

Abstract
Interferon (IFN) signaling is crucial for antiviral immunity. While type I IFN signaling is mediated by STAT1, STAT2, and IRF9, type II IFN signaling requires only STAT1. Here, we studied the roles of these signaling factors in the host response to systemic infection with lymphocytic choriomeningitis virus (LCMV). In wild-type (WT) mice and mice lacking either STAT2 or IRF9, LCMV infection was nonlethal, and the virus either was cleared (WT) or established persistence (STAT2 knockout [KO] and IRF9 KO). However, in the case of STAT1 KO mice, LCMV infection was lethal and accompanied by severe multiorgan immune pathology, elevated expression of various cytokine genes in tissues, and cytokines in the serum. This lethal phenotype was unaltered by the coabsence of the gamma interferon (IFN-γ) receptor and hence was not dependent on IFN-γ. Equally, the disease was not due to a combined defect in type I and type II IFN signaling, as IRF9 KO mice lacking the IFN-γ receptor survived infection with LCMV. Clearance of LCMV is mediated normally by CD8+ T cells. However, the depletion of these cells in LCMV-infected STAT1 KO mice was delayed, but did not prevent, lethality. In contrast, depletion of CD4+ T cells prevented lethality in LCMV-infected STAT1 KO mice and was associated with a reduction in tissue immune pathology. These studies highlight a fundamental difference in the role of STAT1 versus STAT2 and IRF9. While all three factors are required to limit viral replication and spread, only STAT1 has the unique function of preventing the emergence of a lethal antiviral CD4+ T-cell response.
INTRODUCTION
Interferons (IFN) are important mediators of innate and adaptive antiviral immune responses (reviewed in references 86 and 92). They are grouped into the type I IFN (IFN-I) family, which includes alpha interferon (IFN-α) and IFN-β; the type II IFN (IFN-II) family, with IFN-γ being the only member; and the type III IFN family, which consists of the IFN-λs. All IFN-Is bind to a common heterodimeric cell surface receptor, termed IFNAR, that induces the phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT2. Activated STAT1 and STAT2 form a trimolecular complex with interferon regulatory factor 9 (IRF9). This complex, termed interferon-stimulated gene factor 3 (ISGF3), binds to interferon-stimulated response elements (ISREs) to regulate the transcription of IFN-I-regulated genes. Although the IFN-IIIs bind to a distinct heterodimeric receptor (interleukin 28 receptor α [IL-28Rα]/IL-10Rβ), they also signal predominantly through the ISGF3 complex (reviewed in reference 8). In contrast, binding of IFN-γ to its unique receptor, IFNGR, results in the phosphorylation of STAT1 and formation of STAT1 homodimers that recognize gamma-activated sequences (GAS) present in the promoter regions of IFN-II-regulated genes. Furthermore, overlap in signal transduction between the two IFN families exists: IFN-Is can induce the formation of STAT1 homodimers and stimulate GAS-driven gene expression (43, 47, 59, 67, 96, 109), while IFN-γ signaling can result in the formation of a modified ISGF3 complex that binds to ISREs (73). In addition, IFN-I, as well as IFN-II, can activate additional signaling pathways (reviewed in references 41 and 99). However, the nature of these pathways and their biological significance is not yet well understood.
Following infection with many viruses, IFN-Is are rapidly secreted by various cell types, with plasmacytoid dendritic cells (pDCs) among the most potent producers (11, 28, 97). IFN-Is induce the production of several innate antiviral molecules aimed at inhibiting infection of cells, viral replication, and virus spread. As a consequence, disruption of the IFN-I system severely compromises host antiviral defense (reviewed in references 5, 19, 38, and 98). In addition to these direct effects, IFN-Is link the innate and adaptive immune responses. They are important for the maturation of antigen-presenting cells and expression of major histocompatibility complex class I (MHC-I) and MHC-II molecules, and they also activate T and B cells and promote IFN-γ production in CD8+ T cells (5, 93).
Lymphocytic choriomeningitis virus (LCMV) is a member of the family Arenaviridae and has its natural reservoir in rodents (27, 61, 90, 102). In humans, LCMV is a rare cause of meningoencephalitis in adults and fetuses (15, 26, 89, 106, 107), but more recently, it has been associated with lethal infection in transplant organ recipients (36). In adult immunocompetent wild-type (WT) mice, peripheral infection with the neurotropic strain LCMV-Armstrong (LCMV-Arm) causes only mild clinical signs of disease. In contrast, intracranial (i.c.) infection with the virus results in a lethal neurological disease. This disease, termed lymphocytic choriomeningitis (LCM), is characterized by seizures and mononuclear cell infiltrates in the meninges and choroid plexus (4, 12). As LCMV is a noncytopathic virus, LCM is the consequence of the strong host immune response against the virus (reviewed in reference 52), which ultimately causes brain edema and herniation (68). The development of LCM is dependent on CD8+ effector T cells (12, 24, 29–31, 37, 79) but does not require CD4+ T helper cells (24, 58). Following peripheral infection, LCMV is cleared within 10 to 12 days postinfection, and virus elimination is mediated by virus-specific CD8+ T cells (2, 20, 57, 66, 87, 111), but not CD4+ T cells or antiviral antibodies (2, 66).
Recognition of LCMV through the innate pattern receptors RIG-I and MDA5 (108) induces, in an IRF7-dependent manner, the production of IFN-Is within 24 to 48 h (25, 62, 65, 71, 108). IFN-Is are critical to limiting LCMV replication (56), and they stimulate clonal expansion, proliferation, and survival of antiviral CD8+ T cells (3, 55, 74). Although the cellular source of IFN-I production in LCMV infection is still controversial, various cell types, including DCs and macrophages, may contribute to its production (42, 50, 62, 65, 72). While development of LCM is independent of IFN-γ (48, 75), IFNAR-deficient mice do not develop LCM following i.c. infection with LCMV-Arm (74). In these mice, an antiviral cytotoxic T lymphocyte (CTL) response is not detectable, and LCMV persists for a prolonged time (88). Although the role of IFN-III in the infection of mice with LCMV is not well investigated, one study reported that, in contrast to IFN-α, treatment with IFN-λ2 did not result in a reduced virus load (7), suggesting that IFN-III might play no crucial role in the murine host defense against LCMV. However, further studies are needed to clarify the role of IFN-III in the disease.
Despite the central roles of STAT1, STAT2, and IRF9 in IFN-I signaling, there has been only limited investigation of the roles that these factors play in LCMV infection. Similar to IFNAR/IFGNR double-deficient mice (80), STAT1 knockout (KO) mice have impaired IFN-I and IFN-II signaling, as well as increased viral titers, following peripheral infection with LCMV (71). In STAT1 KO mice, this is accompanied by cachexia, increased numbers of IFN-γ-producing NK cells (71), and proliferation of CD8+ T cells within the first 3 to 5 days postinfection (40), suggesting dysregulation of the innate immune response. This is further supported by the observation that STAT1 KO mice develop a lethal disease in the absence of classical LCM following i.c. inoculation with LCMV-Arm (83) and contrasts with the mild clinical course in IFNAR-deficient mice (74). In contrast to STAT1 KO mice, STAT2 KO mice do not develop lethal disease following i.c. infection with LCMV (83), and similar to IFNAR KO mice, viral titers are significantly higher in STAT2-deficient mice than in WT mice (71). However, the role of IRF9 in LCMV infection has so far not been studied. The aim of the current study was to further delineate the role of IFN signaling in the host response to LCMV infection using various mouse strains with defects in IFN-I and/or IFN-II signaling and to determine the mechanism of lethal disease previously reported for STAT1 deficiency (83).
MATERIALS AND METHODS
Animals.
STAT1 KO mice (33) were provided originally by Joan Durbin, Nationwide Children's Hospital, Columbus, OH. STAT2 KO mice (84) were provided by Christian Schindler, Columbia University, New York, NY. IRF9 KO mice (54) were obtained from Riken Bioresource Center, Japan, and IFN-γ receptor-deficient (IFNGR KO) mice (48) were purchased from Jackson Laboratories (Bar Harbor, ME). Double-gene-deficient mice (IRF9 KO × IFNGR KO and STAT1 KO × IFNGR KO mice) were produced by interbreeding, and all genotypes were verified by PCR analysis of tail DNA. All mice used were on the C57BL/6 background and maintained under pathogen-free conditions in the animal facility of the School of Molecular Bioscience, University of Sydney. Ethical approval for the use of all mice in this study was obtained from the University of Sydney Animal Care and Ethics Committee.
LCMV infection.
All mice were aged between 8 and 16 weeks at the time of infection. For virus inoculation, mice were given either i.p. or i.c. injection of phosphate-buffered saline (PBS) plus 1% fetal bovine serum (FBS) containing 1,000 PFU (in 200 μl) or 250 PFU (in 20 μl) of LCMV-Armstrong 53b, respectively. Sham-infected mice, which served as controls, received the same volume of PBS plus 1% FBS without virus. Prior to i.c. injection, mice were anesthetized with 100 μg ketamine and 1 μg xylazine per g bodyweight.
Tissue collection and immunohistochemistry.
To determine pathological changes, mice were euthanized at the times shown, and the brains and organs (lung, liver, spleen, and kidney) were removed and fixed overnight in ice-cold 4% paraformaldehyde in PBS (pH 7.4). Following paraffin embedding, tissue sections (5 μm) were prepared and stained with hematoxylin and eosin (H&E). For immunohistochemistry, tissue was embedded in Tissue Tek (Sakura Finetek, Zoeterwoude, Netherlands) and snap-frozen in liquid-nitrogen-cooled isopentane. Immunohistochemistry was performed as described elsewhere (46). The primary antibodies used were against CD4, CD8 (1:50 dilution; BD Pharmingen, Ryde, Australia), 7/4 (1:50 dilution; Serotec, Raleigh, NC), and Mac1 (1:20 dilution; ATCC clone TIB 128). Sections were incubated successively with a biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) and streptavidin-coupled horseradish peroxidase (Vector Laboratories). Diaminobenzidine was applied as the peroxidase substrate, and sections were briefly counterstained with hematoxylin. The stained sections were examined under a DM4000B bright-field microscope (Leica, Wetzlar, Germany), and images were captured using a Spot Flex camera and Spot V4.5 software (Diagnostic Instruments).
Multiplex assay for cytokines.
To determine cytokine levels in serum, a Luminex assay for 23 cytokines (Bioplex Mouse Cytokine 23-Plex Panel; Bio-Rad, Gladesville, Australia) was performed according to the manufacturer's instructions. Briefly, serum (3 independent samples per genotype and condition) was diluted 1:3 in mouse serum diluent (Bio-Rad) and incubated with antibody-coupled beads and then biotinylated antibody, followed by incubation with streptavidin-phycoerythrin (PE). The samples were analyzed using a Luminex 100 system (Bio-Rad) and Bio-Plex Manager software (Bio-Rad). A significant increase or decrease in the level of a particular cytokine was present if the mean of the values of the samples from infected mice showed at least a 2-fold change compared with the uninfected samples and if the values were significantly different as determined by a Mann-Whitney U test.
RPA.
Total RNA was isolated from snap-frozen tissue using TriReagent (Sigma-Aldrich, Castle Hill, Australia) according to the manufacturer's instructions. RNase protection assays (RPAs) were performed and analyzed as described previously (9). Multiprobe sets were used for RPAs to detect the LCMV nucleoprotein (LCMV-NP) RNA (91) and various cytokine mRNAs (10, 45). For the detection of IL-17 mRNA and IL-23p19 mRNA, probes were generated as described previously (82). Target sequences for the IL-17 probe were nucleotides 77 to 327 (GenBank accession no. NM_010552), and for the IL-23p19 probe, they were nucleotides 304 to 583 (GenBank accession no. NM_031252).
LCMV plaque assay.
Vero cells (6 × 105 cells/well) were plated overnight in six-well plates. Tissues were weighed and homogenized in 1 ml ice-cold PBS. Serial dilutions (1:10, 1:100, and 1:1,000) were made with PBS. The medium was removed from the Vero cells, and 200 μl of diluted tissue lysate was added to each well. In addition, a serial dilution from 108 to 100 PFU/ml of LCMV was used as a positive control. After 1 h of incubation at 37°C with 5% CO2, the lysate was removed, and 2% carboxymethyl cellulose (Sigma-Aldrich) mixed 1:1 with 2× Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS was layered on top of the cells. The cells were incubated at 37°C with 5% CO2 for 5 days and then fixed with 10% formalin in PBS for 2 h at room temperature. The cells were washed with H2O, and stained with 0.1% (wt/vol) crystal violet in 20% (vol/vol) ethanol for 30 min, rinsed again with H2O, and air dried. Plaques were counted in each well, and the number of PFU per g tissue was calculated.
In vivo depletion of CD4+ and CD8+ cells.
Anti-CD4 antibodies (GK1.5, clone TIB-207; ATCC, Manassas, VA) and anti-CD8 antibodies (2.43.1, clone TIB-210; ATCC) were produced from hybridoma supernatant as follows. Hybridoma cells were cultured in RPMI 1640 (Life Technologies, Mulgrave, VA, Australia) and 5% fetal bovine serum. The medium was filtered to remove cells, and the proteins were precipitated by adding dropwise an equal volume of saturated ammonium sulfate solution. The precipitate from 2 liters of supernatant was dissolved in 40 ml phosphate-buffered saline and dialyzed for 24 h against 200 volumes of PBS.
For depletion, mice were injected i.p. with 200 μl of either anti-CD4 or anti-CD8 neutralizing antibody solution on the day of infection and days 1, 3, 5, and 7 postinfection. In vivo depletion was confirmed by fluorescence-activated cell sorter (FACS) analysis. For this, approximately 50 to 100 μl tail vein blood was collected, and erythrocytes were lysed using erythrocyte lysis buffer (Invitrogen) according to the manufacturer's instructions. The cells were then blocked with CD16/CD32 and stained for CD4 (anti-CD4–fluorescein isothiocyanate [FITC]) and CD8 (anti-CD8–allophycocyanin [APC]) (all antibodies were from eBioscience, San Diego, CA) and analyzed using a BD FACSCalibur (BD Pharmingen) and FlowJo (Ashland, OR) software.
FACS analysis of leukocytes from liver.
Mice were perfused with PBS, and the liver was removed. The tissue was chopped into small fragments, passed through a 70-μm cell strainer, and digested for 60 min in collagenase and DNase. The cell suspension was washed and resuspended in 30% Percoll (Amersham). The cells were then layered on top of 80% Percoll and centrifuged for 30 min. The interface between the 30% and 80% layers that contained the leukocytes was carefully removed, and the cells were washed in PBS containing 1% FBS and blocked with CD16/CD32. The cells were stained with fluorochrome-coupled antibodies to detect either CD3 (PE-Cy7), CD4 (PE), and CD8 (APC) or CD11b (PE-Cy7), CD45 (PerCp), Ly6G (PE), and Ly6C (FITC). The samples were analyzed using a BD FACSAria (BD Pharmingen) and FlowJo (Ashland, OR) software.
Isolation of leukocytes from lymph nodes and tetramer stain.
At day 6 postinfection, WT and STAT1 KO mice were euthanized, and the inguinal lymph nodes were removed. The lymph nodes were dispersed and passed through a 70-μm cell strainer. The cells were washed in RPMI 1640 medium and counted. For MHC class I tetramer staining, cells were stained with iTAg MHC class I tetramer-streptavidin-PE loaded with LCMV peptide GP33-41 (Beckman Coulter, Fullerton, CA), anti-CD3 (peridinin chlorophyll protein [PerCP] labeled), and anti-CD8 (APC labeled) according to the manufacturer's protocol and stored at 4°C until analysis by FACS. For MHC class II tetramer staining, cells were stained with IAb gp66-77 tetramer-PE (NIH Tetramer Facility, Emory University, Atlanta, GA) for 3 h at 37°C. The cells were then washed and stained with anti-CD3–PerCP and anti-CD4–APC. The cells were fixed with 0.5% paraformaldehyde and stored at 4°C. The samples were analyzed using a BD FACSAria (BD Pharmingen) and FlowJo (Ashland, OR) software. Significance was determined using a one-tailed Mann-Whitney U test.
RESULTS
LCMV infection has a distinct outcome in STAT1-deficient mice.
I.c. infection of WT mice with LCMV-Arm resulted in a characteristic neurological disease with convulsive seizures and death between 7 and 9 days postinfection (Fig. 1A). In contrast, only mild clinical signs, such as reduced activity, were observed in WT mice following i.p. infection (Fig. 1B and C). In agreement with previous studies (48, 75, 80, 83), IFN-γ signaling deficiency had no impact on the clinical course following i.c. or i.p. infection (Fig. 1A and B) compared with WT mice. In contrast, all STAT2 KO (Fig. 1A and B) and IRF9 KO (Fig. 1) mice survived i.c. and i.p. infection and developed only moderate clinical signs with reduced activity, ruffled fur, and weight loss. Previous studies have indicated that i.c. infection of STAT1 KO mice results in slightly delayed death due to a wasting disease rather than the convulsive seizures characteristic of LCM (83). In our study, we not only confirmed these findings (Fig. 1A), but also demonstrated that i.p. infection of STAT1 KO mice resulted in 100% lethality after 8 to 12 days, associated with marked progressive weight loss (Fig. 1B and C). Similar to IRF9 KO mice, weight loss in STAT1 KO mice started at around day 3 postinfection and reached more than 20% of the initial weight by day 7 postinfection. Furthermore, from day 5 postinfection, weight loss was significantly greater in STAT1 KO mice than in IRF9 KO mice. Thus, the absence of STAT1 has unique effects on the clinical course following LCMV infection that are distinct from deficiency in STAT2, IRF9, or IFNGR.
Fig 1.

STAT1 deficiency causes a lethal disease in mice infected with LCMV. (A) I.c. infection of WT and IFNGR-deficient mice with LCMV caused LCM with characteristic seizures and death. STAT2 KO and IRF9 KO mice developed transient, mild clinical signs following i.c. infection and survived. In contrast, i.c.-infected STAT1 KO mice developed a progressive lethal wasting disease but no seizures. (B) I.p. infection of WT, IFNGR KO, STAT2 KO, and IRF9 KO mice resulted in mild clinical signs, and none of the mice died. Comparable with i.c. infection, i.p. infection of STAT1 KO mice caused a wasting disease and the death of all animals. (C) In contrast to WT mice, starting at day 4 postinfection, i.p. infection with LCMV resulted in a significant reduction of the body weight, by approximately 10 to 15% in IRF9 KO mice and more than 20% in STAT1 KO mice, by day 7 postinfection. Loss of body weight was significantly greater in STAT1 KO mice than in IRF9 KO mice, and in the latter, body weight stabilized from day 8 postinfection. The data are shown as means ± standard errors of the mean (SEM). ** and ***, P < 0.01 and P < 0.001 compared with WT mice; ###, P < 0.001 compared with IRF9 KO mice, as determined by Mann-Whitney U test.
STAT1 has a key role in both IFN-I and IFN-II signaling. To determine if the combined disruption of IFN-I and IFN-II signaling might account for lethality following infection with LCMV, we generated mice that were deficient for both IRF9 and IFNGR. Peripheral infection with LCMV was not lethal in IRF9 KO × IFNGR KO mice, as the mice developed only moderate clinical signs that were indistinguishable from those seen in similarly infected IRF9 KO or STAT2 KO mice (Fig. 1A and B). This demonstrated that the combined loss of IFN-I and IFN-II signaling per se could not produce the effect of STAT1 deficiency on clinical disease following LCMV infection.
The possibility that STAT1-independent IFN-γ signaling might mediate lethality in STAT1 KO mice was also examined. STAT1 KO × IFNGR KO mice infected i.c. or i.p. with LCMV developed a clinical course of lethal disease similar to that of STAT1 KO mice (Fig. 1A and B), which indicated that the lethal phenotype in LCMV-infected STAT1 KO mice is independent of IFN-γ.
Absence of type I, but not type II, IFN signaling results in increased and widespread virus RNA levels.
We investigated the expression of LCMV-NP RNA as a marker for virus load and spread. Following i.p. infection in WT mice, low levels of LCMV-NP RNA were present in the liver at day 3 and day 6 postinfection and in the kidney at day 6 and day 12 postinfection, but it was not detectable in the brain at either day 3, 6, or 12 postinfection (Fig. 2A). Similarly, LCMV-NP RNA was present in the livers of IFNGR KO mice at day 6 and day 12 postinfection but was not detectable in the brain (Fig. 2C). However, compared with WT mice, viral clearance from the livers of infected IFNGR KO mice was delayed. In contrast, in STAT1 KO and IRF9 KO mice infected i.p. with LCMV, LCMV-NP RNA was readily detectable and increased significantly in the liver, kidney, and brain at day 3 and day 6 postinfection (STAT1 KO and IRF9 KO mice) and day 12 postinfection (IRF9 KO mice) (Fig. 2A) compared with similarly infected WT controls. These findings demonstrate the crucial role of both STAT1 and IRF9 in limiting virus spread and replication following infection with LCMV and show that lethality in the STAT1 KO mice cannot be accounted for by viral load alone.
Fig 2.
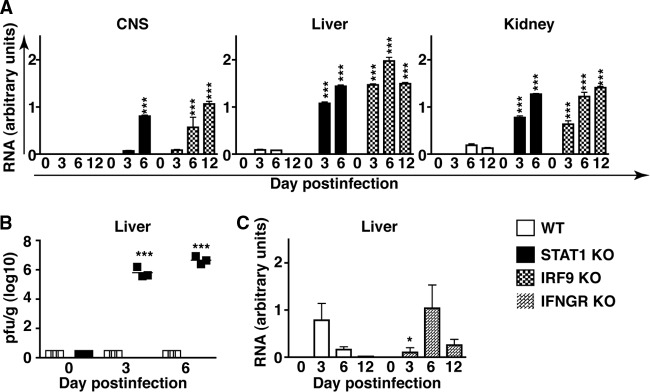
STAT1 and IRF9 are required to limit viral replication and spread and for the elimination of LCMV. (A) WT mice infected i.p. had low levels of LCMV-NP RNA in the liver at days 3 and 6 postinfection and in the kidney at days 6 and 12 postinfection. No viral RNA was detectable in the central nervous system (CNS). High levels of viral RNA were present in the livers, kidneys, and CNS of infected STAT1 KO and IRF9 KO mice. (B) Plaque assays performed on liver lysates from WT and STAT1 KO mice showed no infectious virus detectable by the assay in uninfected WT and STAT1 KO mice or in infected WT mice. In contrast, a high virus titer was present in the livers of day 3 and day 6 infected STAT1 KO mice. (C) Similar to WT mice, LCMV-NP RNA was detectable in the liver, but not the CNS, of IFNGR KO mice. Viral clearance was delayed in IFNGR mice compared with WT mice. Values from RPA were normalized to the housekeeping gene L32 and are shown as mean and SEM. *, P < 0.05, and ***, P < 0.001 compared with the respective time point in WT mice as determined by Mann-Whitney U test.
Increased LCMV RNA levels in STAT1 KO mice correlate with increased virus load.
To assess if the high virus RNA levels in infected IFN-I signaling-deficient mice corresponded to higher levels of infectious virus, we performed plaque assays on liver lysates from uninfected and day 3 and day 6 infected WT and STAT1 KO mice. Lysates from the livers of uninfected WT and STAT1 KO mice did not contain any detectable virus (Fig. 2B). Likewise, liver lysates from day 3 and day 6 infected WT mice did not contain detectable levels of virus, whereas, a high virus titer was found for liver lysates from infected STAT1 KO mice at these times postinfection (Fig. 2B). In concordance with the LCMV RNA levels determined by RPA, a higher LCMV titer was present at day 6 postinfection than at day 3 postinfection in STAT1 KO mice.
Infection of STAT1 KO mice with LCMV results in severe tissue immunopathology.
To determine the basis for the lethal disease caused by LCMV infection in STAT1 KO mice, histological examination was performed on various organs from WT, IFNGR KO, STAT2 KO, IRF9 KO, and STAT1 KO mice infected i.p. with LCMV. The livers (Fig. 3B), kidneys (Fig. 3F), lungs (Fig. 3K), and spleens (Fig. 3O) from WT mice at day 6 postinfection showed no overt pathological changes compared with sham-infected mice (Fig. 3A, E, I, and N, respectively). Comparable with WT mice, organs from LCMV-infected IFNGR KO mice revealed few if any pathological changes (data not shown). At day 6 postinfection, the livers, kidneys, and lungs of IRF9 KO (Fig. 3D, H, and M) and STAT2 KO (data not shown) mice infected i.p. with LCMV showed scattered inflammatory foci. However, the overall tissue architecture was preserved, and no necrosis was present. The spleens of infected IRF9 KO (Fig. 3Q) and STAT2 KO (data not shown) mice showed partial loss of germinal centers. In contrast, at day 6 postinfection, large numbers of leukocytes were present in the livers (Fig. 3C), kidneys (Fig. 3G), and lungs (Fig. 3L) of STAT1 KO mice, and the spleens of these mice showed loss of germinal centers (Fig. 3P). A high number of these infiltrating leukocytes in the livers (Fig. 3R, arrows), kidneys (Fig. 3S, arrows), and lungs (not shown) of infected STAT1 KO mice consisted of polymorphonuclear cells (PMNs). In addition, areas of necrosis were observed in various tissues, including the livers (Fig. 3T) and kidneys (Fig. 3U) of LCMV-infected STAT1 KO mice. In summary, these findings highlight the presence of a generalized and severe multiorgan immune pathology and associated tissue destruction in STAT KO mice infected with LCMV.
Fig 3.

LCMV infection of STAT1 KO mice results in destructive organ pathology. The livers (A), kidneys (E), and lungs (I) of sham-infected STAT1 KO mice showed no overt pathological alterations, and germinal centers present in the spleen appeared normal (N). Comparable findings were obtained for the livers (B), kidneys (F), lungs (K), and spleens (O) from WT mice at day 6 postinfection. In contrast, the livers (C), kidneys (G), and lungs (L) of infected STAT-deficient mice showed prominent mononuclear infiltrates. No organized germinal centers were seen in the spleens (P) of infected STAT1 KO mice. The livers (D, arrowheads), kidneys (H), and lungs (M) from infected IRF9 KO mice contained few foci with infiltrating leukocytes. In the spleens (Q) of infected IRF9 KO mice, germinal centers were less easily discernible than in WT mice but were still identifiable. Higher magnification of tissue sections from the livers (R) and kidneys (S) of infected STAT1 KO mice showed the presence of lymphocytes and polymorphonuclear granulocytes (arrows), as well as focal necrosis, in liver (T, arrows) and kidney (U, arrows; note the normal glomerulus in the top left corner). Original magnifications: A to H, ×20; I to M, ×10; N to Q, ×5; R to U, ×40.
To determine the distribution and phenotype of the leukocytes infiltrating the organs, immunohistochemistry was performed on liver and kidney tissue sections. In the livers and kidneys of sham-infected STAT1 KO mice, only a few CD4+ (Fig. 4A and M) and CD8+ (Fig. 4B and N) T cells and macrophages (Fig. 4C and O) and some neutrophils (Fig. 4D and P) were present. These findings were comparable to those for sham-infected WT mice (data not shown). At day 6 postinfection, the livers (Fig. 4E to H) and kidneys (Fig. 4Q to T) of WT mice showed a moderate increase in infiltrating CD4+ (Fig. 4E and Q) and CD8+ (Fig. 4F and R) T cells, macrophages (Fig. 4G and S), and neutrophils (Fig. 4H and T). In contrast to this, large numbers of CD4+ (Fig. 4I and U) and CD8+ (Fig. 4J and V) T cells, macrophages (Fig. 4K and W), and neutrophils (Fig. 4L and X) were present in the livers and kidneys of LCMV-infected STAT1 KO mice at day 6 postinfection. These findings indicate that a dysregulated immune response to LCMV in STAT1 KO mice may cause peripheral tissue damage and form the basis for the ensuing lethal disease.
Fig 4.

Immunophenotype of infiltrating leukocytes in the livers and kidneys of STAT1 KO mice infected with LCMV. In sham-infected STAT1 KO mice, low numbers of CD4+ T cells (A and M), CD8+ T cells (B and N), macrophages (C and O), and neutrophils (D and P) were present in the liver and kidney, respectively. At day 6 post-i.p. infection, few infiltrating CD4+ (E and Q) and CD8+ (F and R) T cells, macrophages (G and S), and neutrophils (H and T) were seen in the livers and kidneys of WT mice. In contrast, numerous CD4+ (I and U) and CD8+ (J and V) T cells, macrophages (K and W), and neutrophils (L and X) infiltrated the livers and kidneys of infected STAT1 KO mice at day 6 postinfection. Original magnification (all panels), ×20.
To further determine the numbers of infiltrating cells in peripheral organs from WT and STAT1 KO mice infected i.p. with LCMV, FACS analysis was performed on leukocytes isolated from the liver. In uninfected WT and STAT1 KO mice, the numbers and percentages of CD4+ and CD8+ T cells, neutrophils (CD45+, Ly6G+, and CD11b+), and monocytes/macrophages (CD45+, Ly6G−, and CD11b+) were similar (Fig. 5). At day 6 postinfection, the livers of WT mice showed a moderate increase in CD4+ T-cell and macrophage numbers that was significant for CD4+ T cells only in respect to the percentage of live cells compared with uninfected controls. The numbers and percentages of live cells also increased for CD8+ T cells and neutrophils but did not reach statistical significance. In contrast, the numbers and percentages of CD4+ and CD8+ T cells, neutrophils, and macrophages were significantly increased in infected STAT1 KO mice compared with uninfected controls or infected WT mice (Fig. 5).
Fig 5.
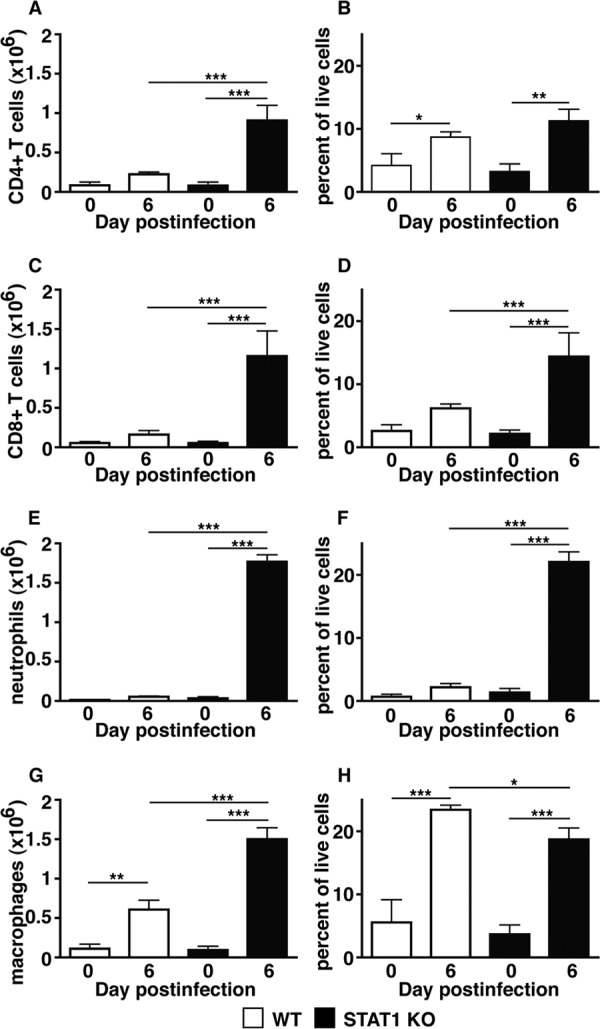
Significantly increased numbers of leukocytes infiltrate the livers of LCMV-infected STAT1 KO mice. Shown is FACS analysis of leukocytes isolated from the livers of WT and STAT1 KO mice at day 6 postinfection. Compared with uninfected WT mice, livers from infected WT mice showed slight increases in the numbers of CD4+ (A) and CD8+ (C) T cells, neutrophils (E), and macrophages (G). In contrast, large numbers of CD4+ (A) and CD8+ (C) T cells, neutrophils (E), and macrophages (G) infiltrated the liver in STAT1 KO mice following infection. Notably, neutrophils were the most frequent cell type, followed by macrophages and CD8+ T cells. (B, D, F, and H) Percentages of populations in respect to all live cells. Data are shown as means and SEM. *, **, and ***, P < 0.05, P < 0.01, and P < 0.001 as determined by Mann-Whitney U test.
Exaggerated cytokine gene expression in organs and cytokine levels in sera of LCMV-infected STAT1 KO mice.
To further characterize the nature of the inflammatory response in LCMV-infected IRF9 KO and STAT1 KO mice, we analyzed the expression of several cytokine and chemokine genes in the liver. In uninfected WT, IRF9 KO, and STAT1 KO mice, the mRNA levels for all cytokines analyzed were similar, with the exception of tumor necrosis factor (TNF) (Fig. 6A) and transforming growth factor beta (TGF-β) (Fig. 6F); the levels of TNF mRNA were significantly lower in IRF9 KO and STAT1 KO mice than in WT mice, while the levels of TGF-β mRNA were significantly lower in STAT1 KO mice than in WT mice. In WT mice infected i.p. with LCMV, increased TNF mRNA levels were seen at days 3 and 6 postinfection (Fig. 6A); IL-1α mRNA levels at day 6 postinfection (Fig. 6B); and TGF-β mRNA levels at days 3, 6, and 12 postinfection (Fig. 6F) were also increased compared with uninfected controls. In WT mice, no significant changes were observed in the mRNA levels for the remaining cytokines analyzed at either day 3 or day 6 (Fig. 6C to J) compared with uninfected controls. In infected IRF9 KO mice, several cytokine mRNA levels were increased compared with uninfected controls: TNF and TGF-β mRNA levels were increased at days 3, 6, and 12 postinfection (Fig. 6A and F); IL-23 p19 mRNA levels at days 3 and 6 postinfection (Fig. 6H); IL-1α and IL-1β mRNA levels at days 3 and 12 postinfection; and IL-17 mRNA levels at day 3 postinfection (Fig. 6J). At day 3 postinfection, WT and IRF9 KO mice showed similar patterns of cytokine gene expression, with the exception of the transiently elevated IL-10, IL-17, and IL-23 p19 mRNA levels in IRF9 KO mice that were not detectable in infected WT mice. Also, TNF mRNA levels were higher in IRF9 KO mice in this early phase than in WT mice. In addition, at day 12 postinfection, TNF, IL-1α, IL-1β, and IL-10 mRNA levels were significantly higher in IRF9 KO mice than in WT mice, which indicated there is a more prolonged response in IRF9 KO mice. For the other cytokine genes examined, there were no significant changes in expression observed following i.p. infection. Together, these findings suggest dysregulation of the antiviral immune response in IRF9 KO mice that affects both the early and later phases of infection.
Fig 6.

Upregulation of various proinflammatory and counterinflammatory cytokine genes in the livers of LCMV-infected WT and STAT1 KO mice. (A to J) Results of densitometric quantification of RPA autoradiographs. The values were normalized to the housekeeping gene L32 and are shown as means and SEM. *, P < 0.05; ** and ##, P < 0.01; *** and ###, P < 0.001 compared with the uninfected control of the same genotype (*) or WT mice (#) as determined by one-way analysis of variance (ANOVA).
In STAT1 KO mice infected i.p. with LCMV, at day 3 postinfection, TNF, IL-1α, IL-1β, and TGF-β mRNAs were increased to levels similar to those found in infected WT and IRF9 KO mice, while IL-23 p19 and IL-17 mRNAs were increased to levels similar to those found in infected IRF9 KO mice (Fig. 6A to J). However, at this time postinfection in STAT1 KO mice, IFN-γ, IL-6, and IL-10 mRNA levels were increased significantly above the levels observed in either WT or IRF9 KO mice. Moreover, in STAT1 KO mice at day 6 postinfection, IL-1α, IL-6, IL-10, IL-23 p19, and IL-17 mRNA levels were reduced significantly compared with day 3 postinfection. Independent of the mouse genotype, no changes were observed in the lymphotoxin β, IL-2, IL-3, IL-4, and IL-5 mRNA levels following i.p. infection with LCMV (data not shown). In summary, the increased expression of various cytokine mRNAs in IRF9 KO and STAT1 KO mice compared with WT mice suggests that IFN-I signaling is important for moderating the early innate antiviral response. Furthermore, the results highlight exaggerated expression of a number of cytokine genes, including IFN-γ, IL-6, and IL-10, in the early phase following LCMV infection in STAT1 KO mice that was distinct from the response seen in IRF9 KO and WT mice.
The possibility that LCMV-infected STAT1 KO mice may be afflicted by a “cytokine storm” was examined next. Sera from uninfected and infected (day 3 and day 6) WT and STAT1 KO mice were analyzed by multiplex assay to determine the levels of several cytokines and chemokines (Fig. 7 and Table 1). Of the 23 cytokines and chemokines analyzed, IL-2, IL-10, IL-12p40, CCL2, CCL4, CCL11, and CXCL1 had lower basal levels in STAT1 KO mice than in WT mice. However, the difference was not significant. At day 3 postinfection, only CCL2 and CCL4 showed a significant increase in WT mice, while IL-2 was significantly reduced (Fig. 7 and Table 1). At day 6 postinfection, CCL2, CXCL1, granulocyte colony-stimulating factor (G-CSF), and IL-5 were increased significantly in the sera of WT mice, while IL-12p70 was decreased significantly. In the sera of infected STAT1 KO mice, CCL2, CCL4, G-CSF, IL-5, IL-6, IL-10, IL-12p70, and IFN-γ were increased significantly at day 3 compared with uninfected controls (Fig. 7 and Table 1). At day 6 postinfection, CCL2, CCL4, G-CSF, and IL-10 remained significantly increased compared with uninfected mice but were lower than on day 3 postinfection, while the levels for IL-12p70 and IFN-γ returned to almost control levels. In contrast to this, the levels of IL-5 and IL-6 in sera of STAT1 KO mice further increased between day 3 and day 6 postinfection. In addition, at day 6 postinfection, IL-2, IL-3, and IL-9 were increased significantly in STAT1 KO mice compared with uninfected mice. The CXCL1 levels were significantly reduced at day 6 in STAT1 KO mice compared with uninfected mice. With the exception of CCL2, all cytokines and chemokines that were significantly increased in infected STAT1 KO mice (day 3 or day 6) were also significantly higher than in the corresponding infected WT mice. These findings demonstrate that LCMV infection of STAT1 KO mice causes a profound increase in several cytokines and chemokines in the serum. Early in the infection, factors important for innate immune cells, including granulocytes and macrophages (e.g., CCL2, CCL4, and G-CSF) dominated, followed later by increased levels of factors (e.g., IL-6, IL-2, IL-3, IL-9, and IL-10) important for T-cell responses.
Fig 7.
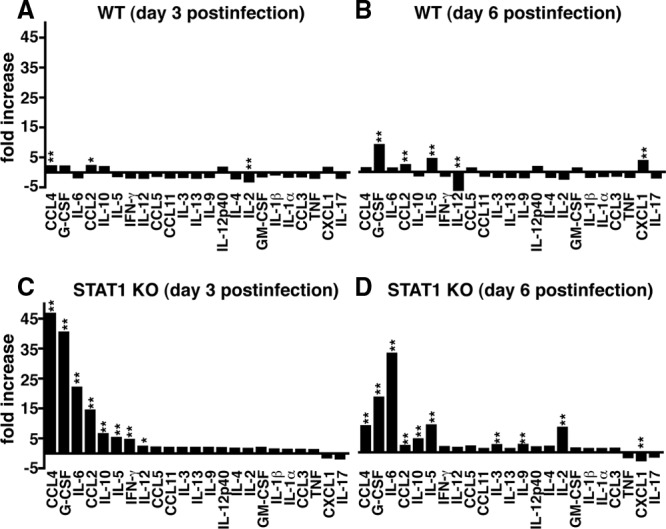
Several cytokines are increased significantly in the sera of infected STAT1 KO mice. In WT mice, a multiplex assay showed small to moderate but significant changes for CCL4, CCL2, and IL-2 levels at day 3 postinfection (A) and for G-CSF, CCL2, IL-5, IL-12, and CXCL1 levels at day 6 postinfection (B). In contrast, at day 3 postinfection (C), the sera of STAT1 KO mice contained significantly higher levels of CCL4, G-CSF, IL-6, CCL2, IL-10, IL-5, IFN-γ, and IL-12 p70 than those of sham-infected controls. (D) At day 6 postinfection, CCL4, G-CSF, IL-6, CCL2, IL-10, IL-5, IL-3, IL-9, and IL-2 were significantly elevated in the sera of LCMV-infected STAT1 KO mice compared with sham-infected mice. In STAT1 KO mice, CXCL1 levels were significantly lower at day 6 postinfection than in the sham-infected controls. *, P < 0.05; **, P < 0.01 compared with the uninfected control as determined by one-way ANOVA.
Table 1.
Levels of cytokines and chemokines in the sera of infected WT and STAT1 KO mice
| Cytokine or chemokine | Level (pg/ml) ±SEM (fold change)a |
|||||
|---|---|---|---|---|---|---|
| WT |
STAT1 KO |
|||||
| 0b | 3 | 6 | 0 | 3 | 6 | |
| IL–1α | 17 ± 1.0 | 12 ± 1.3 (−1.4) | 15 ± 0.6 (−1.2) | 15 ± 0.9 | 17 ± 1.2 (1.5) | 16 ± 1.1 (0.6) |
| IL–1β | 56 ± 9.6 | 37 ± 1.3 (−0.7) | 39 ± 1.9 (−1.4) | 35 ± 5.9 | 46 ± 2.6 (1.3) | 40 ± 0.9 (1.1) |
| IL–2 | 3.3 ± 0.7 | 1.1 ± 0.3 (−2.9) | 1.7 ± 0.1 (−2.0) | 1.6 ± 0.1 | 2.4 ± 0.1 (1.4) | 13.2 ± 2.3 (8.1) |
| IL–3 | 1.6 ± 0.2 | 1 ± 0.1 (−1.5) | 1 ± 0.1 (−1.5) | 0.8 ± 0.2 | 1.4 ± 0.2 (1.8) | 1.8 ± 0.2 (2.4) |
| IL–4 | 2.9 ± 0.3 | 1.5 ± 0.1 (−1.9) | 1.9 ± 0.2 (−1.5) | 1.8 ± 0.1 | 2.8 ± 0.2 (1.6) | 3.3 ± 0.3 (1.87) |
| IL–5 | 7.0 ± 0.4 | 5.9 ± 0.4 (−1.2) | 28 ± 2.3 (4.0) | 7.25 ± 0.8 | 37 ± 1.5 (5.1) | 65 ± 7.6 (9.0) |
| IL–6 | 8.7 ± 2.0 | 5.7 ± 0.2 (−1.5) | 8.9 ± 1.8 (1.0) | 5.8 ± 1.6 | 127 ± 18.0 (21.9) | 191 ± 24.1 (32.9) |
| IL–9 | 21 ± 4.5 | 14 ± 0.7 (−1.5) | 13 ± 1.7 (−1.6) | 13 ± 1.3 | 23 ± 1.0 (1.8) | 32 ± 5.6 (2.5) |
| IL–10 | 28 ± 2.7 | 49 ± 6.0 (1.8) | 27 ± 3.8 (−1.0) | 16 ± 2.6 | 104 ± 7.7 (6.4) | 70 ± 6.9 (4.3) |
| IL–12p40 | 47 ±2.2 | 73 ± 3.7 (1.6) | 72 ± 5.0 (1.5) | 33 ± 2.1 | 59 ± 9.2 (1.8) | 55 ± 7.0 (1.7) |
| IL–12p70 | 21 ± 2.7 | 12 ± 4.0 (−1.8) | 3.8 ± 1.3 (−5.7) | 12 ± 3.5 | 26 ± 2.1 (2.2) | 18 ± 1.8 (1.4) |
| IL–13 | 42 ± 4.3 | 24 ± 3.0 (−1.8) | 28 ± 2.2 (−1.5) | 26 ± 4.6 | 48 ± 4.9 (1.8) | 29 ± 3.2 (1.1) |
| IL–17 | 19 ± 2.1 | 11 ± 0.9 (−1.8) | 12 ± 0.8 (−1.7) | 13 ± 2.6 | 8 ± 1.6 (−1.7) | 9 ± 0.9 (−1.4) |
| CCL11 | 294 ± 9.3 | 177 ± 11.3 (−1.7) | 265 ± 9.4 (−1.1) | 184 ± 12.4 | 340 ± 28.5 (1.9) | 195 ± 21.6 (1.1) |
| G–CSF | 9.6 ± 0.4 | 18 ± 2.2 (1.9) | 81 ± 5.4 (8.4) | 11 ± 1.7 | 432 ± 66.9 (40.3) | 195 ± 28.8 (18) |
| GMc–CSF | 30 ± 1.8 | 23 ± 1.5 (−1.3) | 31 ± 1.0 (1.0) | 26 ± 1.6 | 37 ± 1.9 (1.4) | 35 ± 1.2 (1.3) |
| IFN–γ | 68 ± 7.8 | 40 ± 3.8 (−1.7) | 60 ± 1.9 (−1.1) | 41 ± 6.2 | 184 ± 25 (4.5) | 71 ± 10.1 (1.7) |
| CXCL1 | 22 ± 0.9 | 32 ± 3.1 (1.5) | 74 ± 4.6 (3.3) | 27 ± 3.0 | 22 ± 0.7 (−1.3) | 11 ± 1.3 (−2.6) |
| CCL2 | 79 ± 10.5 | 163 ± 20.2 (2.1) | 160 ± 13.0 (2.0) | 52 ± 5.5 | 738 ± 101.5 (14.3) | 109 ± 6.5 (2.1) |
| CCL3 | 113 ± 6.8 | 87 ± 4.9 (−1.3) | 102 ± 2.9 (−1.1) | 107 ± 14.9 | 124 ± 4.5 (1.2) | 125 ± 5.6 (1.2) |
| CCL4 | 13 ± 2.3 | 26 ± 2.1 (2.0) | 15 ± 1.0 (1.2) | 6.0 ± 1.2 | 279 ± 23.4 (46.6) | 52 ± 6.4 (8.7) |
| CCL5 | 85 ± 5.2 | 77 ± 1.9 (−1.1) | 91 ± 3.8 (1.1) | 73 ± 4.3 | 139 ± 4.7 (1.9) | 139 ± 4.3 (1.9) |
| TNF | 129 ± 21 | 73 ± 5.3 (−1.8) | 85 ± 2.4 (−1.5) | 119 ± 8.9 | 134 ± 19.7 (1.1) | 76 ± 11.6 (−1.6) |
Significant changes compared with uninfected mice are underlined.
Day postinfection.
GM, granulocyte-macrophage.
STAT1 KO mice have increased percentages of LCMV-specific CD4+ and CD8+ T cells.
To assess further the nature of the antiviral T-cell response in LCMV-infected WT and STAT1 KO mice, we analyzed for the presence of LCMV-specific CD4+ and CD8+ T cells that recognized MHC class II or MHC class I tetramers loaded with LCMV peptides, respectively. Less than 0.3% of CD4+ T cells bound MHC class II tetramers loaded with an irrelevant control peptide independent of the genotype or infection with LCMV (Fig. 8A and D). In WT mice, LCMV infection did not result in a significant increase in CD4+ T cells that recognized the LCMV-specific MHC class II tetramer at day 6 postinfection (Fig. 8B and D). In contrast, in infected STAT1 KO mice at day 6 postinfection, there was a significant increase in CD4+ T cells that recognized the LCMV-specific tetramer compared with uninfected STAT1 KO mice. Although there was an increase in CD8+ T cells that recognized LCMV-specific MHC class I tetramers in LCMV-infected WT mice compared with uninfected mice, it was not statistically significant (Fig. 8C and D). On the contrary, in STAT1 KO mice, infection with LCMV resulted in a significant increase in CD8+ T cells that recognized the virus peptide-specific tetramer. These results show that, compared with WT mice, STAT1 deficiency results in a significant early expansion of CD4+ and CD8+ T cells that are specific for LCMV.
Fig 8.

Increased percentages of virus-specific CD4+ and CD8+ T cells in LCMV-infected STAT1 KO mice. (A and D) Representative FACS results from lymph node cells stained with CD3, CD4, and control MHC class II tetramer showed no difference between WT and STAT1 KO mice sham injected or infected with LCMV. (B and D) There was no significant increase in CD4+ T cells from infected WT mice compared with uninfected WT mice that recognized LCMV GP 66-77 MHC class II. In contrast, significantly more CD4+ T cells from infected STAT1 KO mice than from uninfected STAT1 KO mice bound the LCMV GP66-77 MHC class II tetramer. (C and D) Similarly, significantly more CD8+ T cells from infected STAT1 KO mice bound the LCMV GP33-41 MHC class I than uninfected mice, whereas there was no difference between infected and uninfected WT mice. *, P < 0.05 as determined using the one-tailed Mann-Whitney U test.
The lethal disease caused by LCMV infection of STAT1 KO mice is mediated by CD4+ and not CD8+ T cells.
It is well documented that the development of LCM or the clearance of LCMV in adult WT mice infected i.c. or i.p. is mediated by antiviral CD8+ T cells (2, 29, 58, 105, 110). Furthermore, our results suggest that STAT1 deficiency is associated with an increased expansion of LCMV-specific CD8+ and CD4+ T cells. Therefore, we next examined whether CD8+ or CD4+ T cells play a role in the lethal disease in LCMV-infected STAT1 KO mice by depleting these cells following administration of specific neutralizing monoclonal antibodies. By FACS analysis of blood, it was shown that almost complete depletion of either CD4+ or CD8+ cells was achieved, with less than 0.5% positive cells remaining (Fig. 9A). In WT mice infected i.p. with LCMV, depletion of either CD4+ or CD8+ T cells had no impact on the clinical course (Fig. 9B). On the other hand, depletion of CD8+ T cells, but not CD4+ T cells, from WT mice infected i.c. prevented lethal LCM, confirming the effective depletion of CD8+ T cells (Fig. 9D). In contrast, depletion of CD8+ T cells in STAT1 KO mice infected either i.p. (Fig. 9C) or i.c. (Fig. 9E) with LCMV delayed significantly, but did not prevent, lethal disease. However, the depletion of CD4+ T cells in STAT1 KO mice infected i.p. or i.c. with LCMV resulted in complete protection from lethal disease, with all mice surviving >18 days or 35 days, respectively (Fig. 9C and E). The CD4+ T-cell-depleted, LCMV-infected STAT1 KO mice developed mild to moderate clinical signs, such as reduced spontaneous activity and ruffled fur around days 7 to 9 postinfection. By 2 weeks postinfection, no clinical signs were observable. Importantly, nondepleted and CD4+ T-cell-depleted LCMV-infected STAT1 KO mice showed similar loss of body weight (data not shown).
Fig 9.

CD4+ T cells are required for lethal disease in LCMV-infected STAT1 KO mice. (A) Compared with Ig-treated controls (left), mice administered neutralizing monoclonal antibodies against CD8 (middle) or CD4 (right) showed less than 0.5% remaining CD8+ and CD4+ cells, respectively. Depletion of CD4+ T cells had no impact on the survival of WT mice infected i.p. or i.c. with LCMV (B and D) but prevented lethal disease in STAT1 KO mice independent of the route of infection (C and E). In contrast, depletion of CD8+ T cells rescued WT mice infected i.c. with LCMV (D) but did not alter the clinical course of WT mice infected i.p (B) or of STAT1 KO mice infected i.c. (E) with LCMV. (C) Depletion of CD8+ T cells significantly (**, P < 0.05) increased the survival time of STAT1 KO mice infected i.p. with LCMV but did not prevent lethal disease. Prominent mononuclear infiltrates and tissue destruction were evident in the livers (F), kidneys (G), and lungs (H) of STAT1 KO mice infected i.p. with LCMV. In contrast, in similarly infected STAT1 KO mice that were depleted of CD4+ T cells, the livers (I), kidneys (J), and lungs (K) showed only minor mononuclear infiltrates and no obvious organ destruction.
While the livers, kidneys, and lungs of STAT1 KO mice infected i.p. with LCMV showed extensive leukocyte infiltration and concomitant tissue destruction (Fig. 3C, F, and I and 9F to H), CD4+ T-cell depletion in similarly infected STAT1 KO mice resulted in considerably reduced leukocyte infiltration and preservation of the tissue (Fig. 9I to K). In summary, these findings show that CD4+ T cells mediate the severe tissue immunopathology and death, but not weight loss, observed in STAT1 KO mice.
DISCUSSION
IFNs are critical for the innate and adaptive host responses against viral infection (92, 98). IFN-I-regulated gene expression depends largely on the presence of the canonical signaling molecules STAT1, STAT2, and IRF9, while IFN-II signaling is dependent largely on STAT1. Here, we studied the consequences of disrupted IFN-I and IFN-II signaling for the infection of mice with LCMV. Our findings revealed that the effects of STAT1 deficiency on the outcome of LCMV infection are distinct from either STAT2 or IRF9 deficiency and independent of IFN-II signaling. Despite increased innate immune responses, all three IFN-I signaling-deficient mice failed to control infection and developed signs of disease. However, only LCMV infection of mice deficient for STAT1 caused lethal disease that was associated with widely disseminated tissue immune pathology. This shows that STAT1 has a major additional protective function in LCMV infection. We further demonstrated that this protective function involves the prevention of a lethal CD4+ T-cell response that mediates extensive and catastrophic organ damage.
Not surprisingly, increased susceptibility of STAT1 KO mice to viral infection is not restricted to LCMV and has been observed for a variety of other viruses (17, 34, 44, 53, 69, 85, 94, 112). However, no common pathological mechanism appears to exist, as studies have suggested various causes ranging from a role for innate immune cells, such as monocytes, NK cells, and neutrophils (53, 77, 94), to a bias toward a Th2 response (34, 44, 112) and increased IL-17 levels (44). Moreover, the increased susceptibility of STAT1-deficient mice to virus infection is not universal (39, 95), demonstrating that the effects of STAT1 deficiency depend on the nature of the virus and its interaction with the host.
Our results showed that the effects of STAT1 deficiency in LCMV infection were detrimental and distinct from those of either IFN-I, IFN-II, or combined IFN-I and IFN-II signaling deficiency. The survival of IRF9 KO and STAT2 KO mice, as well as IRF9 KO × IFNGR KO mice, following infection with LCMV is concordant with previous observations reported for IFNAR-deficient mice (74, 83) and IFNAR × IFNGR double-deficient mice (100). Significantly, IFN-γ signaling is not only largely dispensable in the host response to LCMV (14, 80), but as we have shown here, is also not essential for the development of lethal disease in infected STAT1 KO mice.
Although IRF9 KO and STAT2 KO mice survived infection with LCMV, infection in these IFN-I signaling-deficient animals was not totally innocuous. Thus, like STAT1 KO mice, STAT2- and IRF9-deficient mice had signs of disease with marked weight loss that in IRF9 KO mice coincided with the increased tissue expression of many proinflammatory cytokine genes. This suggests that IFN-I signaling in the early stages of LCMV infection plays a major role in dampening the innate immune response. Consistent with this role in moderating innate immunity to LCMV infection, IFN-Is are known to be produced within 1 to 2 days following infection (25, 62, 65, 70, 71, 108) and are critical for the early antiviral response (23, 64, 100, 104).
Despite virus dissemination and the enhanced innate immune response in infected STAT2-, IRF9-, and STAT1-deficient mice, only STAT1-deficient animals exhibited lethality. In contrast to IRF9 KO mice, STAT1 KO mice developed a destructive and lethal immune response, demonstrating that STAT1 has a unique and critical role in preventing the emergence of a lethal immune response. This response to LCMV infection in STAT1 KO mice was characterized by florid leukocyte infiltrates in several organs and a “spillover” of several chemokines and cytokines into the serum. The distinctiveness of the immune response in STAT1 KO mice compared with IRF9 KO mice is further evident from the qualitative differences in cytokine expression. To what extent these qualitative differences in cytokine expression might contribute to the death or survival of STAT1 KO and IRF9 KO mice, respectively, remains to be elucidated. Notably, the leukocyte infiltrates in the STAT1 KO mice contained significant numbers of PMNs and monocytes/macrophages. In preliminary studies, we have found that antibody-mediated depletion of neutrophils had no effect on the outcome of disease in STAT1 KO mice (I. L. Campbell and M. J. Hofer, unpublished observation), arguing against a dominant role for these innate immune cells in the lethal disease.
The apparent lack of a central contribution by innate immune cells to lethal disease in LCMV-infected STAT1 KO mice suggested possible involvement of the adaptive immune response. Consistent with a role for the adaptive immune response, a high number of CD4+ and CD8+ T cells were present in the inflammatory infiltrates. As noted above, in WT mice, CD8+ T cells have a critical role in virus clearance or mediating LCM following i.p. or i.c. infection, respectively. Although we found anti-LCMV-specific CD8+ T cells were present, these cells were not essential for development of the lethal disease in infected STAT1 KO mice. In fact, the inability of the STAT1-, STAT2-, and IRF9-deficient mice to clear LCMV and the failure of STAT2 and IRF9-deficient mice infected intracranially with LCMV to develop LCM fits well with the reported reduced CTL activity and limited expansion of CD8+ T cells in IFNAR-deficient mice (3, 74). The basis for such a defect in the antiviral CD8+ T-cell response is currently unknown. Infection of mice with LCMV clone 13 causes functional exhaustion of CD8+ T cells mediated by coinhibitory T-cell receptors, such as PD-1 and LAG3 (13, 18) or the cytokine IL-10 (35), resulting in viral persistence. It is reasonable to suggest that similar mechanisms may contribute to functional exhaustion of T cells in IFN signaling-deficient mice, but this remains to be determined. However, the absence of significantly increased tissue IL-10 mRNA levels in LCMV-infected IRF9 KO mice argues against a role for IL-10 in the ineffective immune response in these animals.
Significantly, our results clearly demonstrated that CD4+ T cells were essential mediators of lethality in the LCMV-infected STAT1-deficient mice. This contrasts with the situation in WT mice, where CD4+ T cells are dispensable for virus clearance following peripheral infection with LCMV-Arm (1, 66, 87) or development of lethal LCM (58). Notably, however, these cells are required for maintaining CTL activity and ultimately elimination of virus in mice infected chronically with LCMV clone 13 (66). Furthermore, in LCMV-infected β2-microglobulin-deficient mice, CD4+ T cells are the principal effector cells that mediate wasting (32, 51). Interestingly, CD4+ T cells were not required for the weight loss and sickness behavior in the LCMV-infected STAT1 KO mice. Together, these findings indicate that, depending on the setting, the role of CD4+ T cells in the infection of mice with LCMV varies.
The reason for the expansion of pathogenic LCMV-specific CD4+ T cells in STAT1 KO mice infected with LCMV is unclear, but it is unlikely to be the consequence of an incapacitated CD8+ T-cell response or defective IFN-I signaling per se, since IRF9- and STAT2-deficient mice did not develop a destructive immune response. In recent years, regulatory T cells have been identified as being important in preventing the development of pathogenic CD4+ T-cell responses (reviewed in reference 60), and failure to generate these cells could account for the destructive T-cell response. However, the role of STAT1 in the development of regulatory T cells is unclear. While some studies reported that STAT1 is required for the effective generation of these cells (78, 81), others showed that STAT1 deficiency promoted (21, 22, 63, 101) or had no impact on (49, 76) the development of CD4+ regulatory T cells, and the role of regulatory T cells in the context of LCMV infection remains to be clarified.
The phenotype of the effector CD4+ T cells in our study also remains unresolved and is the subject of ongoing studies. Previous studies on RSV and severe acute respiratory syndrome (SARS) coronavirus reported a switch from a Th1 to a Th2 CD4+ T-cell response in the absence of STAT1 (34, 44, 112). Notably, the preponderance of granulocytes in the infiltrates of LCMV-infected STAT1 KO mice suggested that a similar mechanism may be involved. However, the tissue cytokine gene expression profiles did not reveal significant changes in the expression of classical Th2 cytokine genes. On the other hand, in several autoimmune disease models, STAT1 deficiency skewed the CD4+ T-cell differentiation toward a Th17 response (6, 16, 76, 103). Whether such skewing occurs in virus-infected STAT1 KO mice has not been reported. In the present study, tissue IL-17 mRNA levels were increased in both STAT1 and IRF9 KO mice infected with LCMV, suggesting that activation of the CD4+ Th17 lineage might occur as a more general response to the absence of IFN-I signaling.
In summary, our findings show that the canonical IFN-I signaling molecules STAT1, STAT2, and IRF9 are fundamental in the host response against LCMV. First, all three signaling factors are essential in limiting initial viral replication and spread, reflecting the well-established key role of canonical IFN-I signaling in antiviral defense. Second, all three signaling factors are involved in regulating the intensity of the early innate immune response to viral infection. Absence of each factor alone leads to an enhanced innate response, highlighting a more general contribution of IFN-I signaling to the regulation of the innate immune response triggered by LCMV infection. However, qualitative differences in the cytokine gene expression profile suggest there are different roles for these IFN-I signaling molecules in the regulation of the innate host response to LCMV. Third, the development of CD4+ T-cell-dependent lethal disease in LCMV-infected STAT1 mice, but not in IRF9 or STAT2 KO mice, points to a unique role for STAT1 in the regulation of the adaptive immune response to LCMV. Together, our findings highlight the complexity of IFN-I signaling in the regulation of innate and adaptive antiviral immunity and further demonstrate the potent and divergent biological effects of noncanonical IFN signaling pathways.
ACKNOWLEDGMENTS
This work was supported in part by project grant 512407 to I.L.C. from the National Health and Medical Research Council of Australia. M.J.H. was a postdoctoral fellow from the Deutsche Forschungsgemeinschaft (DFG HO3298/1-1). W.L. was the recipient of an Australian Postgraduate Scholarship.
We thank Laura Parker for technical assistance and Claire Thompson for helpful comments on the manuscript.
Footnotes
Published ahead of print 11 April 2012
REFERENCES
- 1. Ahmed R, Butler LD, Bhatti L. 1988. T4+ T helper cell function in vivo: differential requirement for induction of antiviral cytotoxic T-cell and antibody responses. J. Virol. 62:2102–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmed R, King CC, Oldstone MB. 1987. Virus-lymphocyte interaction: T cells of the helper subset are infected with lymphocytic choriomeningitis virus during persistent infection in vivo. J. Virol. 61:1571–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aichele P, et al. 2006. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 176:4525–4529 [DOI] [PubMed] [Google Scholar]
- 4. Allan JE, Doherty PC. 1990. Binding of monoclonal antibodies and T cell effector function in vivo. Hybridoma 9:9–15 [DOI] [PubMed] [Google Scholar]
- 5. Alsharifi M, Mullbacher A, Regner M. 2008. Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 86:239–245 [DOI] [PubMed] [Google Scholar]
- 6. Amadi-Obi A, et al. 2007. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat. Med. 13:711–718 [DOI] [PubMed] [Google Scholar]
- 7. Ank N, et al. 2006. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 80:4501–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ank N, West H, Paludan SR. 2006. IFN-lambda: novel antiviral cytokines. J. Interferon Cytokine Res. 26:373–379 [DOI] [PubMed] [Google Scholar]
- 9. Asensio VC, Campbell IL. 1997. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 71:7832–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Asensio VC, Kincaid C, Campbell IL. 1999. Chemokines and the inflammatory response to viral infection in the central nervous system with a focus on lymphocytic choriomeningitis virus. J. Neurovirol. 5:65–75 [DOI] [PubMed] [Google Scholar]
- 11. Asselin-Paturel C, et al. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150 [DOI] [PubMed] [Google Scholar]
- 12. Baenziger J, Hengartner H, Zinkernagel RM, Cole GA. 1986. Induction or prevention of immunopathological disease by cloned cytotoxic T cell lines specific for lymphocytic choriomeningitis virus. Eur. J. Immunol. 16:387–393 [DOI] [PubMed] [Google Scholar]
- 13. Barber DL, et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687 [DOI] [PubMed] [Google Scholar]
- 14. Bartholdy C, Christensen JP, Wodarz D, Thomsen AR. 2000. Persistent virus infection despite chronic cytotoxic T-lymphocyte activation in gamma interferon-deficient mice infected with lymphocytic choriomeningitis virus. J. Virol. 74:10304–10311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barton LL, Mets MB, Beauchamp CL. 2002. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am. J. Obstet. Gynecol. 187:1715–1716 [DOI] [PubMed] [Google Scholar]
- 16. Batten M, et al. 2006. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat. Immunol. 7:929–936 [DOI] [PubMed] [Google Scholar]
- 17. Bente DA, et al. 2010. Pathogenesis and immune response of Crimean-Congo hemorrhagic fever virus in a STAT-1 knockout mouse model. J. Virol. 84:11089–11100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blackburn SD, et al. 2009. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Borden EC, et al. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6:975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Byrne JA, Oldstone MB. 1984. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus: clearance of virus in vivo. J. Virol. 51:682–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caretto D, Katzman SD, Villarino AV, Gallo E, Abbas AK. 2010. Cutting edge: the Th1 response inhibits the generation of peripheral regulatory T cells. J. Immunol. 184:30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang JH, Kim YJ, Han SH, Kang CY. 2009. IFN-gamma-STAT1 signal regulates the differentiation of inducible Treg: potential role for ROS-mediated apoptosis. Eur. J. Immunol. 39:1241–1251 [DOI] [PubMed] [Google Scholar]
- 23. Christensen JE, et al. 2009. Fulminant lymphocytic choriomeningitis virus-induced inflammation of the CNS involves a cytokine-chemokine-cytokine-chemokine cascade. J. Immunol. 182:1079–1087 [DOI] [PubMed] [Google Scholar]
- 24. Christensen JP, Marker O, Thomsen AR. 1994. The role of CD4+ T cells in cell-mediated immunity to LCMV: studies in MHC class I and class II deficient mice. Scand. J. Immunol. 40:373–382 [DOI] [PubMed] [Google Scholar]
- 25. Dalod M, et al. 2002. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 195:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deibel R, Schryver GD. 1976. Viral antibody in the cerebrospinal fluid of patients with acute central nervous system infections. J. Clin. Microbiol. 3:397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deibel R, Woodall JP, Decher WJ, Schryver GD. 1975. Lymphocytic choriomeningitis virus in man. Serologic evidence of association with pet hamsters. JAMA 232:501–504 [PubMed] [Google Scholar]
- 28. Diebold SS, et al. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 424:324–328 [DOI] [PubMed] [Google Scholar]
- 29. Dixon JE, Allan JE, Doherty PC. 1987. The acute inflammatory process in murine lymphocytic choriomeningitis is dependent on Lyt-2+ immune T cells. Cell. Immunol. 107:8–14 [DOI] [PubMed] [Google Scholar]
- 30. Doherty PC, Allan JE, Lynch F, Ceredig R. 1990. Dissection of an inflammatory process induced by CD8+ T cells. Immunol. Today 11:55–59 [DOI] [PubMed] [Google Scholar]
- 31. Doherty PC, Ceredig R, Allan JE. 1988. Immunogenetic analysis of cellular interactions governing the recruitment of T lymphocytes and monocytes in lymphocytic choriomeningitis virus-induced immunopathology. Clin. Immunol. Immunopathol. 47:19–26 [DOI] [PubMed] [Google Scholar]
- 32. Doherty PC, Hou S, Southern PJ. 1993. Lymphocytic choriomeningitis virus induces a chronic wasting disease in mice lacking class I major histocompatibility complex glycoproteins. J. Neuroimmunol. 46:11–17 [DOI] [PubMed] [Google Scholar]
- 33. Durbin JE, Hackenmiller R, Simon MC, Levy DE. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443–450 [DOI] [PubMed] [Google Scholar]
- 34. Durbin JE, et al. 2002. The role of IFN in respiratory syncytial virus pathogenesis. J. Immunol. 168:2944–2952 [DOI] [PubMed] [Google Scholar]
- 35. Ejrnaes M, et al. 2006. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 203:2461–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fischer SA, et al. 2006. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354:2235–2249 [DOI] [PubMed] [Google Scholar]
- 37. Fung-Leung WP, Kundig TM, Zinkernagel RM, Mak TW. 1991. Immune response against lymphocytic choriomeningitis virus infection in mice without CD8 expression. J. Exp. Med. 174:1425–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garcia-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882 [DOI] [PubMed] [Google Scholar]
- 39. Gil MP, et al. 2001. Biologic consequences of Stat1-independent IFN signaling. Proc. Natl. Acad. Sci. U. S. A. 98:6680–6685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gil MP, Salomon R, Louten J, Biron CA. 2006. Modulation of STAT1 protein levels: a mechanism shaping CD8 T-cell responses in vivo. Blood 107:987–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gough DJ, Levy DE, Johnstone RW, Clarke CJ. 2008. IFNgamma signaling—does it mean JAK-STAT? Cytokine Growth Factor Rev. 19:383–394 [DOI] [PubMed] [Google Scholar]
- 42. Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. 2005. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 22:247–257 [DOI] [PubMed] [Google Scholar]
- 43. Haque SJ, Williams BR. 1994. Identification and characterization of an interferon (IFN)-stimulated response element-IFN-stimulated gene factor 3-independent signaling pathway for IFN-alpha. J. Biol. Chem. 269:19523–19529 [PubMed] [Google Scholar]
- 44. Hashimoto K, et al. 2005. Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J. Allergy Clin. Immunol. 116:550–557 [DOI] [PubMed] [Google Scholar]
- 45. Hobbs MV, et al. 1993. Patterns of cytokine gene expression by CD4+ T cells from young and old mice. J. Immunol. 150:3602–3614 [PubMed] [Google Scholar]
- 46. Hofer MJ, Carter SL, Muller M, Campbell IL. 2008. Unaltered neurological disease and mortality in CXCR3-deficient mice infected intracranially with lymphocytic choriomeningitis virus-Armstrong. Viral Immunol. 21:425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofer MJ, Li W, Lim SL, Campbell IL. 2010. The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9. J. Neurosci. 30:1149–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang S, et al. 1993. Immune response in mice that lack the interferon-gamma receptor. Science 259:1742–1745 [DOI] [PubMed] [Google Scholar]
- 49. Huber M, et al. 2008. IL-27 inhibits the development of regulatory T cells via STAT3. Int. Immunol. 20:223–234 [DOI] [PubMed] [Google Scholar]
- 50. Jung A, et al. 2008. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 82:196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kamperschroer C, Quinn DG. 2002. The role of proinflammatory cytokines in wasting disease during lymphocytic choriomeningitis virus infection. J. Immunol. 169:340–349 [DOI] [PubMed] [Google Scholar]
- 52. Kang SS, McGavern DB. 2008. Lymphocytic choriomeningitis infection of the central nervous system. Front. Biosci. 13:4529–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW., 4th 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578 [DOI] [PubMed] [Google Scholar]
- 54. Kimura T, et al. 1996. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells 1:115–124 [DOI] [PubMed] [Google Scholar]
- 55. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202:637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lang PA, et al. 2010. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 52:25–32 [DOI] [PubMed] [Google Scholar]
- 57. Lehmann-Grube F, Lohler J, Utermohlen O, Gegin C. 1993. Antiviral immune responses of lymphocytic choriomeningitis virus-infected mice lacking CD8+ T lymphocytes because of disruption of the beta 2-microglobulin gene. J. Virol. 67:332–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leist TP, Cobbold SP, Waldmann H, Aguet M, Zinkernagel RM. 1987. Functional analysis of T lymphocyte subsets in antiviral host defense. J. Immunol. 138:2278–2281 [PubMed] [Google Scholar]
- 59. Li X, Leung S, Qureshi S, Darnell JE, Jr, Stark GR. 1996. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-alpha. J. Biol. Chem. 271:5790–5794 [DOI] [PubMed] [Google Scholar]
- 60. Liu H, Leung BP. 2006. CD4+CD25+ regulatory T cells in health and disease. Clin. Exp. Pharmacol. Physiol. 33:519–524 [DOI] [PubMed] [Google Scholar]
- 61. Lledo L, Gegundez MI, Saz JV, Bahamontes N, Beltran M. 2003. Lymphocytic choriomeningitis virus infection in a province of Spain: analysis of sera from the general population and wild rodents. J. Med. Virol. 70:273–275 [DOI] [PubMed] [Google Scholar]
- 62. Louten J, van Rooijen N, Biron CA. 2006. Type 1 IFN deficiency in the absence of normal splenic architecture during lymphocytic choriomeningitis virus infection. J. Immunol. 177:3266–3272 [DOI] [PubMed] [Google Scholar]
- 63. Ma H, et al. 2011. Absence of Stat1 in donor CD4 T cells promotes the expansion of Tregs and reduces graft-versus-host disease in mice. J. Clin. Invest. 121:2554–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mack EA, Kallal LE, Demers DA, Biron CA. 2011. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. MBio 2:e00169–11 doi:10.1128/mBio.00169-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Malmgaard L, Salazar-Mather TP, Lewis CA, Biron CA. 2002. Promotion of alpha/beta interferon induction during in vivo viral infection through alpha/beta interferon receptor/STAT1 system-dependent and -independent pathways. J. Virol. 76:4520–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Matloubian M, Concepcion RJ, Ahmed R. 1994. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 68:8056–8063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Matsumoto M, et al. 1999. Activation of the transcription factor ISGF3 by interferon-gamma. Biol. Chem. 380:699–703 [DOI] [PubMed] [Google Scholar]
- 68. Matullo CM, O'Regan KJ, Hensley H, Curtis M, Rall GF. 2010. Lymphocytic choriomeningitis virus-induced mortality in mice is triggered by edema and brain herniation. J. Virol. 84:312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Meier KC, Gardner CL, Khoretonenko MV, Klimstra WB, Ryman KD. 2009. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 5:e1000614 doi:10.1371/journal.ppat.1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Merigan TC, Oldstone MB, Welsh RM. 1977. Interferon production during lymphocytic choriomeningitis virus infection of nude and normal mice. Nature 268:67–68 [DOI] [PubMed] [Google Scholar]
- 71. Miyagi T, et al. 2007. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204:2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Montoya M, Edwards MJ, Reid DM, Borrow P. 2005. Rapid activation of spleen dendritic cell subsets following lymphocytic choriomeningitis virus infection of mice: analysis of the involvement of type 1 IFN. J. Immunol. 174:1851–1861 [DOI] [PubMed] [Google Scholar]
- 73. Morrow AN, Schmeisser H, Tsuno T, Zoon KC. 2011. A novel role for IFN-stimulated gene factor 3II in IFN-gamma signaling and induction of antiviral activity in human cells. J. Immunol. 186:1685–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Muller U, et al. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921 [DOI] [PubMed] [Google Scholar]
- 75. Nansen A, et al. 1998. Role of interferon-gamma in the pathogenesis of LCMV-induced meningitis: unimpaired leucocyte recruitment, but deficient macrophage activation in interferon-gamma knock-out mice. J. Neuroimmunol. 86:202–212 [DOI] [PubMed] [Google Scholar]
- 76. Neufert C, et al. 2007. IL-27 controls the development of inducible regulatory T cells and Th17 cells via differential effects on STAT1. Eur. J. Immunol. 37:1809–1816 [DOI] [PubMed] [Google Scholar]
- 77. Nguyen KB, et al. 2002. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169:4279–4287 [DOI] [PubMed] [Google Scholar]
- 78. Nishibori T, Tanabe Y, Su L, David M. 2004. Impaired development of CD4+ CD25+ regulatory T cells in the absence of STAT1: increased susceptibility to autoimmune disease. J. Exp. Med. 199:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Oldstone MB, et al. 1999. Use of a high-affinity peptide that aborts MHC-restricted cytotoxic T lymphocyte activity against multiple viruses in vitro and virus-induced immunopathologic disease in vivo. Virology 256:246–257 [DOI] [PubMed] [Google Scholar]
- 80. Ou R, Zhou S, Huang L, Moskophidis D. 2001. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J. Virol. 75:8407–8423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ouaked N, et al. 2009. Regulation of the foxp3 gene by the Th1 cytokines: the role of IL-27-induced STAT1. J. Immunol. 182:1041–1049 [DOI] [PubMed] [Google Scholar]
- 82. Ousman SS, Campbell IL. 2005. Regulation of murine interferon regulatory factor gene expression in the central nervous system determined by multiprobe RNase protection assay. Methods Mol. Med. 116:115–134 [DOI] [PubMed] [Google Scholar]
- 83. Ousman SS, Wang J, Campbell IL. 2005. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J. Virol. 79:7514–7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Park C, Li S, Cha E, Schindler C. 2000. Immune response in Stat2 knockout mice. Immunity 13:795–804 [DOI] [PubMed] [Google Scholar]
- 85. Pasieka TJ, Lu B, Leib DA. 2008. Enhanced pathogenesis of an attenuated herpes simplex virus for mice lacking Stat1. J. Virol. 82:6052–6055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pestka S, Krause CD, Walter MR. 2004. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202:8–32 [DOI] [PubMed] [Google Scholar]
- 87. Rahemtulla A, et al. 1991. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 353:180–184 [DOI] [PubMed] [Google Scholar]
- 88. Ritchie KJ, et al. 2004. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 10:1374–1378 [DOI] [PubMed] [Google Scholar]
- 89. Roebroek RM, Postma BH, Dijkstra UJ. 1994. Aseptic meningitis caused by the lymphocytic choriomeningitis virus. Clin. Neurol. Neurosurg. 96:178–180 [DOI] [PubMed] [Google Scholar]
- 90. Rousseau MC, Saron MF, Brouqui P, Bourgeade A. 1997. Lymphocytic choriomeningitis virus in southern France: four case reports and a review of the literature. Eur. J. Epidemiol. 13:817–823 [DOI] [PubMed] [Google Scholar]
- 91. Sandberg K, et al. 1994. Altered tissue distribution of viral replication and T cell spreading is pivotal in the protection against fatal lymphocytic choriomeningitis in mice after neutralization of IFN-alpha/beta. J. Immunol. 153:220–231 [PubMed] [Google Scholar]
- 92. Sen GC, Ransohoff RM. 1993. Interferon-induced antiviral actions and their regulation. Adv. Virus Res. 42:57–102 [DOI] [PubMed] [Google Scholar]
- 93. Seo YJ, Hahm B. 2010. Type I interferon modulates the battle of host immune system against viruses. Adv. Appl. Microbiol. 73:83–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shornick LP, et al. 2008. Airway epithelial versus immune cell Stat1 function for innate defense against respiratory viral infection. J. Immunol. 180:3319–3328 [DOI] [PubMed] [Google Scholar]
- 95. Shresta S, et al. 2005. Critical roles for both STAT1-dependent and STAT1-independent pathways in the control of primary dengue virus infection in mice. J. Immunol. 175:3946–3954 [DOI] [PubMed] [Google Scholar]
- 96. Shuai K, Schindler C, Prezioso VR, Darnell JE., Jr 1992. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science 258:1808–1812 [DOI] [PubMed] [Google Scholar]
- 97. Siegal FP, et al. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science 284:1835–1837 [DOI] [PubMed] [Google Scholar]
- 98. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264 [DOI] [PubMed] [Google Scholar]
- 99. van Boxel-Dezaire AH, Rani MR, Stark GR. 2006. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 25:361–372 [DOI] [PubMed] [Google Scholar]
- 100. van den Broek MF, Muller U, Huang S, Aguet M, Zinkernagel RM. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 69:4792–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. VanDeusen JB, et al. 2006. STAT-1-mediated repression of monocyte interleukin-10 gene expression in vivo. Eur. J. Immunol. 36:623–630 [DOI] [PubMed] [Google Scholar]
- 102. Vanzee BE, et al. 1975. Lymphocytic choriomeningitis in university hospital personnel. Clinical features. Am. J. Med. 58:803–809 [DOI] [PubMed] [Google Scholar]
- 103. Villarino AV, Gallo E, Abbas AK. 2010. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J. Immunol. 185:6461–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wacher C, et al. 2007. Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J. Virol. 81:860–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Walsh CM, et al. 1994. Immune function in mice lacking the perforin gene. Proc. Natl. Acad. Sci. U. S. A. 91:10854–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Warkel RL, et al. 1973. Fatal acute meningoencephalitis due to lymphocytic choriomeningitis virus. Neurology 23:198–203 [DOI] [PubMed] [Google Scholar]
- 107. Wright R, et al. 1997. Congenital lymphocytic choriomeningitis virus syndrome: a disease that mimics congenital toxoplasmosis or cytomegalovirus infection. Pediatrics 100:E9 doi:10.1542/peds.100.1.e9 [DOI] [PubMed] [Google Scholar]
- 108. Zhou S, et al. 2010. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 84:9452–9462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zimmermann A, et al. 2005. A cytomegaloviral protein reveals a dual role for STAT2 in IFN-{gamma} signaling and antiviral responses. J. Exp. Med. 201:1543–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zinkernagel RM, Doherty PC. 1979. MHC-restricted cytotoxic T cells: studies on the biological role of polymorphic major transplantation antigens determining T-cell restriction-specificity, function, and responsiveness. Adv. Immunol. 27:51–177 [DOI] [PubMed] [Google Scholar]
- 111. Zinkernagel RM, Doherty PC. 1997. The discovery of MHC restriction. Immunol. Today 18:14–17 [DOI] [PubMed] [Google Scholar]
- 112. Zornetzer GA, et al. 2010. Transcriptomic analysis reveals a mechanism for a prefibrotic phenotype in STAT1 knockout mice during severe acute respiratory syndrome coronavirus infection. J. Virol. 84:11297–11309 [DOI] [PMC free article] [PubMed] [Google Scholar]