

Abstract
The human cytomegalovirus (HCMV) US12 gene family comprises a set of 10 contiguous genes (US12 to US21), each encoding a predicted seven-transmembrane protein and whose specific functions have yet to be ascertained. While inactivation of individual US12 family members in laboratory strains of HCMV has not been found to affect viral replication in fibroblasts, inactivation of US16 was reported to increase replication in microvascular endothelial cells. Here, we investigate the properties of US16 further by ascertaining the expression pattern of its product. A recombinant HCMV encoding a tagged version of the US16 protein expressed a 33-kDa polypeptide that accumulated with late kinetics in the cytoplasmic virion assembly compartment. To elucidate the function(s) of pUS16, we generated US16-deficient mutants in the TR clinical strain of HCMV. According to previous studies, inactivation of US16 had no effect on viral replication in fibroblasts. In contrast, the US16-deficient viruses exhibited a major growth defect in both microvascular endothelial cells and retinal pigment epithelial cells. The expression of representative IE, E, and L viral proteins was impaired in endothelial cells infected with a US16 mutant virus, suggesting a defect in the replication cycle that occurs prior to IE gene expression. This defect must be due to an inefficient entry and/or postentry event, since pp65 and viral DNA did not move to the nucleus in US16 mutant-infected cells. Taken together, these data indicate that the US16 gene encodes a novel virus tropism factor that regulates, in a cell-specific manner, a pre-immediate-early phase of the HCMV replication cycle.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous opportunistic pathogen that rarely causes symptomatic diseases in healthy, immunocompetent individuals. In contrast, HCMV infections can lead to life-threatening diseases in individuals with acquired or developmental deficiencies in innate and adaptive immunity. Indeed, HCMV is the leading viral cause of congenital infections and causes significant morbidity and mortality in transplant recipients (3, 21, 26).
A hallmark of HCMV pathogenesis is its ability to productively replicate in an exceptionally broad range of target cells. Of the HCMV-susceptible cell types, epithelial cells, fibroblasts, smooth muscle cells, and endothelial cells are the predominant targets for productive viral replication (26, 39).
Signs of HCMV infection have been found in microvascular endothelial cells of capillaries and venules of various organs, including the salivary glands, the gastrointestinal tract, the liver, the kidneys, the lungs, and the brain (2, 39). The ability of the virus to infect the vascular endothelium is thought to be crucial in HCMV pathogenesis (1, 10, 31, 38, 39). In fact, the location of infected endothelial cells (ECs) at the interface between the circulation and organ tissues promotes the hematogenous dissemination of HCMV and the development of HCMV-associated organ infections in acute disease (1, 3). Moreover, persistent HCMV replication in endothelial cells induces inflammatory responses in infected tissues and the development of vascular damage that in turn may initiate the cascade of events eventually resulting in the development of HCMV-associated vascular diseases (1, 3, 10, 20).
Like endothelial cells, epithelial cells are a major target of HCMV in vivo. Indeed, HCMV is not only able to enter a new host through the mucosal epithelium of the gastrointestinal, respiratory, or genitourinary tract (3, 38), but also, infected epithelial cells of alveoli, salivary glands, kidneys, and the gastrointestinal tract are the main sources of infectivity of their corresponding excretions, thus contributing to host-to-host transmission through these body fluids (2, 39, 40).
The ability of HCMV to infect these cell types is determined by viral tropism factors, such as those encoded by the UL locus (pUL) (UL128, UL130, and UL131A) of the HCMV genome. Indeed, mutagenesis or deletion of any open reading frame (ORF) within this region impairs HCMV tropism for endothelial and epithelial cells, dendritic cells, and monocytes as virus transfers to granulocytes (16, 18, 33, 42, 45). Functional versions of the pUL proteins have been found only in fresh clinical virus isolates naturally endowed with a broad cell tropism. Their adaptation to fibroblast cell culture leads to mutations of these genes and the subsequent inability to infect endothelial and epithelial cells. Thus, it has been suggested that the wild-type (wt) pUL proteins may exert suppressive effects on HCMV replication in fibroblasts (7). Accordingly, the pUL genes have been found to be inactivated in laboratory strains of HCMV; indeed, the inability of the Toledo, Towne, and AD169 strains to enter endothelial and epithelial cells has been found to be associated with inactivating mutations within the UL128, UL130, and UL131A ORFs, respectively (18, 20, 31).
The pUL proteins are virion proteins that assemble onto the extracellular portion of the envelope heterodimer gH/gL to form a pentameric complex (gH/gL/pUL). The gH/gL/pUL complex mediates HCMV entry into endothelial and epithelial cells, most likely by regulating interaction with specific entry receptors and the release of incoming viral capsids from endocytic vesicles. The gH/gL/pUL complex is not necessary, however, for entry into fibroblasts (1, 25, 34, 39, 46). Hence, it has been suggested that the association of gH/gL with the pUL proteins switches the entry pathway from direct fusion at neutral pH at the plasma membrane in fibroblasts to the endocytic, low-pH-dependent pathway observed in endothelial and epithelial cells (20, 33, 38). Thus, the gH/gL complex can form either a heterotrimeric association with gO or an alternative complex with pUL proteins. Interaction with gO has been shown to promote the release of infectious virions that are competent for entry into fibroblasts via fusion at the plasma membrane, whereas interaction with pUL proteins mediates the tropism for endothelial and epithelial cells and cell entry via the endocytic pathway (1, 20, 37, 45). However, it has been reported recently that gO of the TR strain acts as a molecular chaperone by promoting the export of gH/gL from the endoplasmic reticulum to the Golgi apparatus and its incorporation into virion envelopes and that a TR gO-null virus in which the virion particles show a severe reduction in gH/gL content compared to TRwt virions is unable to enter fibroblasts and endothelial and epithelial cells (35, 48). These findings thus suggest a revised version of the prevailing model of HCMV entry where gH/gL, not gH/gL/gO, promotes entry into fibroblasts and where both gH/gL and gH/gL/pUL are required for entry into endothelial and epithelial cells (48).
The presence of further virus-encoded factors that may affect viral replication in endothelial and epithelial cells has been suggested by the results of whole-genome functional profiling of a bacterial artificial chromosome (BAC)-based clone derived from the Towne laboratory strain (12). In this genome-wide screening, viral mutants bearing deletions of individual members of both the US22 and US12 gene families were found to express a growth phenotype in either endothelial or epithelial cells that was different from that of the parental strain. Of the US12 family genes, a mutant with a deletion in the US16 gene, encoding a predicted seven-transmembrane (7TM) protein (22), was found to replicate normally in fibroblasts and in epithelial cells but to produce enhanced growth when infection was assayed in microvascular endothelial cells (12). Although a functional interaction of the US16 gene with the gH/gL/pUL complex was not addressed in the study, this finding led to the suggestion that US16 may encode a cell-type-specific temperance factor that moderates replication in endothelial cells, thereby enhancing virus survival and facilitating long-term persistent infections (12, 20). This interpretation, however, is complicated by the fact that the laboratory strain used in the study, the Towne strain, was subsequently shown to bear an inactivating point mutation (a double-T-nucleotide insertion) at the 3′ end of the UL130 ORF that generates a −1 frameshift, replacing the 3′-most 12 codons with a 26-codon extension (11, 18). This mutation has been shown to reduce the protein's stability and its incorporation into the virion envelope (30). The lack of an adequate amount of functional UL130 is thought to prevent the formation of gH/gL/pUL complexes in the endoplasmic reticulum and their subsequent export to the cell surface, thus conferring severely reduced tropism of the Towne strain for endothelial and epithelial cells (1, 18, 30).
Characterization of the pattern of US16 expression and its functional impact on viral replication in the context of a clinical isolate of HCMV (fully competent for infection of endothelial and epithelial cells) remains to be determined. This ambiguity surrounding the role of US16 prompted us to investigate its function in the replication cycle of a clinical isolate in different cell types. Here, we provide evidence that inactivation of the US16 ORF in the low-passage-number clinical strain TR (27, 33, 40) impairs viral replication in different types of endothelial cells, as well as in epithelial cells. Adsorption of the US16-deficient HCMV onto endothelial cells was not significantly different from that of the parental virus. In contrast, the cell-type-specific growth defect was related to inefficient virus entry and/or a postentry event, as demonstrated by the lack of pp65 and viral-DNA movement to the nucleus in cells infected with a virus devoid of US16. Thus, these results suggest that the US16 gene encodes a novel virus tropism factor that regulates, in a cell-specific manner, a phase of the HCMV replication cycle occurring after virion attachment but prior to the release of the viral genome into the nucleus.
MATERIALS AND METHODS
Oligonucleotides.
All oligonucleotides for PCR, mutagenesis, and sequencing were obtained from Invitrogen (Table 1 shows all oligonucleotide sequences).
Table 1.
Oligonucleotides used for cloning, BAC mutagenesis, and IE mRNA analysis
| Primer designation | Sequence (5′ to 3′)a |
|---|---|
| US16-galK forward | CGTTCTCTGGAAACGGCTGCTCTGTCCGAAAACCAGTTCCGAACGAAAATCCTGTTGACAATTAATCATCGGCA |
| US16-galK reverse | CCCCACGGATCTCGCGCCTTAGACGCGCGGTCATATAGCCTCCGGCTGTCTCAGCACTGTCCTGCTCCTT |
| US16-stop forward | TCATGGTGCGCTGCGTTCTCTG |
| US16-stop reverse | CCCCACGGATCTGCCTTAGACGCGCGGTCATATAGCCTCCGGCTGTCATGGGTCTGCGCTTTCCCACCGCGAtctagaGACAGATCGTGTTTCG |
| US16-REV reverse | CCCCACGGATCTGCCTTAGACGCGCGGTCATATAGCCTCCGGCTGTCATGGGTCTGCGCTTTCCCACCGCGACCCAGCGACAGATCGTGTTTCG |
| US16-HA forward | CTAAAAGTCCCCCCACGGATCTCG |
| US16-HA R1 | CTAAGCGTAGTCTGGGACGTCGTATGGGTATCCTCCTCCCTGAAAATACAGGTTTTCTCCTCCGGGCGAGAGGGTGGAC |
| US16-HA R2 | CGTTCTCTGGAAACGGCTGCTCTGTCCGAAAACCAGTTCCGAACGAAAATCTAAGCGTAGTCTGGGACGTC |
| US16-ORF forward | GGGCGAGAGGGTGGAC |
| US16-ORF reverse | GGTCTGCGCTTTCCCACC |
| US16-F forward | CAAGCCAGACTGCGGGT |
| US16-F reverse | CGAACCATGGCGTAACGG |
| US15-ORF forward | CAGCTTGTCAGAGGAAAAGTAGG |
| US15-ORF reverse | GAAGAGAAAAAGGGTTTCAGGTACC |
| US17-ORF forward | CGCCATGGTTCGCGTGAG |
| US17-ORF reverse | TCTCCGAACTCAGAGGCCAC |
| galK forward | CCTGTTGACAATTAATCATCGGCA |
| galK reverse | TCAGCACTGTCCTGCTCCTT |
| IE1 forward | CAAGTGACCGAGGATTGCAA |
| IE1 reverse | CACCATGTCCACTCGAACCTT |
| IE2 forward | TGACCGAGGATTGCAACGA |
| IE2 reverse | CGGCATGATTGACAGCCTG |
| β-Actin forward | CAAAAGCCTTCATACATCTC |
| β-Actin reverse | TCATGTTTGAGACCTTCAA |
Lowercase boldface letters indicate restriction enzyme sites.
Cells and culture conditions.
Low-passage-number human embryonic lung fibroblasts (HELFs) were grown as monolayers in minimum essential medium (MEM) (Gibco/BRL) supplemented with 10% fetal calf serum (FCS) (Gibco/BRL), 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. Human dermal microvascular endothelial cells (HMVECs) (CC-2543) were obtained from Clonetics and cultured in endothelial growth medium (EGM) corresponding to endothelial basal medium (EBM) (Clonetics, San Diego, CA) containing 2% FCS, human recombinant vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), human epidermal growth factor (hEGF), insulin growth factor 1 (IGF-1), hydrocortisone, ascorbic acid, heparin, gentamicin, and amphotericin B (1 mg/ml each). The cells were seeded onto culture dishes coated with 0.2% gelatin. Experiments were carried out with cells at passages 4 to 8. Human umbilical vein endothelial cells (HUVECs) were isolated by trypsin treatment of umbilical cord veins and cultured as described for HMVECs. Experiments were carried out with cells at passages 2 to 5. Lymphatic endothelial cells (LECs) were isolated from human lymph node specimens by collagenase/dispase digestion and purified by two rounds of immunomagnetic-bead selection for the endothelial marker CD31 and the LEC-specific podoplanin marker, respectively. LECs were cultured on collagen type I-coated wells with EGM containing a VEGF subfamily with a cysteine-rich domain (VEGF-C; 25 ng/ml), as previously described (14). All experiments described were performed between the 3rd and 5th in vitro passages.
The retinal epithelial cell line ARPE-19 (ATCC CRL-2302) was cultured in a 1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) (Sigma) and Ham's F-12 medium (Invitrogen) containing 10% FCS with 15 mM HEPES, 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate.
Viruses.
The wild-type TR virus was reconstituted by transfecting HELFs with the corresponding BAC, TR-BAC (a generous gift from Michael Jarvis and Jay Nelson). HCMV TR was derived from an ocular specimen (41) and, after a few passages on fibroblasts, was cloned into a BAC (27, 33). Reconstitution of TR-BAC in fibroblasts generated infectious virus that retained the ability to infect endothelial and epithelial cells, as well as monocytes and macrophages (33).
TR mutants containing mutations in the US16 gene were generated by a two-step replacement strategy using the galk recombineering method, as previously described (6, 47). Briefly, TR-BAC was electroporated into Escherichia coli SW102 (a gift from N. Copeland). To generate TRΔUS16-BAC (Fig. 1), the galK ORF was amplified from pgalK (a gift from N. Copeland) by PCR using the US16-galk primer set (Table 1). At the 3′ ends of the forward and reverse primers, specific sequences (24 and 20 bp, respectively) dictate the amplification of the galK cassette, and their 5′-end 50-bp tails are homologous to the sequences flanking the US16 gene between nucleotides 9245 and 10274 of the TR-BAC complete sequence (GenBank AC146906). Following PCR, the 1,331-bp PCR product was digested with DpnI to remove any plasmid template and gel purified. In order to accomplish the homologous recombination, approximately 50 ng of DNA was electroporated into SW102 bacteria harboring TR-BAC. Cells were then plated on minimal medium (M63) agar plates containing 0.2% galactose and chloramphenicol and incubated at 32°C for 5 days. The colonies that appeared were streaked twice on MacConkey agar plates containing 0.2% galactose and chloramphenicol, which produced bright red bacterial colonies. Several single Gal-positive TRΔUS16-BAC colonies were further characterized for US16 replacement by PCR and sequencing and used to initiate the counterselection step.
Fig 1.

HCMV genome and structure of the US16 mutant viruses. (A) Schematic representation of the HCMV US16 gene region and the modifications that were introduced into the US16 ORF. In TRΔUS16, the US16 ORF was replaced with the galactose kinase marker (galK). In TRUS16stop, a single-nucleotide change was introduced into codons 9, 10, and 11 of the US16 ORF. These changes created a stop codon in the 10th codon, as well as a unique restriction site for XbaI. In TRUS16-REV, the changes introduced into codons 9, 10, and 11 of TRUS16stop were reversed to the wt sequence, thus repairing the whole US16 ORF. TRUS16-HA was generated from TRΔUS16 by reintroducing the US16 ORF fused with the coding sequence for an HA epitope tag at its C terminus. Recombinant BACs were examined for the desired mutation by PCR, restriction digestion analysis, and sequencing. (B) PCR and restriction digestion analysis of viral DNA from infectious RV. Viral DNA was genome purified from RVTRwt (lanes 1, 5, 9, and 13), TRΔUS16 (lanes 2, 6, and 10), TRUS16stop (lanes 3, 7, 11, and 14), TRUS16-HA (lanes 4, 8, 12, and 15), and TRUS16-REV (lanes 16, 17, 18, and 19). PCR was performed using the US16-F primer set (lanes 1, 2, 3, 4, and 16), the US16-ORF primer set (lanes 5, 6, 7, 8, and 17), or the galK primer set (lanes 9, 10, 11, 12, and 18) (Table 1). The sizes of the PCR products were as follows: 1,228 bp for the galK primer set; 924 bp for the US16-ORF primer set; 1,249 bp for the US16-F primer set from RVTRwt, RVTRUS16stop, and RVTRUS16-REV; and 1,309 bp for the US16-F primer set from RVTRUS16-HA. To confirm the successful introduction of the desired stop codon in TRUS16stop, PCR fragments from RVTRwt, RVTRUS16stop, RVTRUS16-HA, and RVTRUS16-REV amplified using the US16-F primer set were digested with the XbaI restriction enzyme (lanes 13, 14, 15, and 19, respectively). Lanes M, molecular markers.
In order to generate TRUS16-HA-BAC and TRUS16stop-BAC (Fig. 1), the galK cassette in TRΔUS16-BAC was replaced in the second step with the appropriate US16-modified gene cassette. The US16-hemagglutinin (HA) cassette was amplified by PCR from TR-BAC using the US16-HA primer set (Table 1) in two-step PCR. In the first step (primers US16-HA F and US16-HA R1), the HA epitope was added to the C-terminal end of the US16 ORF. In the second step (primers US16-HA F and US16-HA R2), the 3′-homology arm used in the first selection step was added to the 3′ end of the US16-HA segment. The US16stop cassette was amplified by PCR from TR-BAC using the US16stop primer set (Table 1). The US16stop reverse primer contains a nucleotide change in each of codons 9, 10, and 11 of the US16 ORF, generating a stop codon (TAG) in codon 10. The PCR products (US16-HA or US16stop) were then digested with DpnI and gel purified, and 200 ng of product was electroporated into SW102 cells harboring the TRΔUS16-BAC clone. In order to select for bacteria lacking the galK gene, the transformed bacteria were plated on M63 agar plates containing 0.2% 2-deoxygalactose (DOG) with glycerol as the sole carbon source and chloramphenicol (47). Gal-negative colonies were characterized for replacement of galK sequences with the mutated US16 versions by PCR amplification of the whole segment, followed by restriction enzyme analysis and sequencing. Two independent TRUS16-HA and TRUS16stop BAC clones were prepared and characterized to ensure that their phenotypes did not result from an off-target mutation.
To generate the revertant TRUS16-REV-BAC (Fig. 1), the US16stop cassette in TRUS16stop BAC was replaced with the galK ORF amplified from pgalK using the US16-galk primer set as described above. Then, in the second recombineering step, the galK cassette was replaced with the US16-REV cassette amplified from TRUS16stop BAC using the US16-stop forward and the US16-REV reverse primers (Table 1). The US16-REV reverse primer contains nucleotide changes in each of codons 9, 10, and 11 of the US16-stop reverse primer that restore the wt codons CGC, TGG, and GTC, respectively. Gal-negative colonies were then characterized for replacement of galK sequences with the US16-REV cassette by PCR amplification of the whole segment, followed by restriction enzyme analysis and sequencing.
Infectious recombinant viruses (RV) TRUS16-HA, TRΔUS16, TRUS16stop, TRUS16-REV, and TRwt were reconstituted by transfection of the corresponding BACs into HELFs. The different HCMV BACs and a plasmid expressing HCMV pp71 (a gift from T. Shenk) were cotransfected by Lipofectamine 2000 (InVitrogen). The transfected HELFs were then cultured until a marked cytopathic effect was observed.
Viral stocks were then prepared by infecting HELFs at a virus-to-cell ratio of 0.01. The cells were incubated in MEM supplemented with 1% heat-inactivated FCS and cultured until a marked cytopathic effect was observed. Stocks were then prepared from sonicated cells, which were centrifugally clarified and frozen at −80°C in MEM containing 20% fetal bovine serum (FBS). Virus titers were determined by plaque assay on HELFs. All infectious RV stocks were routinely checked for the desired change by PCR and restriction digestion analysis (Fig. 1B).
To determine the viral replication kinetics, HELFs, HMVECs, or ARPE-19 cells were infected with TR-BAC-derived viruses at a multiplicity of infection (MOI) of 0.1 PFU/cell. Mock-infected control cultures were exposed to an equal volume of mock-infecting fluid. Virus adsorptions were carried out for 2 h at 37°C. For all experiments, the time at which the virus was first added to the cells was considered time zero. Following infection, cultures were maintained in growth medium for various times postinfection (p.i.). Thereafter, the cells and supernatants were harvested and disrupted by sonication. Viral titers were then measured by an IEA (IE1 plus IE2) indirect immunoperoxidase staining procedure on HELFs, as previously described (15).
Radiolabeled virus particles were produced as previously described (33). Briefly, HELF monolayers were infected with RVTRwt or RVTRUS16stop at an MOI of 1 PFU/cell, and [3H]thymidine (25 μCi/ml; PerkinElmer NEN) was added 24 h later. Radiolabeled virions were concentrated and partially purified from culture supernatants collected 8 days p.i. by centrifugation at 50,000 × g for 1 h through a 20% sorbitol cushion (35). Following resuspension, radioactivity was determined by liquid scintillation, and the amounts of RVTRwt and RVUS16stop virions were normalized to equivalent genome copy numbers by real-time PCR as described below.
Immunofluorescence.
Immunofluorescence analysis of viral antigens was performed as previously described (4, 23) using the rat monoclonal antibody (MAb) anti-HA (clone 3F10; Roche) conjugated to fluorescein isothiocyanate (FITC) and mouse MAbs against IEA (IE1 plus IE2) (clone E13; Argene Biosoft), UL99 (pp28) (clone CH19; Virusys), UL83 (pp65) (clone 3A12; Virusys), and gB (clone CH28; Virusys). The binding of primary antibody was detected with Texas Red-conjugated goat anti-mouse Ig antibodies (Molecular Probes). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole). Samples were observed under a Zeiss Axiovert 25 fluorescence microscope equipped with AxioVision 4.8 software. The infection rate was calculated as the ratio of antigen-positive cells to total cells.
The intracellular localization of viral proteins was examined by an Olympus FV300 inverted laser scanning confocal microscope, and images were captured using FluoView 300 software (Olympus Biosytems).
Immunoblotting.
At the indicated times p.i., whole-cell protein extracts were prepared from infected HELFs or HMVECs by resuspending pelleted cells in Laemmli sample buffer (50 mM Tris-Cl, pH 6.8, 100 mM dithiothreitol [DTT], 2% SDS, 10% glycerol, 1× protease inhibitor cocktail [Sigma P8340]). After boiling at 95°C for 5 min, soluble proteins were collected by centrifugation at 15,000 × g for 10 min. Supernatants were analyzed for protein concentrations with a Bio-Rad Dc protein assay kit (Bio-Rad Laboratories) and stored at −80°C. Proteins were separated by 12% SDS-polyacrylamide gel electrophoresis (50 μg of protein per lane) and then transferred to Immobilon-P membranes (Millipore). The filters were blocked in a solution of 5% nonfat dry milk, 10 mM Tris-Cl (pH 7.5), 100 mM NaCl, and 0.1% Tween 20 and then immunostained with mouse MAbs against IEA (IE1 plus IE2) (diluted 1:250), UL44 (clone CH16; Virusys; diluted 1:2,000), UL83 (pp65; diluted 1:1,000), UL99 (pp28; diluted 1:2,000), and gB (diluted 1:1,000) or with rat anti-HA MAb (clone 3F10; Roche) conjugated to horseradish peroxidase (diluted 1:250). Mouse MAbs against actin (MAB1501R; Chemicon; diluted 1:2,000), tubulin (clone TUB 2.1; Sigma; diluted 1:2,000), and golgin-97 (sc-59820; Santa Cruz; diluted 1:500) were used as controls for cellular protein loading. Immunocomplexes were detected with sheep anti-mouse immunoglobulin antibodies conjugated to horseradish peroxidase (Amersham) and visualized by enhanced chemiluminescence (Super Signal; Pierce).
Nuclear and cytoplasmic extracts were prepared with a Nuclear Extract Kit (Active Motif). The purity of nuclear and cytoplasmic extracts was assessed by immunoblotting with mouse MAbs against nuclear ribonucleoprotein A2/B1 (RNPA2) (clone DP3B3; Abcam; diluted 1:1,000) and tubulin, respectively.
Immunoblot analysis of extracellular virus particles was performed with partially purified virions prepared as previously described (35, 48). Briefly, cell culture supernatants derived from HCMV-infected cells and collected at 8 days p.i. were clarified by centrifugation at 6,000 × g for 20 min, and then viral particles were partially purified by centrifugation at 50,000 × g for 1 h through a 20% sorbitol cushion (35). The virus pellets were lysed in 50 mM Tris-Cl, pH 6.8, 2% SDS, 2% β-mercaptoethanol, and 1× protease inhibitor cocktail (48), and the proteins were fractionated by 12% SDS-polyacrylamide gel electrophoresis.
Attachment assay.
Adsorption of radiolabeled HCMV virions was performed as previously described (24). In brief, prechilled HMVEC monolayers were infected with equivalent amounts of precooled [3H]thymidine-labeled RVTRwt or RVUS16stop virions (normalized to equivalent genome copy numbers by real-time PCR). Cultures were centrifuged at 800 × g for 1 h at 4°C and incubated for 2 h at 4°C. The cells were then washed extensively with cold phosphate-buffered saline (PBS) to remove unbound virus, and the radioactivity associated with cell lysates was determined by liquid scintillation.
Quantitative viral nucleic acid analysis.
Viral DNA was purified from RV stocks according to the procedure described by Feng et al. (13).
To determine the number of viral DNA genomes per microgram of cellular reference DNA (18S rRNA gene), at the indicated times p.i., infected HMVECs were harvested and lysed in a digestion buffer containing proteinase K and extracted using phenol-chloroform, and the DNA was ethanol precipitated as previously described (5). To quantify the amount of viral DNA, samples were subjected to real-time PCR using the previously described probe and primers to amplify a segment of the IE1 gene (5, 22, 43). Briefly, 15 ng of DNA from the samples was added to the Real Master Mix Probe ROX kit containing 5 mM Mg2+ (Applied Biosystems), the oligonucleotide primers, and TaqMan dually labeled IE1 (5′ fluorescein 6-carboxyfluorescein [FAM], 3′ 6-carboxytetramethylrhodamine [TAMRA] quencher) probe (Applied Biosystems), as previously described (43). After activation of AmpliTaq Gold for 10 min at 95°C, the samples underwent 50 cycles of 15 s at 95°C and 1 min at 62°C carried out in an Mx 3000 P (Stratagene). HCMV DNA copy numbers were normalized to the amount of human 18S rRNA gene (Assay-on-Demand 18S, assay no. HS99999901_s1; Applied Biosystems) amplified per reaction. Standard curves were constructed using values from the serially diluted genomic DNA mixed with an IE1-encoding plasmid (17).
Real-time reverse transcription (RT)-PCR analysis was performed on an Mx 3000 P (Stratagene) using SYBR green as a nonspecific PCR product fluorescent label. After HCMV infection, total cellular RNA was extracted using the NucleoSpin RNA kit (Macherey-Nagel). RNA (1 μg) was then retrotranscribed at 42°C for 60 min in PCR buffer (1.5 mM MgCl2) containing 5 μM random primers, 0.5 μM dNTP, and 100 U of Moloney murine leukemia virus reverse transcriptase (Ambion) in a final volume of 20 μl. cDNAs (2 μl) (or water as a control) were amplified in duplicate by real-time RT-PCR using the Brilliant SYBR green QPCR Master Mix (Stratagene) in a final volume of 25 μl. Primer sequences for assessing IE1, IE2, and β-actin mRNA levels are listed in Table 1. Following an initial denaturing step at 95°C for 2 min to activate 0.75 unit of Platinum Taq DNA polymerase (Invitrogen), the cDNAs were amplified for 30 cycles (95°C for 1 min, 58°C for 1 min, and 72°C for 1 min). For quantitative analysis, the log change in fluorescence was plotted against the cycle number, and a threshold (CT) was set for the changes in fluorescence at a point in the linear-PCR amplification phase. The CT values for each gene were normalized to the CT values for β-actin using the ΔCT equation. The level of target RNA, normalized to the endogenous β-actin reference and relative to the 12-h infected cells, was calculated by the comparative CT method and the 2−ΔΔCt equation.
Statistical analysis.
Results are expressed as the mean and standard deviation (SD) for three independent experiments. Data were analyzed for significance using one-way analysis of variance (ANOVA) with Bonferroni posttest correction for multiple comparisons. A P value of ≤0.05 was considered significant.
RESULTS
pUS16 is a late protein expressed in the cytoplasm of infected cells.
The expression pattern of the protein encoded by the US16 gene had not previously been characterized and was thus important to establish at the outset. To this end, since no US16-specific antibody was available, we generated an infectious RV derivative from an infectious BAC clone containing the genome of the clinical isolate TR (27, 33, 40). The derivative RVTRUS16-HA (Fig. 1) expresses the US16 ORF as a fusion protein with an HA epitope tag sequence at its carboxyl terminus (4). The expression of the tagged pUS16-HA protein was then examined by immunoblot analysis of cell total protein lysates prepared at different times p.i. from HELFs infected with RVTRUS16-HA (Fig. 2A) and using an MAb directed against the HA epitope. The expression of a single protein band with an apparent molecular mass of about 33 kDa was detected starting from 48 h p.i. and remained until 96 h p.i. The predicted size of pUS16-HA (36 kDa) fits well with that of the band detected by the anti-HA MAb. To confirm further the temporal kinetic class of pUS16 expression, RVTRUS16-HA-infected HELFs were grown in the continuous presence of foscarnet (PFA), and protein extracts were prepared at 72 h p.i. In this extract, pUS16-HA was not detected, confirming that the US16 gene is expressed with true late (L) gene kinetics (Fig. 2A). The expression levels of IEA (IE1 and IE2), UL44, and UL99 were assessed as controls for representative immediate-early (IE), early (E), and true late (L) HCMV proteins, respectively.
Fig 2.

The US16 gene of HCMV encodes a late cytoplasmic protein. (A) Kinetics of pUS16-HA protein expression in infected cells. HELFs were grown to subconfluence and then infected with HCMV RVTRUS16-HA (MOI, 1 PFU/cell). At the indicated times p.i., total protein cell extracts were prepared, fractionated by SDS-PAGE (50 μg protein/lane), and analyzed by immunoblotting with the anti-HA, anti-IEA, anti-UL44, or anti-UL99 MAb, as described in Materials and Methods. The immunodetection of tubulin with a MAb was performed as an internal control. Cell extracts were isolated from mock-infected cells; cells infected for 24, 48, 72, and 96 h; or cells infected and treated with PFA (200 μg/ml) for 72 h. (B) Localization of US16-HA protein in the cytoplasmic assembly compartment of HCMV-infected HELFs. HELFs were grown to subconfluence and then infected with HCMV RVTRUS16-HA (MOI, 0.1 PFU/cell). At 96 h p.i., the cells were fixed, permeabilized, and stained for pUS16-HA (green), pUL99 (red), or gB (red). Immunofluorescence experiments were repeated three times, and representative results are presented. (C) pUS16-HA is not present in extracellular virus particles. RVTRUS16-HA particles were partially purified from clarified culture supernatants by centrifugation through a 20% sorbitol cushion. Protein extracts from virions (V) and infected cells (C) were then fractionated by SDS-PAGE and analyzed by immunoblotting with the anti-HA, anti-pp65, anti-gB, or anti-UL99 MAb. Immunodetection of golgin-97, a commonly used marker for the TGN, was performed as a control.
The intracellular location of pUS16-HA was then investigated by immunofluorescence using the anti-HA MAb as a probe. As seen in Fig. 2B, in HELFs infected for 96 h with RVTRUS16-HA, the pUS16-HA protein showed a cytoplasmic staining pattern. A similar cytoplasmic pattern was also observed in HELFs transiently transfected with a pUS16 expression vector (data not shown). Moreover, pUS16-HA colocalized with glycoprotein B (gB), as well as with the UL99-encoded pp28 virion tegument protein, suggesting that it accumulates within the cytoplasmic virion assembly compartments of infected cells.
To investigate the presence of pUS16-HA in virions, extracellular virus particles harvested from RVTRUS16-HA-infected HELFs were partially purified by centrifugation through a 20% sorbitol cushion (35) and compared by immunoblotting to RVTRUS16-HA-infected cell extracts. As expected, the tagged protein was observed in cell extracts (Fig. 2C). In contrast, pHUS16-HA was not detected in RVTRUS16-HA extracellular virions. The expression of pp28, pp65, and gB was assessed as a control for representative tegument and envelope proteins. Golgin-97, a protein associated with the cytoplasmic face of the trans-Golgi network (TGN) and observed in the virion assembly compartment (9), was analyzed as a control and was not detected in the partially purified extracellular particles.
Taken together, these results indicate that pUS16 is a true late protein that localizes in the cytoplasmic assembly compartment but is not a structural constituent of virions.
US16-deficient viruses fail to replicate in endothelial cells.
To investigate the contribution of pUS16 to HCMV replication in different cell types, we generated pUS16-deficient viruses from the clinical isolate TR. Virus derived from this BAC clone is able to infect endothelial and epithelial cells, as well as monocytes and macrophages (33), thus retaining the broad cell tropism of clinical isolates of HCMV. In the derivative TRΔUS16, the US16 coding region of TR-BAC was replaced with a galK marker cassette (47) (Fig. 1). The replacement of US16 was confirmed by PCR (Fig. 1B) and sequencing analyses. Infectious RVs were subsequently reconstituted from two independent TRΔUS16 BAC clones (3.3. and 5.3) by transfecting HELFs and subsequently analyzing their growth properties.
To determine the viral growth kinetics of the mutant viruses, HELFs or HMVECs were infected with one of the RVs TRwt, TRΔUS16-3.3, TRΔUS16-5.3, or the revertant RVTRUS16-REV at an MOI of 0.1 PFU/cell and titrated for infectious virus at 0, 2, 4, 6, 8, 10, and 12 days p.i. The replication kinetics of RVTRΔUS16-5.3 in fibroblasts displayed only a minimal defect in comparison with RVTRwt and RVTRUS16-REV (Fig. 3). In contrast, RVTRΔUS16-5.3 exhibited an approximately 5-log-unit decrease in titer in HMVECs on days 8, 10, and 12 p.i. compared with RVTRwt (Fig. 3). Similar results were obtained with the RV produced from the TRΔUS16-3.3 clone (data not shown). The replication kinetics of the US16 revertant virus (RVTRUS16-REV) (Fig. 1) in HMVECs was similar to that of TRwt, indicating that the replication defect of the ΔUS16 viruses in endothelial cells was due to the specific disruption of the US16 coding region.
Fig 3.

Growth kinetics of ΔUS16, US16stop, and US16-REV viruses in fibroblasts and in endothelial cells. HELFs or HMVECs were infected with the parental RVTRwt, RVTRΔUS16 (clone 5.3), RVTRUS16stop, or RVTRUS16-REV (MOI, 0.1 PFU/cell). The extent of virus replication was then assessed by titrating the infectivity of supernatants of cell suspensions on HELFs using the IE antigen indirect immunoperoxidase staining technique (15). The data shown are the averages of three experiments ± SD.
To further support these observations, we generated another mutant, termed TRUS16stop, in which 3 bp of the US16 coding sequence was changed to create a stop codon near the start of the US16 ORF (Fig. 1). When examined in HMVECs, RVTRUS16stop displayed a severe growth defect, as seen in the RVTRΔUS16 deletion mutant (Fig. 3), confirming that the failure to replicate in endothelial cells almost certainly stems from the inability of US16-deficient viruses to express the US16 gene product.
Then, to rule out the possibility that the defective phenotype of the pUS16-deficient viruses in endothelial cells resulted from modifications to US16 that may have altered the expression patterns of the flanking US12 genes, the content of US15 and US17 mRNAs was determined in HELFs infected with RVTRwt, RVTRΔUS16-5.3, or RVTRUS16stop. Quantitative real-time RT-PCR indeed demonstrated that the expression of the neighboring US12 genes was not affected by modifications introduced in US16 mutant viruses (data not shown).
Altogether, these data indicate that the US16 gene of a clinical HCMV isolate encodes a protein that is needed for efficient infection and productive replication in endothelial cells.
HCMV lacking US16 exhibits a very early defect in endothelial cells.
To investigate which stage of the HCMV infectious cycle is compromised in endothelial cells by the lack of pUS16, we examined the progression of the replicative cycle of a US16-deficient virus by monitoring the expression of representative HCMV proteins. For this purpose, total cell extracts were prepared at various times p.i. from HELFs or HMVECs that had been infected with RVTRwt or RVUS16stop. The extracts were then analyzed for their contents of IE, E, and L proteins by immunoblotting with specific antibodies, and the expression levels of IEA (IE1 and IE2), UL44, and UL99 were assessed as controls for IE, E, and L proteins, respectively (Fig. 4A). All these proteins accumulated with the same kinetics in HELFs infected with either TRwt or RVUS16stop. In contrast, the expression of IEA, UL44, and pp28 was substantially inhibited in HMVECs infected with the US16-deficient virus (Fig. 4A), demonstrating that the inactivation of pUS16 caused a defect in virus replication that occurs prior to the expression of IE proteins.
Fig 4.

The replicative cycle of US16-deficient viruses in endothelial cells is blocked at a stage prior to the expression of IE genes. (A) Expression of representative IE, E, and L proteins in endothelial cells infected with TR or TRΔUS16 viruses. HMVECs were grown to subconfluence and then mock infected (M) or infected with the parental RVTRwt or RVUS16stop (MOI, 1 PFU/cell). At the indicated times p.i., total cell extracts were prepared and analyzed by immunoblotting with anti-IEA, anti-UL44, or anti-UL99 MAb as described in Materials and Methods. Actin immunodetected with a MAb served as an internal control. (B) The US16 gene product is required for IE gene expression in endothelial cells. HMVECs were infected with RVTRwt or RVUS16stop (MOI, 0.1 PFU/cell). Total RNA was isolated at the indicated time p.i. and reverse transcribed. Real-time RT-PCR was carried out with the appropriate IE1, IE2, and β-actin primers to quantify the expression levels of IE1 and IE2 mRNA. For each time point, IE1 and IE2 mRNA levels were normalized according to the expression of the actin gene. The results were then analyzed using a standard-curve model, and the levels of IE1 and IE2 mRNAs were normalized to the levels of endogenous β-actin mRNA. The value at each time point was normalized to the value observed with cells infected with RVTRwt for 12 h, which was set at 1. Data are shown as means and SD. ***, P ≤ 0.001 versus calibrator sample.
To test whether the lack of IE protein expression in endothelial cells infected with the US16-deficient virus was due to an impairment of RNA accumulation, mRNA was extracted from HMVECs that had been infected with either the RVTRwt or RVUS16stop virus and analyzed by real-time RT-PCR for IE1 and IE2 mRNA content. Figure 4B shows that, at all time points analyzed, RVUS16stop failed to express a significant level of either IE1 or IE2 mRNA.
Next, to rule out the possibility that the lack of IE gene expression in endothelial cells was due to a defect restricted to HMVECs, different types of endothelial cells were infected with RVTRwt, RVTRΔUS16, RVTRUS16stop, or RVTRUS16-REV. The frequency of HCMV infection was then determined at 24 h p.i. by evaluating the expression of IEA by immunofluorescence analysis (36). As expected, following the infection of HMVECs with US16-deficient viruses, the frequency of IEA detection (Fig. 5) was dramatically lower than that observed for TRwt (0.4% on average for both RVTRΔUS16 and RVTRUS16stop). Moreover, very few of the cells became infected by US16-deficient viruses in cultures of HUVECs (less than 0.5% on average for both RVTRΔUS16 and RVTRUS16stop) or primary human LECs (less than 0.2% on average for both RVTRΔUS16 and RVTRUS16stop) compared to cultures of the same cell type infected with either the parental RVTRwt or the revertant RVTRUS16-REV (from 30% to 50%, depending on the endothelial cell type examined). As for HMVECs, both the last two types of endothelial cells have been shown to support the productive replication of clinical HCMV strains (14, 33). Thus, these results sustain the view that pUS16 plays an essential role in an event occurring prior to the expression of the IE genes in various types of endothelial cells.
Fig 5.

Lack of expression of IE proteins in different types of endothelial cells infected with US16 mutant viruses. HELFs, HMVECs, HUVECs, or LECs were infected with RVTRwt, RVTRΔUS16, RVTRUS16stop, or RVTRUS16-REV at an MOI of 0.1 PFU/cell. At 24 h p.i., cells were fixed, permeabilized, and stained with an anti-IEA (IE1 plus IE2) MAb. Images of ECs infected with US16-deficent viruses were purposely chosen to include positive nuclei to show virus addition, since random fields were on average negative for IEA staining. Immunofluorescence experiments were repeated three times, and representative results are presented (magnification, ×10).
HCMV US16-null viruses are defective for an entry and/or a postentry event in endothelial cells.
To test whether inactivation of the US16 ORF affects the first step of the HCMV infectious cycle in endothelial cells, i.e., virus adsorption, we used [3H]thymidine-labeled RVTR and RVTRUS16stop virus particles that had been generated in HELFs and partially purified by centrifugation (33). Equivalent amounts of radiolabeled RVTR and RVUS16stop virions (normalized to equivalent genome copy numbers by real-time PCR) were incubated with HMVECs alone or in the presence of heparin (as a positive control for inhibition of viral adsorption) for 2 h at 4°C (a condition known to allow virus adsorption only). The cells were then washed extensively, and the radioactivity associated with the cells was measured. As shown in Fig. 6, the level of TRUS16stop virion binding was not significantly different from that observed for TRwt, indicating the successful attachment of both viruses to HMVECs. As expected, heparin treatment prevented virus adsorption. Thus, these results indicate that HCMV attachment to endothelial cells is not significantly affected by the absence of the US16 protein.
Fig 6.

Inactivation of US16 does not impair virion attachment to endothelial cells. HMVECs were infected with equal numbers of [3H]thymidine-labeled RVTR or RVUS16stop virion particles at 4°C in the absence (−) or presence (+) of heparin (30 μg/ml) for 2 h to allow virus adsorption only. The cells were then washed extensively, cell extracts were prepared, and the associated radioactivity was determined by liquid scintillation. The amount of radiolabeled RVTRwt that bound to HMVECs was arbitrarily set to 100 (calibrator sample). Data are shown as means and SD. *, P ≤ 0.05; **, P ≤ 0.01 versus calibrator sample.
Next, we performed a virion content delivery assay to investigate whether the steps subsequent to HCMV adsorption are affected by the lack of pUS16. The highly abundant pp65 protein of the HCMV tegument rapidly localizes to the nuclei of infected cells following membrane fusion and viral entry. Therefore, the nuclear accumulation of pp65 delivered into cells by infecting virions can be used to assess the entry of US16-deficient viruses into endothelial cells. When TRwt or US16-deficient viruses were used to infect HELFs, equivalent numbers of pp65-positive nuclei were observed at 8 h p.i. (Fig. 7), indicating the successful entry of the RVTRΔUS16 and RVTRUS16stop virions and the dissociation of pp65 from the capsids. The infection of HMVECs with TRwt or TRUS16-REV also resulted in the presence of pp65 in a significant number of nuclei (Fig. 7). In contrast, the nuclear accumulation of pp65 was not observed in HMVECs infected with the US16-deficient mutant viruses (Fig. 7), indicating a functional defect in their ability to enter and/or to disassemble the capsids in endothelial cells.
Fig 7.

The UL83-encoded pp65 tegument protein does not accumulate in the nuclei of endothelial cells infected with US16-deficient viruses. HMVECs were infected with RVTR, RVTRΔUS16, RVTRUS16stop, or RVTRUS16-REV (MOI, 0.1 PFU/cell). At 8 h p.i., the cells were fixed, permeabilized, and stained with an anti-UL83 (pp65) MAb. Immunofluorescence experiments were repeated three times, and representative results are presented (magnification, ×10).
To verify the movement of the viral genomes into the nucleus, nuclear and cytoplasmic fractions were prepared from HMVECs infected for 4 h with equivalent amounts of RVTRwt or RVUS16stop virions (normalized to equivalent genome copy numbers by real-time PCR) (13). Successful cell fractionation was demonstrated by assessing the distribution of cellular tubulin and nuclear ribonucleoprotein RNPA2 (Fig. 8A). According to immunofluorescence data, a significant amount of the input pp65 delivered into cells by infecting RVTRwt virions localized to the nuclear fraction within 4 h, while it was almost absent in the nuclear fraction of cells infected with RVUS16stop virions (Fig. 8A). The presence of the input viral genome in the nuclear fraction was then assayed by real-time PCR using primers specific for the IE1 ORF, and the amount of input DNA was normalized by monitoring the cellular 18S gene. As can be seen in Fig. 8B, the amount of viral DNA present in the nuclei of HMVECs infected with RVUS16stop was significantly lower than that of the nuclear fraction prepared from cells infected with RVTRwt (Fig. 8B).
Fig 8.
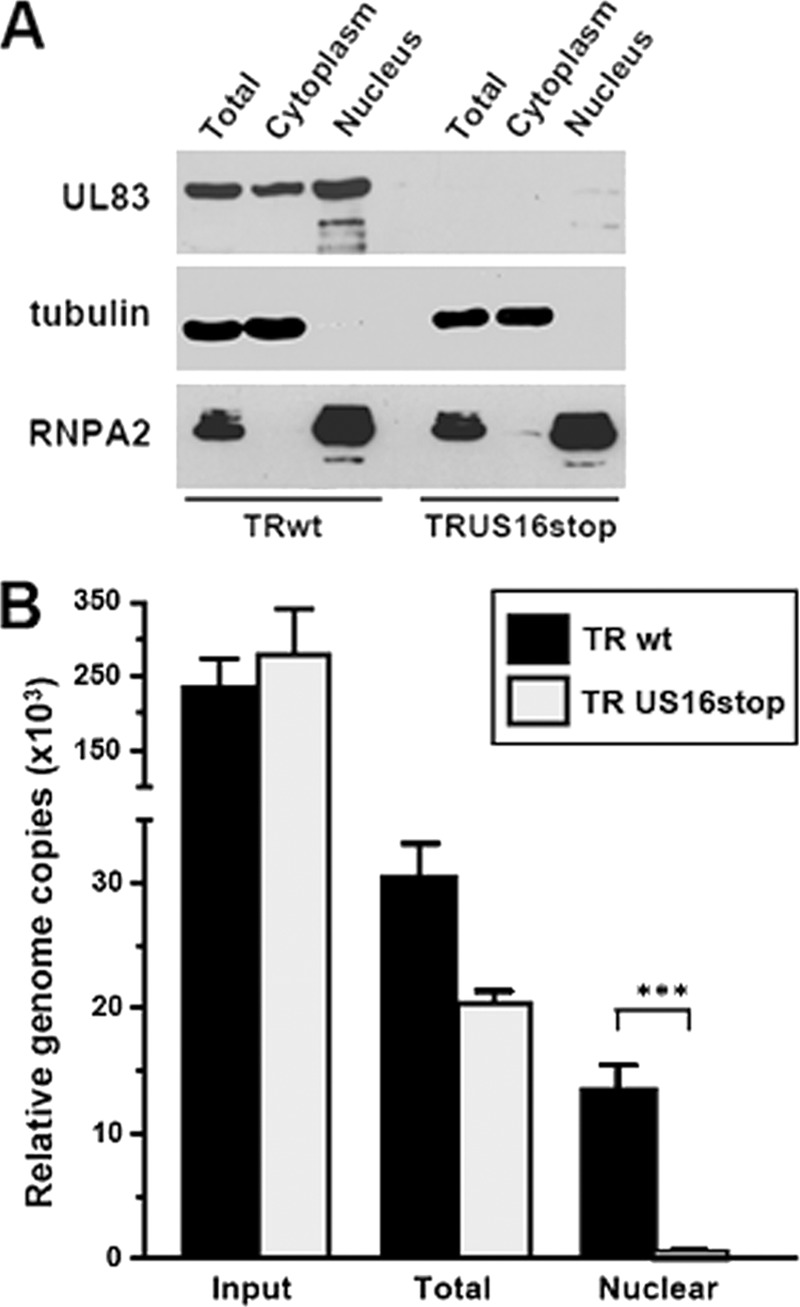
The HCMV ΔUS16 genome does not move to the nucleus in endothelial cells. (A) Separation of nuclear and cytoplasmic fractions. HMVECs were infected with equal numbers of RVTRwt or RVUS16stop virion particles. At 4 h p.i., infected cells were harvested, and nuclear and cytoplasmic extracts were prepared. Equal amounts of protein from the total cell lysate, the nuclear fraction, and the cytoplasmic fraction were then assayed for their pp65 content by immunoblotting. The purity was determined by immunoblotting for the nuclear ribonucleoprotein RNPA2 and tubulin. (B) Viral DNA in the nuclear fraction of HMVECs infected with RVTR or RVUS16stop. HMVECs were infected and fractionated as described for panel A. The amounts of viral DNA present in the nuclear fractions of infected cells were then quantified by real-time PCR using primers specific for the IE1 ORF and normalized to levels of the endogenous 18S gene. The data shown are the averages of three experiments plus SD. ***, P ≤ 0.001 compared to the amount of viral DNA measured in total extracts of cells infected with RVTRwt.
Since the disassembly of pp65 from capsids and the release of viral DNA into the nucleus are defective in endothelial cells infected with HCMV lacking the US16 gene, we can conclude that the US16 protein is required in these cells at an early stage of the HCMV infectious cycle and that its function is related to viral entry and/or to an event subsequent to entry but prior to the release of the viral genome into the nucleus.
pUS16 is required for infection of epithelial cells.
To further examine the role of pUS16 in HCMV infection of other relevant cell types, we infected primary retinal pigmented epithelial cells (ARPE-19 cells) with RVTRwt, RVTRΔUS16, RVTRU16stop, or the revertant RVTRUS16-REV at an MOI of 0.1 PFU/cell and titrated for infectious virus at 0, 2, 4, 6, 8, 10, and 12 days p.i. Similar to the results obtained for the infection of endothelial cells, the US16-deficient viruses displayed a severe growth defect in these epithelial cells (Fig. 9A). The defect was rescued by the reconstitution of the entire US16 ORF, as in RVTRUS16-REV, whose replication in ARPE-19 cells was similar to that of TRwt, confirming that the defective replication of the US16-deficient viruses in ARPE-19 cells was due to the specific disruption of the US16 coding region.
Fig 9.

US16-deficient viruses are defective for growth in epithelial cells. (A) Growth kinetics of ΔUS16, US16stop, and US16-REV in ARPE-19 cells. ARPE-19 cells were infected with RVTRwt, RVTRΔUS16 (clone 5.3), RVTRUS16stop, or RVTRUS16-REV (MOI, 0.1 PFU/cell). The extent of virus replication was then assessed by titrating the infectivity of supernatants of cell suspensions by standard plaque assay on HELFs. The data shown are the averages of three experiments ± SD. (B) Lack of IE expression and pp65 nuclear accumulation in epithelial cells infected with US16 mutant viruses. ARPE-19 cells were infected with RVTRwt, RVTRΔUS16, RVTRUS16stop, or RVUS16-REV at an MOI of 0.1 PFU/cell. At 8 h p.i. for pp65 and 24 h p.i. for IEA, cells were fixed, permeabilized, and stained with an anti-IEA (IE1 plus IE2) MAb or an anti-UL83 (pp65) MAb. Immunofluorescence experiments were repeated three times, and representative results are presented (magnification, ×10).
Finally, we investigated whether the very early phases of the HCMV replication cycle were defective in epithelial cells infected with HCMV lacking US16, as observed with endothelial cells. To this end, the expression of IEA and the nuclear accumulation of pp65 were monitored in ARPE-19 cells infected with TRwt or US16-deficient virus. Again, the expression of IE proteins and the nuclear accumulation of pp65 were impaired by the lack of functional pUS16 (Fig. 9B).
Taken together, our results argue that pUS16 plays a cell-specific role in the replication cycle of HCMV, a role that is performed during virus entry and/or in an event subsequent to entry but before the expression of IE genes in both endothelial and epithelial cells.
DISCUSSION
This study was undertaken to investigate further the function of the protein encoded by the US16 gene of HCMV. The US16 gene is a member of the US12 gene family, which includes a set of 10 contiguous tandemly arranged genes (US12 to US21) in the unique short (US) region of the HCMV genome (22, 28). Apart from HCMV, US12 family homologs have been identified only in CMV specific to higher primates, such as rhesus CMV (RhCMV) and chimpanzee CMV (CCMV) (22). Although the presence of seven-transmembrane hydrophobic domains in all the US12 ORFs first led to the prediction that they belonged to the 7TM protein superfamily (32), a more recent phylogenetic analysis showed that the US12 family represents a distinct branch of the 7TM superfamily that has little similarity to cellular or HCMV-related G-protein-coupled receptors (GPCRs), such as UL33, UL78, US27, or US28 (22). Indeed, a detailed alignment study analyzing similarities between the US12 homologs of HCMV, RhCMV, and CCMV and other similar proteins (i.e., GPCRs and 7TM proteins) revealed that the closest relatives of the individual US12 members are, in fact, their colinear viral counterparts, indicating that the initial duplication and divergence events that led to the US12 family occurred prior to the divergence of the rhesus and human lineages (22).
Importantly, deletion or inactivation of individual US12 family members or deletion of the entire locus from the genome of HCMV laboratory strains did not affect viral replication in fibroblasts, and this led to the classification of these genes as nonessential for viral growth in cultured cells (9, 12, 49). Thus, it was hypothesized that the US12 proteins may exert regulatory roles during the HCMV infection of specific cell types and/or under different physiological conditions in vivo (9, 24). The high conservation of US12 sequences among clinical isolates supports this view (27). Nonetheless, very little information is available on the expression, localization, and functions of individual US12 members, with the sole exception of the localization of US14, US17, and US18 proteins, for which detailed immunofluorescence analyses have shown the dependence of their expression on viral DNA synthesis and their association with the cytoplasmic virion assembly zone, as well as with components of the cellular secretory pathway (8, 9). Although, these US12 proteins have not yet been identified in virions (44), these findings suggest that their functions may be connected with virion maturation and egress (9). Here, we found that pUS16 is expressed as a late protein that colocalizes with cytoplasmic viral assembly zone markers, such as gB and UL99 (Fig. 2B). Thus, the expression kinetics and intracellular localization of pUS16 are similar to those of the other US12 proteins characterized so far. However, we did not observe significant nuclear localization of US16, as has been reported for a US17-derived C-terminal fragment (8). Further clues about the functions of individual US12 family members have been provided from a global functional analysis of the Towne genome (12). Two members of the gene family, namely, US16 and US19, were identified in this study as endothelial-cell-specific temperance factors, since replication of mutants with a deletion in either the US16 or US19 ORF was enhanced in cultures of human microvascular endothelial cells compared to that of the parental wild-type virus (12). These results led to the hypothesis that US16 and US19 facilitate long-term persistent replication in this cell type, rather than acute cytolytic replication, by acting as negative modulators of endothelial cell infection (20). However, this systematic mutagenesis analysis was performed on the genome of the Towne laboratory strain, which was subsequently demonstrated to be unable to infect endothelial cells efficiently, rendering it impossible to understand entirely the role of these US12 genes during the infection of endothelial cells. To investigate further the functional roles of US16 and US19, we have begun the study of their functional properties in the context of replication of a clinical isolate of HCMV in a variety of cell types.
Here, we have demonstrated that the absence of US16 in the clinical TR strain results in a defective growth phenotype in both endothelial and epithelial cells, contrasting with that previously observed with the Towne laboratory strain. Thus, the different virus strains used to investigate the role of US16 are most likely the cause of the contrasting conclusions drawn.
The cell-specific growth defect of US16 mutant viruses was likely due to the mutations made to the US16 locus and not to potential second-site mutations that could have affected neighboring or more distant genes. This conclusion is supported by three lines of evidence. First, two independent BAC clones were used to generate two different RVΔUS16 viruses, and both showed a severe replication defect in endothelial cells. Second, a third US16-deficient virus, the US16stop mutant, bearing a premature interruption of the US16 ORF caused by the insertion of a stop codon at its N terminus (extremely unlikely to interfere with the expression of the adjacent US12 genes), was also unable to replicate in endothelial and epithelial cells. Third, analysis of US15 and US17 mRNA levels by real-time RT-PCR demonstrated that the expression of neighboring genes was not affected by the modifications introduced in the RVΔUS16 and RVUS16stop viruses.
To investigate the basis for the lack of infectivity of the US16 mutant viruses in endothelial and epithelial cells, we examined the progression of their replication cycle by monitoring the expression of representative viral proteins. The expression of IEA, UL44, and UL99 was substantially impaired after infection of endothelial cells with US16-deficient viruses. Since the expression of IE1 and IE2 mRNAs was also abolished (Fig. 4B), these findings suggest that the viral replicative cycle of US16-deficient viruses is blocked at a stage prior to IE gene transcription. Therefore, we tested the hypothesis that the earliest phases of the HCMV replication cycle are affected by the lack of functional pUS16. The absence of significant nuclear delivery of tegument pp65 protein and of incoming viral DNA in both endothelial and epithelial cells infected with the US16-deficient viruses suggested that the cell-specific growth defect likely stems from either inefficient virus attachment or entry or from a postentry event, such as the disassembly of virus particles, capsid translocation to the nuclear pores, and release of the viral genomes into the nucleus. Defective adsorption of mutant HCMV lacking functional US16 to the surfaces of these cell types seemed unlikely because the AD169 laboratory strain was shown to adsorb efficiently to the surfaces of epithelial cells (33) in spite of the absence of the US16 gene product in extracellular virions (44). This hypothesis was confirmed by the binding efficiencies that radiolabeled RVTRwt and RVTRUS16 stop virions showed after their incubation with endothelial cells, indicating that inactivation of US16 does not significantly affect the adsorption stage. The severe reduction in the infectivity of the US16 mutant viruses could therefore be a direct consequence of deficiencies in entry fusion and/or in a postentry event. In this regard, the defective growth phenotype of the US16 mutant viruses in endothelial and epithelial cells seems to overlap that of clinical isolates of HCMV with targeted mutations in the pUL genes that enable entry into these cell types (18, 33). This observation therefore leads one to hypothesize that the lack of pUS16 might affect the assembly of a correct gH/gL/pUL complex and thus the efficiency of entry during the subsequent infectious cycle in endothelial and epithelial cells. A direct role of pUS16 in entry into endothelial and epithelial cells can be excluded by the absence of detectable amounts of the protein in extracellular virus particles partially purified from culture supernatants of RVTRUS16-HA-infected HELFs (Fig. 2C), which suggests that pUS16 can modulate some early entry-related events, though it is not incorporated into virions. In this scenario, pUS16 could interact with other HCMV-encoded proteins required for assembly and/or envelopment of particles within the assembly compartment, promoting the incorporation of adequate levels of envelope components, such as gH/gL/pUL, that are required for the acquisition of virion competence for entry into endothelial and epithelial cells. The localization of pUS16 to the virion assembly compartment might indicate its involvement in the phase of virion maturation and/or egress. To verify this hypothesis, the levels of the gH/gL/pUL complex in extracellular virions produced by US16-deficient viruses and the interactions of pUS16 with other virus- or cell-encoded proteins are being investigated in ongoing studies. Alternatively, the defective phenotype of the US16-null viruses might not be due to an impact on the gH/gL/pUL complex. Thus, pUS16 might regulate some aspect(s) of the virion assembly and/or egress by exerting an as yet unidentified signaling function within the assembly compartment, as was hypothesized recently for pUS27, another HCMV-encoded 7TM protein, whose inactivation reduces the ability of clinical isolates of HCMV to spread efficiently through extracellular virus particles without affecting the direct cell-to-cell spread (29). However, irrespective of the molecular mechanisms, a role of US16 in virion assembly and/or egress is sustained further by the partial restoration of the endothelial cell tropism of a US16-deficient virus that has been observed in preliminary complementation experiments in which the gene was supplied in trans by adenovirus-mediated transduction of HMVECs (A. Luganini, S. Landolfo, and G. Gribaudo, unpublished data).
During coevolution with its host, HCMV has developed the ability to infect a wide variety of cell types by exploiting different combinations of cell receptors, entry pathways, envelope glycoproteins, and requirements for viral tropism factors (1, 19). Tropism factors regulate viral infection and replication across a wide range of cell types by determining the conditions under which cells become infected. The present study characterizes the US16 ORF of a clinical isolate of HCMV as a novel tropism factor required for infection of endothelial and epithelial cells. This discovery advances our knowledge of the molecular complexities that underlie HCMV cell tropism. Future studies are needed to determine the mechanism(s) by which pUS16 affects, in a cell-specific manner, virion infectivity during the very early stages of replication.
ACKNOWLEDGMENTS
We are indebted to Michael Jarvis and Jay Nelson (VGTI, Oregon Health Science University) for providing TR-BAC. We also thank Tom Shenk (Princeton University) for pCGN71, Neal Copeland (NCI, Frederick, MD) for the E. coli SW102 strain and pgalK, Arnaldo Caruso (University of Brescia) for lymphatic endothelial cells, and Andrea Gallina (University of Milan) for the ARPE-19 cells. We are grateful to Wolfram Brune (Heinrich Pette Institute, Hamburg, Germany) for helpful discussions.
This work was supported by grants from the Italian Ministry for University and Scientific Research (Research Programs of Significant National Interest, PRIN 2007 and 2008) to G.G. and S.L., the Piedmont Region (Ricerca Sanitaria Finalizzata 2008 and 2009 to G.G., M.D.A., and S.L.), and FIRB-Futuro in Ricerca 2008 (to M.D.A.).
Footnotes
Published ahead of print 11 April 2012
REFERENCES
- 1. Adler B, Sinzger C. 2009. Endothelial cells in HCMV infection: one host cell out of many or a crucial target for virus spread? Thromb. Haemost. 102:1057–1063 [DOI] [PubMed] [Google Scholar]
- 2. Bissinger AL, Sinzger C, Kaiserling E, Jahn G. 2002. Human cytomegalovirus as a direct pathogen: correlation of multiorgan involvement and cell distribution with clinical and pathological findings in a case of congenital inclusion disease. J. Med. Virol. 67:200–206 [DOI] [PubMed] [Google Scholar]
- 3. Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470 [DOI] [PubMed] [Google Scholar]
- 4. Brune W, Nevels M, Shenk T. 2003. Murine cytomegalovirus m41 open reading frame encodes a Golgi-localized antiapoptotic protein. J. Virol. 77:11633–11643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caposio P, Luganini A, Hahn G, Landolfo S, Gribaudo G. 2007. Activation of the virus-induced IKK/NF-κB signalling axis is critical for the replication of human cytomegalovirus in quiescent cells. Cell. Microbiol. 9:2040–2054 [DOI] [PubMed] [Google Scholar]
- 6. Caposio P, Luganini A, Bronzini M, Landolfo S, Gribaudo G. 2010. The Elk-1 and serum response factor binding sites in the major immediate-early promoter of human cytomegalovirus are required for efficient viral replication in quiescent cells and compensate for inactivation of the NF-κB sites in proliferating cells. J. Virol. 84:4481–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dargan DJ, et al. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 91:1535–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Das S, Skomorovska-Prokvolit Y, Wang FZ, Pellet PE. 2006. Infection-dependent nuclear localization of US17, a member of the US12 familiy of human cytomegalovirus-encoded seven-transmembrane proteins. J. Virol. 80:1191–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Das S, Pellet PE. 2007. Members of the HCMV US12 family of predicted heptaspanning membrane proteins have unique intracellular distributions, including association with the cytoplamsic virion assembly complex. Virology 361:263–273 [DOI] [PubMed] [Google Scholar]
- 10. Digel M, Sinzger C. 2006. Determinant of endothelial cell tropism of human cytomegalovirus, p. 445–464 In Reddehase MJ. (ed), Cytomegaloviruses. Molecular biology and immunology. Caister Academic Press, Wymondham, Norfolk, United Kingdom [Google Scholar]
- 11. Dolan A, et al. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301–1312 [DOI] [PubMed] [Google Scholar]
- 12. Dunn W, et al. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng X, Schroer J, Yu D, Shenk T. 2006. Human cytomegalovirus pUS24 is a virion protein that functions very early in the replicative cycle. J. Virol. 80:8371–8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fiorentini S, et al. 2011. Human cytomegalovirus productively infects lymphatic endothelial cells and induces a secretome that promotes angiogenesis and lymphangiogenesis through interleukin-6 and granulocyte-macrophage colony-stimulating factor. J. Gen. Virol. 92:650–660 [DOI] [PubMed] [Google Scholar]
- 15. Gerna G, et al. 1992. Monitoring of ganciclovir sensitivity of multiple human cytomegalovirus strains coinfecting blood of an AIDS patient by an immediate-early antigen plaque assay. Antivir. Res. 19:333–345 [DOI] [PubMed] [Google Scholar]
- 16. Gerna G, et al. 2005. Dendritic cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 86:275–284 [DOI] [PubMed] [Google Scholar]
- 17. Gribaudo G, et al. 2002. Human cytomegalovirus infection induces cellular thymidylate synthase gene expression in quiescent fibroblasts. J. Gen. Virol. 83:2983–2993 [DOI] [PubMed] [Google Scholar]
- 18. Hahn G, et al. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 78:10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Isaacson MK, Juckem LK, Compton T. 2008. Virus entry and innate immune activation. Curr. Top. Microbiol. Immunol. 325:85–100 [DOI] [PubMed] [Google Scholar]
- 20. Jarvis MA, Nelson JA. 2007. Human cytomegalovirus tropism for endothelial cells: not all endothelial cells are created equal. J. Virol. 81:2095–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Landolfo S, Gariglio M, Gribaudo G, Lembo D. 2003. The human cytomegalovirus. Pharmacol. Therapeut. 98:269–297 [DOI] [PubMed] [Google Scholar]
- 22. Lesniewski M, Das S, Skomorovska-Prokvolit Y, Wang FZ, Pellet PE. 2006. Primate cytomegalovirus US12 gene family: a distinct and diverse clade of seven-transmembrane proteins. Virology 354:286–298 [DOI] [PubMed] [Google Scholar]
- 23. Luganini A, Caposio P, Landolfo S, Gribaudo G. 2008. Phosphorothioate-modified oligodeoxynucleotides inhibit human cytomegalovirus replication by blocking virus entry. Antimicrob. Agents Chemother. 52:1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luganini A, et al. 2010. Peptide-derivatized dendrimers inhibit human cytomegalovirus infection by blocking virus binding to cell surface heparan sulfate. Antivir. Res. 85:532–540 [DOI] [PubMed] [Google Scholar]
- 25. Mocarski ES. 2007. Betaherpesvirus genes and their functions, p 202–228 In Arvin AM, et al. (ed), Human herpesviruses: biology, therapy and immunoprophylaxis. Cambridge Press, Cambridge, England: [PubMed] [Google Scholar]
- 26. Mocarski ES, Jr, Shenk T, Pass RF. 2006. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins Co., Philadelphia, PA [Google Scholar]
- 27. Murphy E, et al. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy E, Shenk T. 2008. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 325:1–19 [DOI] [PubMed] [Google Scholar]
- 29. O'Connor CM, Shenk T. 2011. Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not direct cell-to-cell spread. J. Virol. 85:3700–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patrone M, et al. 2005. Human cytomegalovirus UL130 protein promotes endothelial cell infection through a producer cell modification of the virion. J. Virol. 79:8361–8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev. Med. Virol. 20:136–155 [DOI] [PubMed] [Google Scholar]
- 32. Rigoutsos I, et al. 2003. In silico pattern-based analysis of the human cytomegalovirus genome. J. Virol. 77:4326–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ryckman BJ, et al. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82:60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ryckman BJ, Chase MC, Johnson DC. 2010. Human cytomegalovirus TR strain glycoprotein O acts as a chaperone promoting gH/gL incorporation into virions but is not present in virions. J. Virol. 84:2597–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schuessler A, Laib Sampaio K, Sinzger C. 2010. Improvement of a qualitative cell tropism assay for rapid and reliable characterization of human cytomegalovirus mutants. J. Virol. Methods 167:218–222 [DOI] [PubMed] [Google Scholar]
- 37. Scrivano L, Singzer C, Nitschko H, Koszinowski UH, Adler B. 2011. HCMV spread and cell tropism are determied by distinct virus populations. PLoS Pathog. 7:e1001256 doi:10.1371/journal.ppat.1001256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sinzger C. 2008. Entry route of HCMV into endothelial cell. J. Clin. Virol. 41:174–179 [DOI] [PubMed] [Google Scholar]
- 39. Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 325:63–83 [DOI] [PubMed] [Google Scholar]
- 40. Sinzger C, et al. 1995. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are the major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741–750 [DOI] [PubMed] [Google Scholar]
- 41. Smith IL, et al. 1998. Clinical failure of CMV retinitis with intravitreal cidofovir is associated with antiviral resistance. Arch. Ophthalmol. 116:178–185 [DOI] [PubMed] [Google Scholar]
- 42. Straschewski S, et al. 2011. Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J. Virol. 85:5150–5158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka N, et al. 2000. Quantitative analysis of cytomegalovirus load using a real-time PCR assay. J. Med. Virol. 60:455–462 [DOI] [PubMed] [Google Scholar]
- 44. Varnum SM, et al. 2004. Identification of proteins in human cytomegalovirus HCMV particles: the HCMV proteome. J. Virol. 78:10960–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 79:10330–10338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang D, Shenk TT. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36 doi:10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 84:2585–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]