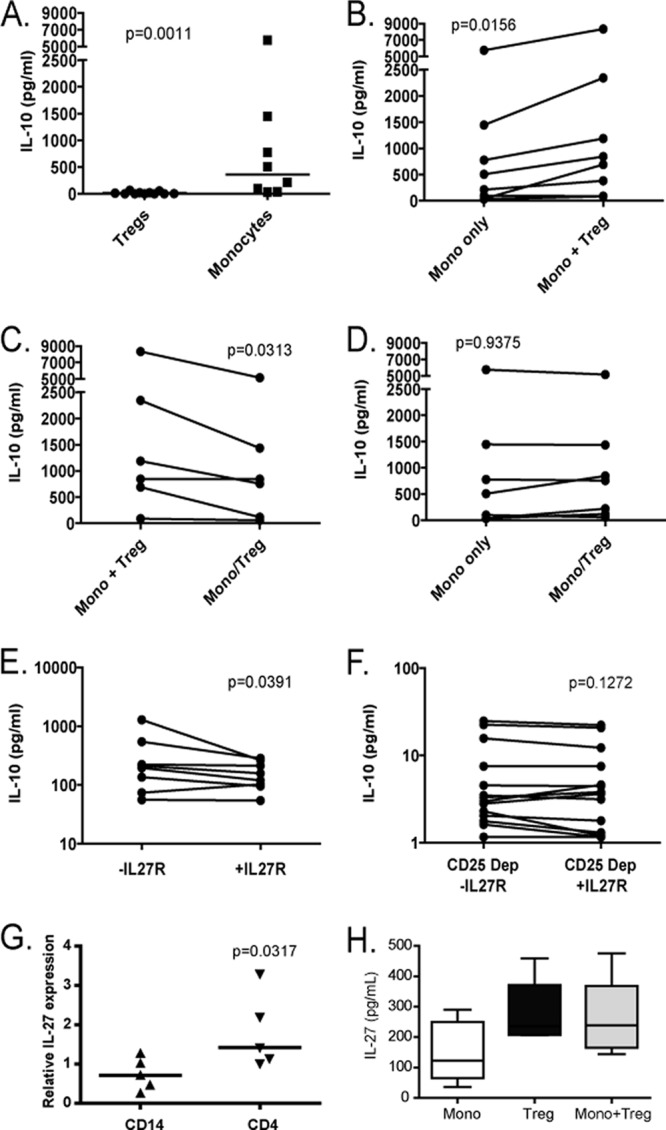
Induction of IL-10 in monocytes involves contact-dependent Treg activity and is also modulated by IL-27. CD14+ monocytes and CD25hi CD127lo Tregs from 8 untreated chronically HIV-1-infected subjects were purified by FACS sorting. Cell subsets were then cultured alone or in cocultures, and IL-10 was measured in supernatants after 3 days of incubation. (A) IL-10 production by Tregs and monocytes cultured alone. (B, C, and D) Monocytes were either cultured alone or cocultured with Tregs in direct contact (Mono+Treg) or separated by a Transwell membrane (Mono/Treg). (E) PBMCs from 8 untreated chronically infected individuals were cultured in the presence or absence of recombinant IL-27 receptor protein (IL-27R), and IL-10 was measured in the supernatants. (F) The same experiments were performed with the same persons after CD25 depletion. (G) Production of IL-27 in sorted CD14 monocytes and CD4 T cells was assessed by quantitative PCR. (H) IL-27 production was assessed in supernatants of purified monocytes cultured alone, purified Tregs cultured alone, or Tregs cultured with monocytes.
IL-27 has been described as a potent inducer of IL-10 production (2, 3, 19). We therefore next incubated LPS-stimulated PBMCs from chronically HIV-1-infected, untreated subjects with IL-27 recombinant receptor protein (IL-27R), which inhibits IL-27 activity by competing with the cell surface ligand. This resulted in decreased IL-10 production relative to that of PBMCs alone (Fig. 5E). Once Tregs were depleted, we did not observe an impact of recombinant IL-27R on IL-10 production (Fig. 5F). We next examined transcription levels of IL-27 and its receptor in CD4+ T cells and monocytes in untreated, chronically HIV-1-infected subjects and HIV-1-negative controls. In viremic HIV-1-infected individuals, CD4+ T cells expressed twice as much IL-27 transcript as CD14+ monocytes (Fig. 5G) and cultured Tregs showed a trend toward greater IL-27 protein production relative to that of monocytes, which remained unchanged when monocytes were cocultured with Tregs (Fig. 5H). In contrast, monocytes and CD4+ T cells from HIV-1-negative controls expressed similar levels of IL-27 mRNA (data not shown). This suggests that the IL-27 pathway plays a role in the regulation of IL-10 production in chronic HIV-1 infection. We then sought to determine whether differential expression of the IL-27 receptor in leukocyte populations of peripheral blood could play a role in our observations. We found that IL-27Rα mRNA was detectable and expressed at similar levels in CD4+ T cells and monocytes and observed no difference between the six HIV-1-infected participants and the six HIV-1-negative controls investigated (data not shown).
DISCUSSION
We describe a novel mechanism by which regulatory T cells control IL-10 production in chronic HIV-1 infection and provide unique mechanistic insight into the role of regulatory T cells in immune exhaustion. In these studies, we investigated the interplay between two different major immunoregulators in HIV-1 infection—the cytokine IL-10 and regulatory T cells—and defined a novel mechanism by which regulatory T cells inhibit HIV-1-specific CD4 T cell responses through upregulation of IL-10 production in monocytes. Our published studies (34) and data from others (36) suggest that monocyte/macrophage subsets are critically involved in the regulation of T cell responses in progressive HIV-1 infection and that elevated IL-10 levels are a hallmark of chronic HIV-1 disease. Although it has been reported that monocytes can produce IL-10, the direct regulation of monocyte IL-10 production by Tregs through a mechanism that requires contact is unique. Previous studies, including our own, have shown broad expression of IL-10 mRNA, yet we identified monocytes as the primary source of IL-10 protein expression by ICS. This difference in transcript and protein expression may be due to the significant degree of posttranslational regulation of IL-10 that has been reported (26, 32, 36, 41). The observation that IL-27 blockade abrogates the Treg-mediated effect on monocyte IL-10 production suggests that IL-27 contributes to this effect. Several reports indicate that monocytes are potently stimulated by IL-27 (21, 31) and that IL-10 production is rapidly upregulated in monocytes by IL-27 stimulation through activation of STAT1 and STAT3 (22). Given our results, we propose that Tregs may induce IL-10 production by secreting IL-27 that acts upon monocytes in a paracrine fashion to stimulate IL-10 production.
IL-10 is thought to primarily affect T cell function by its effects on monocytes/macrophages, and this suggests that Tregs may exhibit a portion of their regulatory effect by stimulating IL-10, which in turn would decrease the effectiveness of monocytes as antigen-presenting cells by downregulating major histocompatibility complex (MHC) class II and CD80/86 and also by decreasing monocyte production of proinflammatory cytokines (12). However, other cellular subsets have also been implicated as producers of IL-10, and infection of mice with LCMV induced a potent upregulation of IL-10 in dendritic cells and B cells but only a mild increase in macrophages (6). Our prior results show that although monocytes express higher levels of IL-10 RNA in progressive HIV-1 infection, they do not do so exclusively: several subsets of leukocytes upregulate IL-10 transcription in the presence of uncontrolled viremia and may provide important contributions to IL-10 production in vivo (5). It is important to note that most untreated, chronically HIV-1-infected individuals examined in this study had relatively conserved CD4 counts and moderate viral loads. Although our results show that in this context monocytes are the main source of IL-10 protein, previous studies (15–17) have identified at more advanced HIV-1 disease stages a subset of IL-10 producing HIV-1-specific CD8 T cells capable of suppressing cytolysis and IL-2 production. These findings in individuals with low CD4 counts and very high viremia further indicate that the activity of the IL-10 pathway depends on the stage of HIV-1 infection. These findings suggest that temporal or pathogen-specific differences in the cellular induction of IL-10 may also be important and that regulation of IL-10 at the posttranscriptional level may occur (23). Thus, in spite of major progress made in the understanding of IL-10 regulation (37), the modulation and impact of this pathway appear to vary considerably according to the infectious agent (8), precluding a straightforward generalization of findings obtained in a given model system.
Our data demonstrate that in the setting of uncontrolled viral replication, “natural” Tregs, defined as CD127lo CD25hi CD4+ T cells, induce IL-10 production in monocytes while inhibiting the proinflammatory cytokine TNF-α by these same cells. In contrast, these effects of Tregs on cytokine production were not observed in subjects who did not respond to IL-10 blockade and in whom Treg depletion did not enhance HIV-1-specific CD4 T cell proliferation. Depletion of Tregs was achieved by using anti-CD25, which may not be entirely specific for Tregs, as CD25 is also upregulated upon T cell activation. However, findings with sorted Tregs defined as CD127lo CD25hi were consistent with our model. Further studies are needed to determine which characteristics of Tregs and/or monocytes in viremic individuals lead to this functional phenotype. Of note, in our assays we did not find evidence for HIV-1-specific “inducible” CD25lo IL-10-secreting Tr1-inducible regulatory T cells, a mechanism of immune impairment that has been demonstrated for other pathogens (9). We also did not find evidence that this Treg-mediated mechanism required PD-1, despite recent evidence that showed that PD-1 signaling on monocytes could upregulate IL-10 production (36).
These studies highlight the diversity of mechanisms by which chronic pathogens modulate immunoregulatory networks to attenuate antigen-specific T cell immunity. The net impact of the IL-10 pathway in HIV-1 infection in vivo is likely complex and dependent on disease stage. The interplay of Tregs with monocytes, with the ensuing upregulation of the IL-10 pathway that we describe in this study, might lead to increased disease progression by limiting host immune responses and impairing pathogen clearance, or given the role of IL-10 in moderating inflammation, it may also have the benefit of limiting immune-mediated damage to the host. The data additionally stress the role of monocytes/macrophages in HIV-1-associated immune impairment and highlight the need for a better understanding of the HIV-1-induced alterations of these cellular subsets. Given that blockade of the IL-10 pathway appears to restore T cell function in HIV-1-infected individuals, targeting this pathway may have potential as a therapeutic to augment HIV-1-specific immune responses in those with chronic infection. Indeed, blockade of the IL-10 pathway in animal models of chronic infection demonstrated a benefit in regard to viral clearance while having few overt signs of induced pathology (6, 13). Further studies are needed, however, to determine in greater detail the mechanisms of IL-10 regulation and action in infectious diseases and the distinctive alterations of this pathway related to specific pathogens.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bruce D. Walker for his help with scientific input and his critical review of the manuscript, the clinical and laboratory staff at the Massachusetts General Hospital, and all study participants for their invaluable role in this project.
This work was supported by the National Institutes of Health (grant 1K08 AI084546-01 to D. Kwon, RO1 HL092565 to D. Kaufmann, and P01 AI-080192 to D. Kaufmann and D. Kavanagh) and the Harvard University Center for AIDS Research (NIH P30 AI060354). The authors have no conflicting financial interests.
Footnotes
Published ahead of print 11 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Ancuta P, et al. 2001. Opposite effects of IL-10 on the ability of dendritic cells and macrophages to replicate primary CXCR4-dependent HIV-1 strains. J. Immunol. 166:4244–4253 [DOI] [PubMed] [Google Scholar]
- 2. Anderson CF, Stumhofer JS, Hunter CA, Sacks D. 2009. IL-27 regulates IL-10 and IL-17 from CD4+ cells in nonhealing Leishmania major infection. J. Immunol. 183:4619–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Batten M, et al. 2008. Cutting edge: IL-27 is a potent inducer of IL-10 but not FoxP3 in murine T cells. J. Immunol. 180:2752–2756 [DOI] [PubMed] [Google Scholar]
- 4. Blackburn SD, Wherry EJ. 2007. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 15:143–146 [DOI] [PubMed] [Google Scholar]
- 5. Brockman MA, et al. 2009. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 114:346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brooks DG, et al. 2006. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 12:1301–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carrington M, Nelson G, O'Brien SJ. 2001. Considering genetic profiles in functional studies of immune responsiveness to HIV-1. Immunol. Lett. 79:131–140 [DOI] [PubMed] [Google Scholar]
- 8. Couper KN, Blount DG, Riley EM. 2008. IL-10: the master regulator of immunity to infection. J. Immunol. 180:5771–5777 [DOI] [PubMed] [Google Scholar]
- 9. Couper KN, et al. 2008. IL-10 from CD4CD25Foxp3CD127 adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog. 4:e1000004 doi:10.1371/journal.ppat.1000004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai WJ, Köhler G, Brombacher F. 1997. Both innate and acquired immunity to Listeria monocytogenes infection are increased in IL-10-deficient mice. J. Immunol. 158:2259–2267 [PubMed] [Google Scholar]
- 11. Denis M, Ghadirian E. 1993. IL-10 neutralization augments mouse resistance to systemic Mycobacterium avium infections. J. Immunol. 151:5425–5430 [PubMed] [Google Scholar]
- 12. de Waal Malefyt R, et al. 1991. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 174:915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ejrnaes M, et al. 2006. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 203:2461–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El-Far M, et al. 2008. T-cell exhaustion in HIV infection. Curr. HIV/AIDS Rep. 5:13–19 [DOI] [PubMed] [Google Scholar]
- 15. Elrefaei M, Barugahare B, Ssali F, Mugyenyi P, Cao H. 2006. HIV-specific IL-10-positive CD8+ T cells are increased in advanced disease and are associated with decreased HIV-specific cytolysis. J. Immunol. 176:1274–1280 [DOI] [PubMed] [Google Scholar]
- 16. Elrefaei M, et al. 2009. TGF-beta and IL-10 production by HIV-specific CD8+ T cells is regulated by CTLA-4 signaling on CD4+ T cells. PLoS One 4:e8194 doi:10.1371/journal.pone.0008194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elrefaei M, et al. 2007. HIV-specific IL-10-positive CD8+ T cells suppress cytolysis and IL-2 production by CD8+ T cells. J. Immunol. 178:3265–3271 [DOI] [PubMed] [Google Scholar]
- 18. Erikstrup C, et al. 2007. Reduced mortality and CD4 cell loss among carriers of the interleukin-10-1082G allele in a Zimbabwean cohort of HIV-1-infected adults. AIDS 21:2283–2291 [DOI] [PubMed] [Google Scholar]
- 19. Fitzgerald DC, et al. 2007. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat. Immunol. 8:1372–1379 [DOI] [PubMed] [Google Scholar]
- 20. Graziosi C, et al. 1994. Lack of evidence for the dichotomy of TH1 and TH2 predominance in HIV-infected individuals. Science 265:248–252 [DOI] [PubMed] [Google Scholar]
- 21. Guzzo C, Che Mat NF, Gee K. 2010. Interleukin-27 induces a STAT1/3- and NF-kappaB-dependent proinflammatory cytokine profile in human monocytes. J. Biol. Chem. 285:24404–24411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iyer SS, Ghaffari AA, Cheng G. 2010. Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J. Immunol. 185:6599–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang KH, Im SH. 2005. Differential regulation of the IL-10 gene in Th1 and Th2 T cells. Ann. N. Y. Acad. Sci. 1050:97–107 [DOI] [PubMed] [Google Scholar]
- 24. Kaufmann DE, et al. 2007. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 8:1246–1254 [DOI] [PubMed] [Google Scholar]
- 25. Kaufmann DE, Walker BD. 2009. PD-1 and CTLA-4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J. Immunol. 182:5891–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kishore R, Tebo JM, Kolosov M, Hamilton TA. 1999. Cutting edge: clustered AU-rich elements are the target of IL-10-mediated mRNA destabilization in mouse macrophages. J. Immunol. 162:2457–2461 [PubMed] [Google Scholar]
- 27. Knapp S, et al. 2003. Interleukin-10 promoter polymorphisms and the outcome of hepatitis C virus infection. Immunogenetics 55:362–369 [DOI] [PubMed] [Google Scholar]
- 28. Kwon DS, Kaufmann DE. 2010. Protective and detrimental roles of IL-10 in HIV pathogenesis. Eur. Cytokine Netw. 21:208–214 [DOI] [PubMed] [Google Scholar]
- 29. Miyazoe S, et al. 2002. Influence of interleukin-10 gene promoter polymorphisms on disease progression in patients chronically infected with hepatitis B virus. Am. J. Gastroenterol. 97:2086–2092 [DOI] [PubMed] [Google Scholar]
- 30. Naicker DD, et al. 2009. Interleukin-10 promoter polymorphisms influence HIV-1 susceptibility and primary HIV-1 pathogenesis. J. Infect. Dis. 200:448–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pflanz S, et al. 2002. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity 16:779–790 [DOI] [PubMed] [Google Scholar]
- 32. Powell MJ, Thompson SA, Tone Y, Waldmann H, Tone M. 2000. Posttranscriptional regulation of IL-10 gene expression through sequences in the 3′-untranslated region. J. Immunol. 165:292–296 [DOI] [PubMed] [Google Scholar]
- 33. Reed SG, et al. 1994. IL-10 mediates susceptibility to Trypanosoma cruzi infection. J. Immunol. 153:3135–3140 [PubMed] [Google Scholar]
- 34. Rodríguez-García M, et al. 2011. Expression of PD-L1 and PD-L2 on human macrophages is up-regulated by HIV-1 and differentially modulated by IL-10. J. Leukoc. Biol. 89:507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rouse BT, Sarangi PP, Suvas S. 2006. Regulatory T cells in virus infections. Immunol. Rev. 212:272–286 [DOI] [PubMed] [Google Scholar]
- 36. Said EA, et al. 2010. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 16:452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saraiva M, O'Garra A. 2010. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10:170–181 [DOI] [PubMed] [Google Scholar]
- 38. Shin H, Wherry EJ. 2007. CD8 T cell dysfunction during chronic viral infection. Curr. Opin. Immunol. 19:408–415 [DOI] [PubMed] [Google Scholar]
- 39. Shin HD, et al. 2000. Genetic restriction of HIV-1 pathogenesis to AIDS by promoter alleles of IL10. Proc. Natl. Acad. Sci. U. S. A. 97:14467–14472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sozzani S, et al. 1998. Interleukin 10 increases CCR5 expression and HIV infection in human monocytes. J. Exp. Med. 187:439–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stoecklin G, et al. 2008. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J. Biol. Chem. 283:11689–11699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stylianou E, Aukrust P, Kvale D, Müller F, Frøland SS. 1999. IL-10 in HIV infection: increasing serum IL-10 levels with disease progression—down-regulatory effect of potent anti-retroviral therapy. Clin. Exp. Immunol. 116:115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trabattoni D, et al. 2003. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood 101:2514–2520 [DOI] [PubMed] [Google Scholar]
- 44. Virgin HW, Wherry EJ, Ahmed R. 2009. Redefining chronic viral infection. Cell 138:30–50 [DOI] [PubMed] [Google Scholar]
- 45. Weiss L, et al. 2004. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood 104:3249–3256 [DOI] [PubMed] [Google Scholar]
- 46. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.