

Abstract
Paramyxoviruses initiate entry through the concerted action of the tetrameric attachment glycoprotein (HN, H, or G) and the trimeric fusion glycoprotein (F). The ectodomains of HN/H/G contain a stalk region important for oligomeric stability and for the F triggering resulting in membrane fusion. Paramyxovirus HN, H, and G form a dimer-of-dimers consisting of disulfide-linked dimers through their stalk domain cysteines. The G attachment protein stalk domain of the highly pathogenic Nipah virus (NiV) contains a distinct but uncharacterized cluster of three cysteine residues (C146, C158, C162). On the basis of a panoply of assays, we report that C158 and C162 of NiV-G likely mediate covalent subunit dimerization, while C146 mediates the stability of higher-order oligomers. For HN or H, mutation of stalk cysteines attenuates but does not abrogate the ability to trigger fusion. In contrast, the NiV-G stalk cysteine mutants were completely deficient in triggering fusion, even though they could still bind the ephrinB2 receptor and associate with F. Interestingly, all cysteine stalk mutants exhibited constitutive exposure of the Mab45 receptor binding-enhanced epitope, previously implicated in F triggering. The enhanced binding of Mab45 to the cysteine mutants relative to wild-type NiV-G, without the addition of the receptor, implicates the stalk cysteines in the stabilization of a pre-receptor-bound conformation and the regulation of F triggering. Sequence alignments revealed that the stalk cysteines were adjacent to a proline-rich microdomain unique to the Henipavirus genus. Our data propose that the cysteine cluster in the NiV-G stalk functions to maintain oligomeric stability but is more importantly involved in stabilizing a unique microdomain critical for triggering fusion.
INTRODUCTION
Nipah virus (NiV) is an emergent, highly pathogenic zoonotic agent classified as a risk group 4 pathogen (22). NiV and Hendra virus (HeV) are members of the genus Henipavirus within the subfamily Paramyxovirinae. NiV was originally identified in 1999 as the infectious agent responsible for causing severe encephalitis and respiratory disease in humans and livestock in Malaysia (11, 14, 16). This emergent zoonotic disease has spread to Singapore, India, and Bangladesh, with recurrent outbreaks continuing in Bangladesh almost yearly for the past decade. Cited causes of NiV infections in Bangladesh include drinking contaminated palm date juice and close contacts of individuals with pteropid fruit bats, considered the natural reservoir host (13, 24, 28). What was first seen as a disease with little to no human-to-human transmission is now known to have a human-to-human transmission rate of approximately 50% in recent outbreaks, with mortality rates as high as 70% (23). With no effective treatments available, it is essential to better understand the fundamental mechanisms of infection in order to aid in the design of efficacious therapeutics.
To initiate paramyxovirus entry into target cells, the viral lipid membrane must merge with the cell membrane (17, 18, 36). To overcome the energy barrier required for viral and cell membrane mixing, paramyxoviruses have evolved the use of two surface glycoproteins to drive fusion. One glycoprotein, the attachment protein, functions in recognition and binding to cellular receptors. Receptor binding results in allosteric triggering of the second protein, termed the fusion (F) protein, which is responsible for the fusion of viral envelopes with cell membranes. The F protein shares structural similarities classically described for the class I fusion proteins of influenza virus and HIV. The trimeric mature F protein requires cleavage of the precursor F0 protein into disulfide-linked F1/F2 subunits, which are held in a metastable conformation prior to fusion. Triggering of the F protein during fusion results in drastic structural transitions, first involving the insertion of a hydrophobic fusion peptide into the host membrane and ending in the folding of the N- and C-terminal α-helical heptad repeat (HR) domains within each trimer, in a process termed six-helix bundle formation. The postfusion F conformation is a highly stable and irreversible state.
Paramyxovirus attachment proteins are designated HN, H, or G depending on their hemagglutinin (H) and/or neuraminidase (N) activity. The HN attachment proteins of Newcastle disease virus (NDV) and most paramyxoviruses bind and cleave sialic acid moieties, while H and G proteins bind to protein receptors. Even though the H attachment proteins of morbilliviruses, such as measles virus (MeV), bind to protein-based receptors, they retain hemagglutinin activity, albeit through binding of the protein receptor rather than through sialic acid. Unlike those of other paramyxoviruses, the henipavirus G (HNV-G) attachment proteins are devoid of both hemagglutinin and neuraminidase function but bind to cellular receptors ephrinB2 and ephrinB3 (6, 30, 31). Paramyxovirus attachment proteins are all type II integral membrane proteins that form noncovalently associated tetramers consisting of dimers-of-dimers, which are typically covalently linked within each dimer but not between dimers. The C-terminal globular head or receptor binding domain connects to the N-terminal cytoplasmic tail and transmembrane region through a highly flexible stalk domain. Crystal structures of the NiV-G and HeV-G globular domains reveal the characteristic six-bladed beta-propeller seen in other paramyxovirus HN and H structures (9, 37). Regions of the HN stalk domains for NDV and parainfluenza virus 5 (PIV5), recently crystalized, show a parallel tetrameric α-helical coiled-coil configuration simply termed a four-helix bundle (4HB) (7, 39). Although no structural information exists for the H or G attachment protein stalk domain, secondary-structure predictions and alignments support the existence of a similar α-helical structure (4, 20).
The fact that receptor attachment and fusion activities reside on two separate paramyxovirus proteins indicates that these functions are mechanistically linked. Significant attempts have been made at understanding the role of the attachment protein in triggering the conformation cascade in F associated with fusion with regard to HN and H proteins (15, 33). As studies regarding the mechanism of F triggering by HNV-G proteins emerge, comparison to HN and H systems are beginning to reveal fundamental similarities, as well as differences, in their triggering signals (19).
Identification of the residues or microdomains in each glycoprotein that are required for functional interactions remains a central issue important for full understanding of the molecular mechanisms of paramyxovirus entry. Abundant evidence suggests a critical role for the stalk domain of attachment proteins in F triggering (15). The stalk domain has been shown to self-oligomerize and to form homotypic F-specific interactions. In the past, regions in the globular domain have also been suggested to have direct interactions with F required for fusion triggering; however, recent studies argue against such physical interactions with the H or HN protein. For example, the MeV H stalk domain could tolerate extensions of as much as 50% of its predicted normal length while retaining fusion promotion activity (32). Presumably, these additions heighten the position of the globular head, including regions of the distal stalk domain, beyond any specific contacts with MeV F. In another study using chimeric attachment proteins, the NiV-G head domain could transmit an ephrinB2 protein binding signal to the NDV stalk, instead of the typical carbohydrate binding signal, to promote fusion with NDV F (27). This result confirms previous chimera data regarding the HN stalk domain in specific contacts with homotypic F proteins (15). Similarly, a chimeric protein comprising an NDV HN head domain with a NiV-G stalk was shown to promote fusion with NiV-F, thus supporting a general hypothesis that the site of functional paramyxovirus glycoprotein interactions in the attachment protein localizes to the stalk domain (35).
The 4HB stalk domain structure seen in PIV5 HN has a hydrophobic core region with 7-residue repeat (3, 4) and 11-residue repeat (3-4-4) stretches (7). HNV-G proteins also contain hydrophobic residues with both 7-mer and 11-mer repeat patterns in the membrane-proximal regions of their stalks. While several mutations of hydrophobic isoleucine residues in the HeV-G stalk domain were found to inhibit F triggering, some were fusion permissive, highlighting the potential existence of helical and nonhelical subdomains within the stalk (4). Indeed, the henipavirus attachment protein stalks are approximately 40 amino acids (aa) longer than HN stalks (stalk lengths, 78 aa for PIV5 HN, 74 aa for NDV HN, and 118 aa for HNV-G), and while the HN 4HB may align to regions in G, the extra G stalk residues may confer unique functional properties.
HNV-G proteins contain a distinct cluster of three cysteine residues (C146, C158, C162) located in a distal portion of the stalk domain. Cysteines in the stalk domains of HN/H proteins have a pivotal role in disulfide-linked dimerization. However, the removal of these intermolecular disulfide bonds does not eliminate the fusion-triggering capabilities of either HN or H (26, 34). Stalk extension in the MeV H protein also indicates that the location of these disulfide bonds is not important for triggering, since residues inserted into a central stalk region heighten the positions of the intermolecular bonds yet remain fusogenic (32). In this study, we used mutagenesis to understand the role of cysteine residues in the Nipah virus G stalk. Our present analysis indicates that these cysteines not only play a role in stabilizing the oligomeric state of G through disulfide-linked dimerization but also help maintain G in a pre-receptor-bound conformation. Additionally, the cysteine cluster in a distal section of the G stalk was found to lie near a proline-rich microdomain unique to the henipaviruses among Paramyxovirinae genera. We propose that the most important function of the cysteine cluster is to ensure the proper folding of the proline-rich microdomain, which is critical to the conformational stability associated with F triggering.
MATERIALS AND METHODS
Cells and culture conditions.
All cell lines were maintained in a culture medium supplemented with 10% fetal bovine serum (FBS) at 37°C under 5% CO2. 293T (human embryonic kidney) cells were cultured in Dulbecco's modified Eagle's medium, Vero (African green monkey kidney) cells in alpha minimum essential medium, and CHO-pgsA745 cells in Ham's F-12 medium. CHOpgsA745 is a mutant cell line derived from Chinese hamster ovary (CHO) K1 cells that lack endogenous expression of heparin sulfate proteoglycans. CHO-pgsA745 cells stably expressing ephrinB2 (CHOB2 cells) have been described previously (31). CHOB2 cells were incubated in the presence of G-418 (500 μg/ml) during culture.
Plasmids and antibodies.
Each NiV envelope glycoprotein gene was subcloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA) under the control of a cytomegalovirus (CMV) promoter. Envelope nucleotide sequences of NiV-F and NiV-G were codon optimized and synthesized by GeneArt (Germany) (GenBank accession numbers AY816748 and AY816745, respectively). NiV-G was tagged at the C terminus with hemagglutinin (HA), while NiV-F was tagged with AU1 at the C terminus. The production of rabbit polyclonal anti-G antibodies (806), rabbit polyclonal anti-F antibodies, and rabbit monoclonal anti-G antibodies Mab26 and Mab45 has been described previously (1, 29). The soluble ephrinB2-Fc fusion protein (B2-Fc), containing ectodomain residues 27 to 227 of mouse ephrinB2, was purchased from R&D Systems (Minneapolis, MN).
Construction of cysteine mutant NiV-G glycoproteins.
The HA-tagged NiV-G expression plasmid served as a template for NiV-G stalk domain cysteine site-directed mutagenesis using a QuikChange II kit (Stratagene, Cedar Creek, TX). NiV-G cysteine residues 146, 158, and 162 were individually converted to serine residues. This amino acid substitution results in minimal secondary effects on the protein structure, since the mutation consists of a sulfur-to-oxygen substitution. NIV-G with the cysteine-to-serine change at position 146 is referred to as C1, while NIV-G with a similar change at 158 or 162 is termed C2 or C3, respectively.
Nonreducing gel electrophoresis.
Clarified cell lysates expressing NiV wild-type (WT) or mutant glycoproteins were prepared 24 h after transfection from 293T cells using 1% Triton X-100 in phosphate-buffered saline (PBS) as the lysis buffer. Purified pseudotyped virus displaying NiV-G, C1, or C2 with NiV-F were mixed with lysis buffer directly. NuPAGE LDS sample buffer (Invitrogen) was added to the lysate samples, which were heated at 99°C for 10 min, followed by separation on 4%-to-12% NuPAGE Bis-Tris precast polyacrylamide gels (Invitrogen). NuPAGE antioxidant (Invitrogen) was added to the running buffer in the cathode chamber during gel electrophoresis to prevent the reoxidation of sensitive amino acids. Samples were transferred to polyvinylidene difluoride (PVDF) membranes, and the NiV-G proteins were detected with horseradish peroxidase (HRP)-conjugated anti-HA antibodies (Thermo Scientific, Waltham, MA).
BN-PAGE analysis.
Cell surface-expressed and virus surface-displayed NiV glycoproteins were subjected to analysis by blue native polyacrylamide gel electrophoresis (BN-PAGE). For BN-PAGE analysis of transiently expressed NiV glycoproteins, 293T cells were transfected with NiV-G, C1, C2, or C3 with or without the NiV-F expression plasmid. At 24 h posttransfection, the medium was removed, the cells were washed in PBS, and BN-PAGE sample buffer (Invitrogen) containing digitonin (0.5%) or Triton X-100 (0.25%) was added to the pelleted cells. Cells were lysed for 15 min on ice. After lysis, the samples were centrifuged at 20,000 × g for 30 min at 4°C. Purified pseudotyped virus displaying NiV-G, C1, or C2 with NiV-F were mixed with BN-PAGE sample buffer containing digitonin (0.5%) or Triton X-100 (0.25%) directly. Coomassie blue G-250 was added to the samples, followed by separation on 4%-to-12% native PAGE gels (Invitrogen). Samples were transferred to PVDF membranes, and the NiV-G proteins were detected with anti-HA-HRP antibodies (Thermo Scientific, Waltham, MA), while NiV-F was detected using anti-AU1 antibodies (Thermo Scientific).
Cross-linking.
A 50 μM stock solution of BS3 (bis[sulfosuccinimidyl] suberate) (Thermo Scientific), a membrane-impermeant cross-linking agent, was prepared in PBS. 293T cells expressing wild-type or cysteine mutant NiV-G proteins were removed from tissue culture plates with 10 mM EDTA in PBS, washed with PBS, and incubated with 0.1 mM or 1 mM BS3 or with PBS alone at room temperature for 40 min in microcentrifuge tubes. The cells were then washed twice with 500 μl of PBS, after which they were lysed with lysis buffer (1% Triton X-100, 150 mM NaCl, 40 mM Tris [pH 7.4]). Clarified cell lysates samples were analyzed by Western blot analysis, and anti-HA-HRP antibodies were used to detect WT and cysteine mutant NiV-G proteins.
Cell surface binding assays.
293T cells were seeded ino 6-well plates and were subsequently cotransfected with WT NiV-G, C1, C2, or C3 and NiV-F plasmids. Twenty-four hours posttransfection, the cells were removed from the tissue culture plates and were placed in flow cytometry tubes for cell surface antibody staining. The cell were incubated for 30 min on ice with either rabbit polyclonal anti-G antibodies (806), mouse monoclonal anti-HA antibodies (Covance), soluble B2-Fc fusion protein (R&D Systems), rabbit monoclonal anti-G antibodies (Mab26, Mab45), or rabbit polyclonal anti-F antibodies. Then the cells were washed with a buffer (1% fetal bovine serum in PBS) and were incubated with their respective R-phycoerythrin-conjugated secondary anti-mouse IgG or anti-rabbit IgG antibodies (Invitrogen) for 30 min on ice. The cells were washed again and were fixed with 2% paraformaldehyde, and the data were collected using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Cell surface biotin labeling and coimmunoprecipitation.
WT or mutant NiV-G proteins were either expressed independently or coexpressed with NiV-F in 293T cells on polylysine-coated 6-well tissue culture plates. For control experiments, WT NiV-G and NiV-F were expressed individually. These cells were labeled with EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific) at 0.5 mg/ml in PBS for 30 min at 4°C. All cells were washed in PBS (3 times); then lysed and clarified lysates were prepared by microcentrifugation. The biotin-labeled control samples individually expressing WT NiV-G and NiV-F were combined once the clarified lysates were prepared. Each lysate was incubated with 2 μg of ephrinB2-Fc (R&D Systems) and protein G-coated Dynabeads (Invitrogen). After 2 h of incubation at 4°C, the beads were washed four times with lysis buffer to remove any unbound complexes and were boiled in PBS and 4× NuPage LDS Sample buffer (Invitrogen) under nonreducing conditions for elution. The precipitates were then incubated with streptavidin-agarose (Thermo Scientific) for 2 h at 4°C. Beads were washed 3 times with lysis buffer and were eluted at 99°C for 10 min in PBS and NuPAGE LDS sample buffer containing 10% β-mercaptoethanol. These precipitates were analyzed by Western blotting with anti-AU1 antibodies to detect AU1-tagged NiV-F proteins and with anti-HA-HRP antibodies to detect HA-tagged WT and mutant NiV-G proteins.
Measurements of NiV-F triggering.
The method for measuring the activation of F has been described previously (1). Briefly, CHO cells were transfected with NiV-F and WT or cysteine mutant G expression plasmids, as well as with a green fluorescent protein (GFP) expression plasmid. Eighteen hours posttransfection, the cells were mixed with either CHO (negative control) or CHOB2 cells equivalently, chilled on ice for 2 h, and subsequently incubated for 45 min at 4°C or 37°C in the presence of excess (500 μM) biotinylated HR2 peptide. The controls regarding the specificity and function of HR2 peptide are explained in the supplementary data of reference 1. The cells were then washed with buffer (1% FBS in PBS), fixed in 0.5% paraformaldehyde in wash buffer, and washed again twice with buffer. The biotinylated HR2 peptide bound to F was detected using allophycocyanin (APC)-conjugated streptavidin and was quantified by flow cytometric analysis on GFP-positive cells.
Pseudotyped virus and infection assay.
Pseudotyped NiVG/F viruses were made from VSV-ΔG–mCherry, a recombinant vesicular stomatitis virus (VSV) derived from a full-length cDNA clone of the VSV Indiana serotype in which the G envelope protein has been replaced with mCherry. 293T cells seeded in a 10-cm dish were transfected with 15 μg of NiV F and/or 15 μg of NiV-G (WT, C1, C2, or C3) by using Lipofectamine 2000 (Invitrogen) and were subsequently infected with VSV-ΔG–mCherry, itself pseudotyped with VSV-G. The resulting NiV G/F-pseudotyped viruses were collected at 24 h postinfection. The viral particles were then pelleted via ultracentrifugation in 20% sucrose and resuspended in 100 μl of Opti-MEM (Invitrogen). The virus pseudotyped with WT/F, C1/F, C2/F, C3/F, WT NiV-G only, or NiV-F only was diluted 1:100 in PBS and was then incubated with or without 1 μg of B2-Fc (R&D Systems) at room temperature. After 30 min, the viruses were used to infect Vero cells seeded in a 48-well plate. The mCherry fluorescence was measured after cell lysis in a black 96-well plate using a Tecan M1000 fluorescence plate reader. Infections were performed in triplicate.
RESULTS
Cysteine residues 158 and 162 of NiV-G function in intermolecular disulfide bond formation.
Like other paramyxovirus attachment proteins, the functional oligomeric unit of NiV-G is considered a tetramer composed of two disulfide-linked dimers that associate noncovalently (8, 9, 29). The residues important for disulfide-linked dimer formation have been mapped to stalk domain cysteines for both NDV HN and MeV H. The intermolecular disulfide bond(s) in the NiV-G oligomer is also thought to form from cysteine residues in the stalk domain, since 14 of the 17 possible cysteines in NiV-G participate in the formation of intramolecular bonds within the globular domain (37). The three cysteine residues in the stalk of NiV-G are clustered and membrane distal to a stretch of 11-mer and 7-mer hydrophobic core repeat residues that align well with the 4-helix bundle (4HB) structure recently described for PIV5 (Fig. 1) (7, 39). To elucidate the functional role of each cysteine in the stalk domain of NiV-G, including its involvement in intermolecular disulfide bond formation, we mutated each individual cysteine in the stalk domain to a conserved serine residue. NiV-G proteins with stalk mutations C146S, C158S, and C162S are referred to as C1, C2, and C3, respectively.
Fig 1.
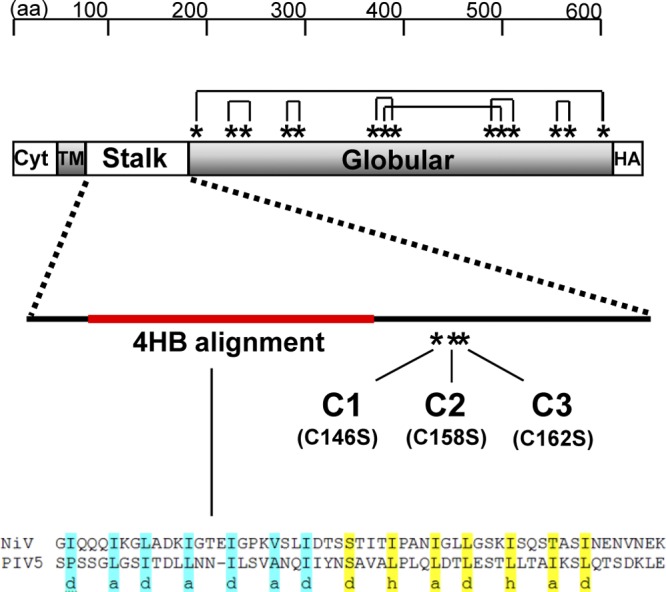
Schematic diagram of the locations of cysteine residues and intramolecular disulfide bonds in the NiV-G protein sequence. The NiV-G attachment protein contains a total of 17 cysteine residues (*): 3 in the stalk region and 14 in the globular region containing the receptor binding domain. The NiV-G crystal structure revealed intramolecular disulfide bond formation between the cysteine residues in the globular domain as shown (9, 37). Three mutant NiV-G constructs (C1, C2, C3) were made by substitution of serine for one of the individual cysteine residues (C146S, C158S, C162S) in the stalk domain. The three stalk cysteine residues are membrane distal to a stretch of 7-mer and 11-mer hydrophobic core repeat residues that aligns with the 4-helix bundle (4HB) structure recently described for PIV5 (red) (7). Residues in the PIV5 4HB crystal structure (PDB file 3TSI) template were aligned to NiV-G stalk domain residues 89 to 145. The 4HB heptad repeats of the PIV5-HN stalk with “a” and “d” positions (highlighted in blue), followed by 11-mer repeats with the “a,” “d,” and “h” positions (highlighted in yellow), are shown. Cyt, cytoplasmic tail; TM, transmembrane domain; HA, hemagglutinin tag.
On a nonreducing sodium dodecyl sulfate (SDS)-PAGE gel, wild type (WT) NiV-G ran as a monomer, a dimer, and a tetramer (Fig. 2, lane 1), as described previously (8, 29). C1 failed to run as a tetramer, while C2 and C3 ran only as monomeric species (Fig. 2, lanes 2 to 4). Under reducing conditions, WT and cysteine mutant NiV-G ran as a single diffuse band with an apparent molecular mass of ∼68 kDa (Fig. 2, lanes 5 to 8). The nonreducing SDS-PAGE data suggest that cysteine residues 158 and/or 162 of NiV-G participate in intermolecular disulfide bond formation. It remains to be determined whether both of these cysteine residues have the ability to form disulfide bonds or whether, because of the proximity of these residues to each other, a mutation in one residue affects the local redox potential and hence the ability of the other to form a disulfide bond.
Fig 2.
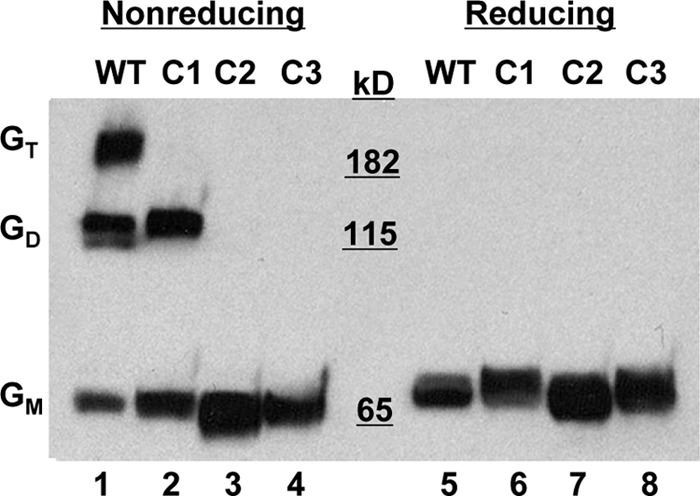
Western blot analysis of cysteine mutants under reducing and nonreducing conditions. 293T cells were transfected with the WT NiV-G plasmid or with NiV-G containing a stalk domain cysteine mutant (C1, C2, or C3). Cell lysates were collected 24 h posttransfection, and proteins were separated by SDS-PAGE under nonreducing (left) or reducing (right) conditions. Anti-HA-HRP detected the HA-tagged NiV-G proteins by Western blot analysis. One experiment representative of more than three repeats is shown. GT, tetramer; GD, dimer; GM, monomer.
NiV-G cysteine stalk domain mutants form loosely assembled tetramers, and tetramers are present on the cell surface.
Because SDS-PAGE migration patterns do not reflect the native oligomeric state, we subjected the mutant and WT NiV-G proteins to native gel electrophoretic analysis. Blue native PAGE (BN-PAGE) has recently been shown to preserve a fragile tetrameric and higher oligomeric state in the MeV H protein (10). The delicate MeV H oligomers were maintained by using the mild detergent digitonin but not during more stringent lysis with Triton X-100. By use of a similar method of BN-PAGE analysis, the WT and cysteine mutant NiV-G proteins were expressed in 293T cells and were solubilized using either digitonin or Triton X-100. To determine whether the presence of NiV-F affected the oligomeric properties of WT or mutant NiV-G proteins, the BN-PAGE analysis included lysates from NiV-F coexpression conditions. The WT NiV-G ran as a slightly diffuse band with a peak intensity just below the 280-kDa marker present with both digitonin and Triton X-100 extraction. The molecular weight of this oligomer is approximately 4 times that of the monomeric NiV-G band analyzed by nonreducing SDS-PAGE conditions (Fig. 2), and it is therefore presumed to represent the NiV-G tetramer. This result indicates that WT NiV-G in its native oligomeric state is a tetramer and that this oligomeric structure was preserved better by BN-PAGE than by SDS-PAGE. Interestingly, each of the cysteine mutants revealed the presence of a single band with the same motility as the WT tetramer when the mutant proteins were extracted by digitonin. However, with Triton X-100 lysis, two discrete bands at 65 kDa and slightly below 160 kDa also appeared in the cysteine mutant proteins, but not in the WT. The molecular weight of these bands indicates G monomers and dimers occurring only with the cysteine mutants. These results suggest that NiV-G tetramers can form without covalent dimerization, as with C2 and C3, albeit weakly compared to WT tetramer formation. Additionally, the oligomeric stability of the WT or cysteine mutant NiV-G proteins was not affected by the presence of NiV-F (Fig. 3A and B, compare lanes 1 and 2, 3 and 4, 5 and 6, and 7 and 8).
Fig 3.
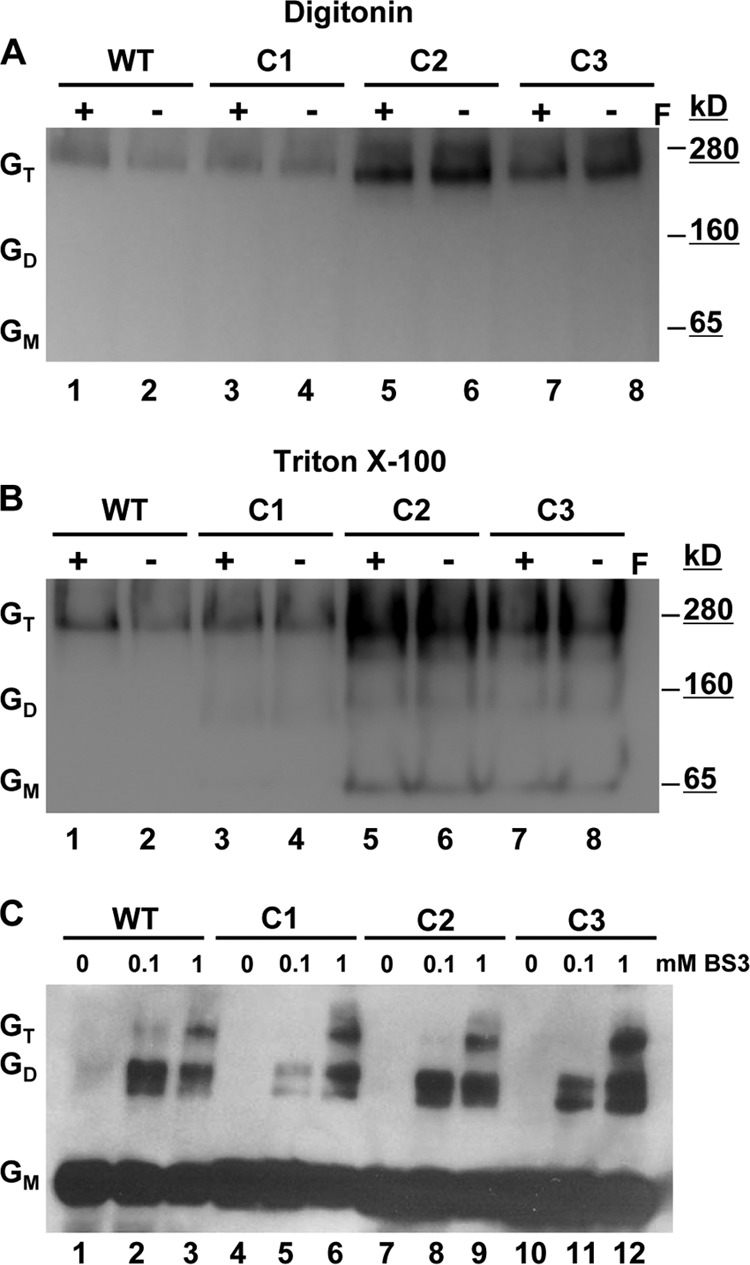
Oligomeric analysis of the cysteine mutant proteins. (A and B) 293T cells expressing WT or cysteine mutant (C1, C2, or C3) NiV-G proteins in the presence (+) or absence (−) of NiV-F (F) were collected 24 h posttransfection. Cells were removed, washed, and then lysed using either digitonin (A) or Triton X-100 (B) and were separated by native BN-PAGE. (C) 293T cells expressing WT NiV-G or the C1, C2, or C3 mutant were also removed, washed, and placed in the presence of the indicated concentrations of the chemical cross-linker BS3. After cell surface fixation with BS3, clarified cell lysates were prepared with a reducing agent and were subjected to Western blot analysis. One experiment representative of three is shown. GT, tetramer; GD, dimer; GM, monomer.
Next, we asked if the tetramers formed with the NiV-G mutants were transported to the cell surface. To detect the presence of oligomeric NiV-G species on the cell surface, we used a noncleavable, membrane-impermeant cross-linker (BS3) previously shown to preserve the oligomeric status of NiV-G and NiV-F (21). WT and mutant NiV-G proteins all ran as monomers (Fig. 3C, lanes 1, 4, 7, and 10) on a reducing SDS-PAGE gel in the absence of any cross-linker but clearly formed dimeric and tetrameric species with increasing concentrations of BS3 (Fig. 3C, lanes 3, 6, 9, and 12). There was no discernible difference between WT NiV-G and the cysteine mutants.
Cysteine mutants are fusion defective.
Cells bearing NiV-G and NiV-F on their surfaces can fuse with neighboring cells displaying the ephrinB2 and/or ephrinB3 receptor, resulting in the formation of syncytia or multinucleated giant cells. To characterize the fusion promotion activities of the NiV-G stalk domain cysteine mutants, receptor-bearing 293T cells were cotransfected with the C1, C2, or C3 stalk mutant or with WT NiV-G along with NiV-F (Fig. 4A). At 24 h posttransfection, the cotransfected 293T cells were qualitatively assessed for multinucleated cells representing syncytium formation; the results revealed that the mutants with cysteine mutations in the NiV-G stalk completely lacked fusion. Vero cells, another fusion-permissive cell line, were also tested, and while abundant fusion was visualized for the WT, the cysteine mutants again were completely fusion defective, displaying confluent monolayers without multinucleated cells even after 4 days posttransfection (Fig. 4B). This fusion-null phenotype is a striking departure from the phenotypes seen with other paramyxovirus systems. Cysteine-to-serine substitutions in the stalk of MeV H reduce but do not prevent fusion (34). Additionally, the attachment proteins of certain strains of human parainfluenza viruses and NDV do not form disulfide-linked dimers, yet these viruses remain infectious (25, 38).
Fig 4.

Fusion promotion and oligomerization activities of NiV-G cysteine mutants coexpressed with NiV-F. (A) 293T cells coexpressing WT or cysteine mutant (C1, C2, or C3) NiV-G proteins and NiV-F were fixed 24 h after transfection with 4% paraformaldehyde for 30 min at room temperature, washed with PBS, and then incubated with 300 nM 4′,6-diamidino-2-phenylindole in PBS for 2 min at room temperature. Cells were washed again with PBS, and cell nuclei were imaged at 40× using an Olympus 1X70 microscope. Multinucleated cell syncytia are indicated by asterisks. (B) Similarly, Vero cells were cotransfected with WT or cysteine mutant (C1, C2, or C3) NiV-G proteins and NiV-F and were imaged 4 days posttransfection. (C and D) Lysates from 293T cells expressing WT, C1, C2, or C3 NiV-G protein with (+) or without (−) NiV-F (F) were subjected to SDS-PAGE analysis under reducing (C) or nonreducing (D) conditions. The samples run under nonreducing conditions were first immunoprecipitated by ephrinB2-Fc. Uncleaved (F0) and cleaved (F1) versions of F are indicated. GT, tetramer; GD, dimer; GM, monomer.
To determine whether the disulfide-linked dimerization patterns seen in Fig. 2 were altered in the cysteine mutants when they were coexpressed with NiV-F, we repeated the SDS-PAGE under coexpression conditions. First, the total expression levels of the stalk mutants compared to that of the WT were analyzed. Whole-cell lysates prepared from transfected 293T cells and analyzed by Western blotting showed that there were no aberrant changes in the total expression levels of WT G, mutant G proteins, or F (Fig. 4C). The whole-cell lysates examined by nonreducing gel electrophoresis conditions revealed that the oligomeric patterns were similar with or without NiV-F coexpression (Fig. 4D, compare lanes 1 and 2, 3 and 4, 5 and 6, and 7 and 8). Taking these findings together, the fusion-deficient phenotype was not due to significant changes in overall expression levels; however, the stability of the tetramer may directly affect fusion promotion.
NiV-G cysteine mutants retain wild-type receptor binding efficiencies but exhibit aberrant exposure of the receptor binding-enhanced (RBE) epitope of Mab45.
Our experiments thus far show that the cysteine mutants can form dimers and tetramers, albeit with a weaker stability than that of oligomers formed by WT NiV-G, and a portion of these oligomers can be detected at the cell surface (Fig. 3C). To determine whether the conformation and cell surface expression levels of the cysteine mutants contribute to their fusion-deficient phenotype, WT NiV-G and the cysteine mutants were individually cotransfected with NiV-F into 293T cells, and cell surface expression was measured by flow cytometry using soluble ephrinB2-Fc, a conformation-independent polyclonal anti-G antibody (806), and a polyclonal anti-F antibody (834). Figure 5A shows that mutant NiV-G proteins C1 to C3 are expressed at 30 to 50% of WT NiV-G levels when measured by either 806 or ephrinB2-Fc binding. The normalized ephrinB2-Fc/806 binding ratio for each cysteine mutant is equivalent to that of WT NiV-G (Fig. 5B), indicating that the cysteine mutants showed no conformational defect with regard to ephrinB2 receptor binding. NiV-F was also coexpressed with the cysteine mutants at levels comparable to those for WT NiV-G (Fig. 5C).
Fig 5.

Cell expression, receptor binding, and conformational analysis of NiV-G cysteine mutants with a monoclonal anti-G antibody. The cysteine mutant or WT NiV-G proteins were coexpressed with NiV-F in 293T cells and were subjected to flow cytometry analysis. (A and C) Polyclonal rabbit anti-G antibodies (806), soluble B2-Fc (A), or polyclonal rabbit anti-F antibodies (C) were used to bind coexpressed proteins. The amount of antibody binding was calculated on the basis of mean fluorescent intensity (MFI) values, and the results for the mutants were normalized as a percentage of WT binding (Norm. MFI). (B) To normalize for cell surface expression, the binding values for B2-Fc and antibody 806 from panel A were calculated as a B2-Fc/806 ratio, with the WT condition set at 100%. (D) Binding analysis of anti-G monoclonal antibody 45 (Mab45) was performed in the presence and absence of soluble B2-Fc in order to determine the receptor binding enhancement (RBE) phenotype. The percentage of change in Mab45 binding was calculated as the ratio of Mab45 binding with versus without ephrinB2 stimulation, multiplied by 100 and subtracted by 100. (E) In addition, Mab45 was used to bind cotransfected proteins, and binding values were normalized as ratios of Mab45 binding to anti-G antibody 806 binding, as described for panel B. Error bars represent standard errors for three experiments conducted in duplicate.
Monoclonal antibody Mab45 recognizes an RBE epitope on NiV-G that is implicated in F triggering. Multiple lines of evidence implicate the coordinated interaction of the RBE epitope and the stalk domain in mediating receptor-induced F triggering (1). Thus, we utilized Mab45 to further examine the conformational integrity of the cysteine mutants. For WT NiV-G, addition of ephrinB2-Fc resulted in a 70% increase in Mab45 binding, indicating exposure of the RBE epitope (Fig. 5D). In contrast, none of the cysteine mutants showed any increase in Mab45 binding upon ephrinB2-Fc binding. Interestingly, in the absence of ephrinB2-Fc, Mab45 bound to mutants C1 to C3 2- to 3-fold better than to WT NiV-G when normalized for expression levels using polyclonal anti-G antibody 806 (Fig. 5E). That is, when the Mab45/806 binding ratio for WT NiV-G was set to 100%, mutants C1 to C3 gave normalized values of 200 to 300%. In aggregate, these results suggest either that the stalk domain cysteines are important for maintaining NiV-G in a pre-receptor-binding conformation or that they regulate the receptor-induced conformational change that is required for F triggering. In either case, mutation of these stalk domain cysteines results in a premature transition to a post-receptor-binding conformation (RBE epitope) that compromises the ability of G to promote fusion.
NiV-G cysteine mutants interact with processed NiV-F on the cell surface.
A current model of paramyxovirus fusion, termed the dissociation or clamp model, proposes that the attachment protein and the prefusion F protein are associated in a complex prior to receptor binding and that upon receptor binding, the attachment protein releases F to allow for the drastic conformational changes F must undergo during fusion (15). Henipaviruses are thought to follow the clamp model of fusion, since G and F can be coimmunoprecipitated in a complex, and there is a reciprocal correlation between the strength of F-G interactions and fusogenicity (3, 5). Therefore, we sought to determine whether the cysteine mutants had any altered interactions with NiV-F. To assess NiV-G mutant and NiV-F interactions on the cell surface, co-IP was performed after cell surface biotinylation with thiol-cleavable Sulfo-NHS-SS-Biotin. Since soluble ephrinB2 (B2-Fc) was shown to bind to both mutant and WT NiV-G proteins (Fig. 5A), we used B2-Fc to pull down the NiV-G protein directly and NiV-F indirectly (Fig. 6). The NiV-G cysteine mutants were either expressed individually or coexpressed with NiV-F, cell surface biotinylated, precipitated with B2-Fc, eluted, and further precipitated with streptavidin-agarose. All streptavidin-agarose precipitates were analyzed by Western blotting using anti-G or anti-F specific antibodies. As shown in Fig. 6A, all NiV-G cysteine mutants coimmunoprecipitated both the unprocessed (F0) and the processed (cleaved) (F1) form of cell surface biotin-labeled NiV-F. As a postlysis control, NiV-G and NiV-F were expressed separately and biotinylated, and the lysates were mixed together before processing identical to that described above (Fig. 6A, lane 9). This control indicated the requirement for NiV-G and NiV-F to be expressed in the same cell prior to IP with B2-Fc for detection of NiV-G/F interactions. In Fig. 6B, equal fractions of all the transfected cell lysates were analyzed before B2-Fc IP for NiV-F and after B2-Fc but before streptavidin-agarose IP for G as a representation of the amount of protein input. By comparison of the amounts of F and G input, the NiV-G cysteine mutants appeared to coimmunoprecipitate with F0/F1 better than wild-type NiV-G, indicating a strengthened F-G interaction for the cysteine mutants. As predicted by the clamp model of paramyxovirus fusion, the strength of F-G interactions displayed by nonfusogenic G proteins is expected to be enhanced over that for wild-type F-G interactions, since fusogenic F-G complexes are thought to dissociate after fusion promotion, while nonfusogenic F-G complexes are unable to disengage. In addition, our results also indicate that the fusion-deficient phenotype of the cysteine mutants does not result from an inability to bind to processed or unprocessed NiV-F.
Fig 6.
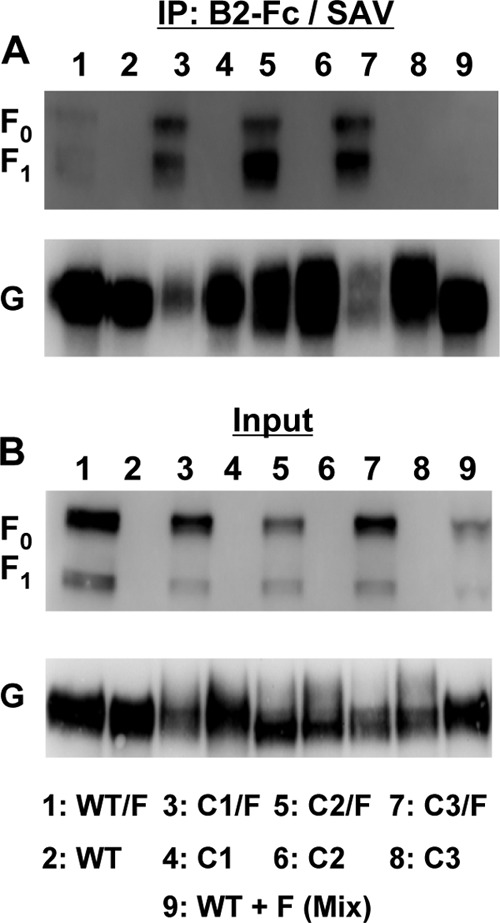
Coimmunoprecipitation of cell surface-expressed NiV-G cysteine mutants with NiV-F. WT, C1, C2, or C3 NiV-G proteins were either expressed separately or coexpressed with NiV-F (WT/F, C1/F, C2/F, C3/F) in 293T cells. The transfected cells were surface biotinylated using Sulfo-NHS-SS-Biotin as described in Materials and Methods. The clarified cell lysates were incubated with B2-Fc to immunoprecipitate (IP) NiV-G and coimmunoprecipitate NiV-F. As a control, WT NiV-G and NiV-F were separately expressed, biotinylated, and combined after lysis but before being subjected to B2-Fc IP [WT + F (Mix)]. The B2-Fc precipitates were eluted under nonreducing conditions and were split into two sample sets. (A) One set was precipitated with streptavidin-agarose (SAV), eluted under reducing conditions, and analyzed by Western blotting using anti-AU1 antibodies to detect the AU1-tagged NiV-F proteins and anti-HA-HRP antibodies to detect the HA-tagged WT and mutant NiV-G proteins. The coimmunoprecipitated uncleaved (F0) and cleaved (F1) versions of NiV-F are shown. (B) The other B2-Fc IP sample set was analyzed for the amount of input NiV-G, before the SAV IP for which results are shown in panel A, by reducing SDS-PAGE and the Western blot methods described above. In addition, all cell lysates before B2-Fc IP were analyzed for the amount of input NiV-F protein by Western blotting. Results of one experiment representative of three are shown.
NiV-G cysteine mutants do not induce NiV-F triggering.
Since the NiV-G cysteine mutants can bind the ephrinB2 receptor and can clearly interact with NiV-F on the cell surface, we hypothesize that the fusion defect of the cysteine mutants is due to their inability to trigger F. Receptor binding to G is thought to induce an allosteric signal that triggers F to undergo a series of conformational changes resulting in the fusion of the viral and host membranes. F triggering involves the formation and exposure of heptad repeat region 1 (HR1), which is coincident with fusion peptide insertion into the target cell membrane. The HR1 and HR2 in this prehairpin intermediate then fold together to form the 6-helix bundle that drives membrane fusion (2). To test our hypothesis, we used a previously established assay for F triggering that relies on the binding of labeled HR2 peptides to the trimeric HR1 core, which is exposed during prehairpin intermediate formation (1, 2). WT or cysteine mutant G proteins were coexpressed with NiV-F in receptor-deficient CHO cells. These NiV-F/G-expressing cells were mixed with CHO cells stably expressing ephrinB2 (CHOB2) and were held at 4°C for 2 h to synchronize receptor binding. The labeled HR2 peptide was then added to the cell mixture, and fusion was induced by shifting the temperature to 37°C. Flow cytometry was then used to quantify the amount of HR2 binding, which was used as an indicator of F triggering, or prehairpin formation. WT NiV-G was able to induce F triggering when stimulated by ephrinB2 at 37°C, while the cysteine mutant NiV-G proteins were unable to trigger F (Fig. 7A). The MFI values for three experiments conducted similarly, as described for Fig. 7A, are summarized for all the NiV-G proteins stimulated with ephrinB2 at 37°C (Fig. 7B). These results suggest that the cysteine mutants are deficient in fusion promotion due to their inability to induce F triggering.
Fig 7.

Measurement of the ability of NiV-G cysteine mutants to induce NiV-F triggering. (A) Receptor-deficient CHO cells expressing WT, C1, C2, or C3 NiV-G proteins were coexpressed with NiV-F and were stimulated with ephrinB2 expressed on CHOB2 cells for 2 h at 4°C. Cell mixtures were kept at 4°C or were brought to 37°C for 45 min to allow for F triggering in the presence of 500 mM biotinylated HR peptide. Control experiments were performed by stimulation with receptor-deficient CHO cells instead of CHOB2 cells to measure background HR2 peptide binding. Filled histograms represent triggering with the CHO cell negative control, while open histograms represent the positive signals obtained using CHOB2 cells as target cells. (B) The absolute MFI values measuring the HR2 binding shown in panel A at 37°C were normalized to the percentage of F triggering with WT NiV-G and are summarized for the F/G (WT), F/C1, F/C2, and F/C3 protein pairs. Data are averages ± standard errors for three experiments.
NiV-G/F-pseudotyped virus containing cysteine stalk mutants also display weakened tetrameric stability, maintain G-F interactions, and are noninfectious.
F-G envelope complexes may have different physicochemical properties when present on the cell surface versus on virions. To determine whether the disulfide-linked dimerization patterns and/or oligomeric properties seen with the cysteine mutants (Fig. 2 and 3) were the same when these mutants were pseudotyped onto VSVΔG-mCherry reporter viruses (29), we repeated the nonreducing SDS-PAGE and BN-PAGE gel analyses on the pseudotyped VSVs shown in Fig. 8. In both the nonreducing (Fig. 8A) and BN-PAGE (Fig. 8B) gel analyses, mutants C1 and C2 exhibited weakened oligomeric stability compared to that of wild-type NiV-G, with mutant C2 being the least stable. The complete lack of dimeric and tetrameric species for the C2 mutant again implicates residue 158 in intermolecular disulfide bond formation. We were unable to obtain high-quality C3-pseudotyped virus. This is most likely due to the combination of lower expression levels and decreased oligomeric stability seen with mutant C3 (Fig. 3B, 4D, and 5A). While we were able to detect C3 mutant expression and coimmunoprecipitation with NiV-F on the cell surface (Fig. 5A and 6A), the efficiency of NiV-G/F pseudotyping onto VSV appears to be sensitive to cell surface expression and may require an expression threshold. Even the slight difference between C1 and C2 cell surface expression (∼50% versus ∼40% [Fig. 5A]) appears to have a significant effect on the amount of C1 versus C2 displayed on VSV (Fig. 8A), highlighting this point.
Fig 8.
Cysteine mutants on pseudotyped virus. Pseudotyped virus was made by allowing VSV-ΔG–mCherry, a recombinant vesicular stomatitis virus, to bud from cells expressing WT or mutant NiV-G and NiV-F on the cell surface. (A and B) The virus was run on an SDS-PAGE gel under nonreducing conditions (A) or on a native PAGE (BN-PAGE) gel with digitonin (Dig) or Triton X-100 (X100) (B) and was detected by Western blot analysis using both anti-HA-HRP for NiV-G and anti-AU1-HRP. Oligomeric complexes are labeled as the tetramer (GT), dimer (GD), and monomer (GM). (C) The virus was immunoprecipitated (IP) with B2-Fc and was detected by Western blot analysis using both anti-HA-HRP for NiV-G and anti-AU1-HRP for NiV-F for co-IP analysis. The input virus in the IP experiment was subjected to Western blot analysis with anti-AU1-HRP. NiV-G is indicated (G), and the uncleaved and cleaved versions of NiV-F are indicated as F0 and F1, respectively. (D) Vero cells were infected with a virus, pseudotyped with WT, C1, C2, or C3 NiV-G and with NiV-F, that had been incubated 30 min at room temperature either with (+) or without (−) B2-Fc. Infectivity was measured using relative fluorescence units (RFU) based on the mCherry fluorescence signal after cell lysis. Error bars represent standard errors for three experiments conducted in triplicate.
To further verify that the NiV-G and NiV-F proteins were able to interact on the virus particle surface, NiV F/G-pseudotyped virus was precipitated with B2-Fc (Fig. 8C), and all precipitates were analyzed by Western blotting to detect NiV-G and coimmunoprecipitated NiV-F. Like cell surface-expressed NiV-G and NiV-F (Fig. 7A), wild-type NiV-G and cysteine mutants C1 and C2 on the pseudotyped viruses were able to coimmunoprecipitate both F0 and F1 (Fig. 8C). If NiV-G and NiV-F were pseudotyped separately and were then mixed together for the IP with B2-Fc, no F was coimmunoprecipitated with G (Fig. 8C, WT+F). Thus, as on the cell surface, this control demonstrates that NiV-G and NiV-F must be present on the same membrane (virus or cell) in order for cognate interactions to occur.
Finally, to determine the fusion-deficient phenotype of the cysteine mutants in the context of virus entry in addition to syncytium formation, we infected Vero cells with the VSVΔG-mCherry reporter virus pseudotyped with the C1 or C2 stalk mutant or with WT NiV-G, along with NiV-F. At 24 h postinfection, the Vero cell lysates were assayed for the amount of mCherry fluorescence, indicative of infectivity levels (Fig. 8D). As expected, none of the viruses pseudotyped with the cysteine mutants showed any infectivity. WT F/G-pseudotyped virus exhibited robust infectivity that was abolished in the presence of ephrinB2-Fc, demonstrating the specificity of the pseudotyped virus infection.
The cysteine residues in the NiV-G stalk are located in a distinct domain essential for F triggering but not for physical interactions with F.
An alignment of the stalk domain for NiV-G and HeV-G (residues 71 to 188) is shown in Fig. 9A. Based on functional, sequence conservation, and structural homology criteria, the stalk domain can be partitioned into 4 subdomains. Domains II and III are more conserved than domains I and IV. Domain II aligns well with the 11-mer and/or partial 7-mer hydrophobic core repeat residues that form the 4HB structure recently described for PIV5 and NDV (Fig. 9B) (7, 39). Indeed, this domain appears to be conserved across attachment proteins from all five Paramyxovirinae genera, although there are significant differences in noncore repeat residues. Domain III is membrane distal to domain II and is absolutely conserved between HeV and NiV. In a previous alanine-scanning study of all isoleucine residues in the stalk of HeV-G (asterisked in Fig. 9A), only mutations at residues I155 and I160 in domain III, near the cysteine residues at positions 146, 158, and 162, showed reduced cell surface expression similar to that seen with our cysteine mutants (4). Therefore, it appears that mutations in this region of the stalk are more likely to affect cell surface expression, suggesting that the functional structure of domain III is particularly sensitive to amino acid substitutions. This is consistent with the unusually large number of prolines (6 out of 28 residues) and cysteines in domain III, which likely limits its conformational flexibility.
Fig 9.

Cysteine residues in the NiV-G stalk are located in a distinct domain important for fusion but not for physical F interactions. (A) The stalk amino acid residues 71 to 188 from the reference sequences of NiV-G and HeV-G were aligned. The locations of predicted subdomains in the stalks in NiV-G and HeV-G are indicated (I to IV) and were partitioned based on the functional characterization of HeV-G point mutations (*) within these domains, where fusion-permissive (green) and fusion-null (red) phenotypes are indicated by colored asterisks, variability in residues (blue letters), and the positions of prolines (highlighted in red) and cysteines (highlighted in yellow). (B) NiV-G stalk residues 101 to 145 (domain II) were aligned to paramyxovirus attachment protein (HN, H and G) stalks representing all five genera within the Paramyxovirinae by anchoring the conserved I112 and P121 residues in NiV-G (highlighted in red). The 4HB heptad repeats of the PIV5-HN stalk with “a” and “d” positions (highlighted in blue), followed by 11-mer repeats with the “a,” “d,” and “h” positions (highlighted in yellow), are shown. Since the heptad pattern is shifted in several paramyxoviruses, the transition point between the 7-mer and the 11-mer may differ among attachment proteins. Sequences of the following viral proteins were used in alignments: Nipah virus G (NiV) (Genbank accession number NC_002728), Hendra virus G (HeV) (NC_001906.2), measles virus H (MeV) (NC_001498.1), canine distemper virus H (CDV) (NC_001921.1), parainfluenza virus 5 (PIV5) (NC_006430.1), Newcastle disease virus HN (NDV) (NC_002617.1), human parainfluenza virus 1 HN (HPIV1) (NC_003461.1), HPIV2 HN (NC_003443.1) and HPIV3 HN (NC_001796.2) (C) 293T cells were cotransfected with NiV-F and either WT NiV-G, the NiV-G I155A mutant (155), or a NiV-G mutant with a deletion of amino acids 146 to 182 (Δ3). Cell lysates were immunoprecipitated (IP) with B2-Fc and were analyzed by Western blotting as described for Fig. 8C. Results of one experiment representative of three are shown.
The homologous 4HB region in the HNV-G stalk is located in domain II, containing residues 100 to 145 (Fig. 9B). Since regions that directly interact with F lie within the NDV and PIV5 4HB (7, 39), which in turn aligns with HNV-G domain II, we hypothesize that F-specific interactions occur within this membrane-proximal region of the G stalk, as opposed to regions above residue 146. To test this hypothesis, we constructed a NiV-G mutant containing a deletion of residues 146 to 182 (domains III and IV). This mutant with a 37-amino-acid deletion in the G stalk (Δ3) could not promote fusion (data not shown); remarkably, however, it was able to coimmunoprecipitate NiV-F as well as, if not better than, wild-type NiV-G (Fig. 9D). Therefore, the cysteine residues in HNV-G subdomain III are essential for F triggering but not for physical F interaction.
DISCUSSION
Here we used a panoply of assays to examine the role of stalk domain cysteine residues in maintaining the structure and function of NiV-G. Cysteine residues of secretory pathway proteins in general can participate in disulfide bond formation during protein folding, which serves to stabilize folding intermediates, as well as the mature protein architecture (12). The NiV-G protein, with a total of 17 cysteine residues, forms 7 intramolecular disulfide bonds in the completely folded six-bladed beta-propeller structure. The three cysteine residues remaining are located in the stalk and have not been characterized previously for their functional role in oligomerization or fusion promotion. As in other paramyxovirus systems, we found that cysteines in the stalk of NiV-G assist in maintaining oligomeric stability by participating in intersubunit disulfide bond formation. However, in contrast to the situation for HN or H attachment proteins, where cysteine mutations in the stalk domain at most attenuate fusion, we found that mutants with mutations of cysteine residues in the NiV-G stalk domain were completely fusion deficient.
The key phenotype associated with the NiV-G stalk domain mutants was enhanced recognition by the conformation-dependent monoclonal antibody Mab45. As demonstrated by Aguilar et al. (1), Mab45 has a receptor binding-enhanced epitope that is postulated to recognize receptor-induced conformational changes in NiV-G involving regions near the base of the globular head domain and the stalk domain. Since Mab45 binds to the NiV-G cysteine mutants 2- to 3-fold better than it binds to WT NiV-G (Fig. 5E), and ephrinB2 binding to these mutants did not enhance Mab45 binding (Fig. 5D), the cysteine mutants presumably transition prematurely to a post-receptor-binding conformation. In a previous alanine-scanning mutagenesis study of all isoleucine residues in the HeV-G stalk (4), only isoleucine mutations in domains II and III (red asterisked residues in Fig. 9A) resulted in fusion deficiency and enhanced binding to conformational antibodies also thought to recognize G in a post-receptor-bound conformational state. Thus, we propose that domains II and III are important for maintaining HNV-G in a quasi-metastable state, in which HNV-G allosterically responds to signals induced by receptor binding to the globular domain in a manner that triggers F.
Extracellular proteins trafficking through the secretory pathway encounter chaperones and enzymes that bind to residues such as cysteines and prolines to aid in slow folding reactions, such as disulfide bridge formation or peptidyl-prolyl isomerization (12). Mutations in what we now refer to as subdomain III (residues 146 to 173) (Fig. 9A) may affect the manner in which proteins assisting in HNV-G folding interact with this stalk region. Specifically, mutations of individual cysteine residues can create nonnative free thiols, which, in turn, can serve as targets for endoplasmic reticulum-resident isomerases. Mutations in subdomain III do not appear to affect the folding of the receptor binding domain, since the levels of ephrinB2 binding normalized to cell surface expression were similar for the cysteine mutants and the WT (Fig. 5B). Additionally, mutations of the NiV-G stalk cysteines did not affect tetramer formation per se, although they did compromise tetrameric stability (Fig. 3A and B and 8B). C158 (C2) and C162 (C3) appear important for forming disulfide-linked dimers, since individual mutations at either position resulted in the proteins running exclusively as monomers on nonreducing SDS-PAGE gels (Fig. 2 and 8A).
It remains unclear whether C158 and C162 participate in the formation of a single or multiple intersubunit disulfide bonds. We speculate that due to the proximity of these two cysteine residues, C158 and C162 first form an intramolecular disulfide bond to protect these reactive thiols during protein folding. In the later stages of G folding, this intramolecular bond would isomerize to form a single or, most likely, double intermolecular disulfide bonds (12). In this scenario, the C2 and C3 mutants, which lack either C158 or C162 thiols, respectively, cannot form the early intramolecular bond between C158 and C162; thus, they are prevented from isomerization and intersubunit disulfide bond formation, an idea consistent with the nonreducing SDS-PAGE results (Fig. 2, 4D, and 8A). Additionally, the absence of tetramers in mutant C1 under nonreducing conditions (Fig. 2, 4D, and 8A) suggests that another intersubunit disulfide bond, which functions to stabilize the tetramer in wild-type G, may exist across NiV-G dimer pairs. Lastly, the fact that mutants C2 and C3 were completely monomeric under nonreducing conditions (Fig. 2, 4D, and 8A) suggests that intersubunit bond formation at position C146 is dependent on the prior formation of the C158/C162 intersubunit bond(s). A model for HNV-G disulfide linkage in the stalk domain is shown in Fig. 10.
Fig 10.
Model for henipavirus G stalk domain disulfide bond formation. (Left) Cysteine residues at positions 158 and 162 in the HNV-G attachment protein are implicated in intersubunit disulfide bond formation and are hypothesized to be protected during NiV-G folding via an intramolecular disulfide bond between C158 and C162. In this scenario, C146 remains a free thiol (S). (Center) Disulfide bond isomerization is predicted to rearrange the intramolecular C158–C162 bond to form double intersubunit disulfide bonds in the HNV-G dimer. (Right) The stability of the HNV-G tetramer observed by nonreducing SDS-PAGE is thought to occur via intersubunit disulfide bond formation through C146. The G protein globular domain is represented as a cylinder and the stalk domain as a stick.
Other than henipaviruses, whose stalks are approximately 40 amino acids longer than those of most HN proteins, morbillivirus attachment proteins also contain extended stalk domains compared to the typical HN stalk (the MeV H stalk has 120 aa). Upon assessment of stalk domain sequences across the Morbillivirus genus, it was apparent that the H stalks could be divided into subdomains similar to HNV-G, as indicated by functional studies of HNV-G proteins (see Fig. S1A in the supplemental material). As with henipavirus G, subdomains I and IV in morbillivirus H are less conserved than subdomains II and III. The H stalk subdomain II also contains the aligned 4HB region shown in Fig. 9B, while subdomain III contains cysteines important for intersubunit disulfide-linked dimerization (34). Interestingly, despite similar stalk lengths, alignment of subdomains III and IV of henipaviruses to those of morbilliviruses highlighted a unique proline-rich microdomain present only in the henipavirus stalk (see Fig. S1B in the supplemental material). Although the exact function of the proline-rich microdomain in the HNV-G stalk remains to be understood, future studies using chimeras of stalk subdomains between MeV-H and NiV-G are likely to be revealing. Since MeV-H and HNV-G differ in their site of receptor engagement (at the side or at the top of the six-bladed beta-propeller for MeV-H and HNV-G, respectively), we speculate that the proline-rich microdomain plays a unique role in transmitting the receptor-induced allosteric signals to effectuate fusion triggering. Future biochemical and chimeric studies may answer these and other questions regarding the similarities and differences in the fusion mechanisms across paramyxoviruses.
In all, we showed that cysteines in the stalk domain of NiV-G are important not only for maintaining the oligomeric stability of G but also for proper folding of a unique subdomain located in the membrane-distal region of the stalk that is necessary for F triggering. This subdomain is critical for maintaining G in its pre-receptor-bound conformation, and although it is essential for F triggering, it is not necessary for stable F interactions (Fig. 9C). These studies supplement our knowledge of the function of the stalk domains of paramyxovirus attachment proteins in F triggering and identify a critical microdomain that can be targeted for therapeutic or vaccine development.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carol Kozina for technical assistance and Sidney Elmer and Natalie Jouravel for thoughtful discussions.
This work was supported by a Laboratory Directed Research and Development Grant given to O.A.N. at Sandia National Laboratories (SNL). SNL is a multiprogram laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000. Additional funding was provided by NIH grant AI069317 to B.L. and a UCLA Undergraduate Research Scholarship to D.C. We also acknowledge support from the Pacific Southwest Regional Center of Excellence (PSWRCE) through a subproject award to B.L. (U54 AI065359).
Footnotes
Published ahead of print 11 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Aguilar HC, et al. 2009. A novel receptor-induced activation site in the Nipah virus attachment glycoprotein (G) involved in triggering the fusion glycoprotein (F). J. Biol. Chem. 284:1628–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguilar HC, Aspericueta V, Robinson LR, Aanensen KE, Lee B. 2010. A quantitative and kinetic fusion protein-triggering assay can discern distinct steps in the Nipah virus membrane fusion cascade. J. Virol. 84:8033–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aguilar HC, et al. 2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 81:4520–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bishop KA, et al. 2008. Residues in the stalk domain of the Hendra virus G glycoprotein modulate conformational changes associated with receptor binding. J. Virol. 82:11398–11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bishop KA, et al. 2007. Identification of Hendra virus G glycoprotein residues that are critical for receptor binding. J. Virol. 81:5893–5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonaparte MI, et al. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. U. S. A. 102:10652–10657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bose S, et al. 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J. Virol. 85:12855–12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bossart KN, et al. 2005. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of Hendra virus. J. Virol. 79:6690–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bowden TA, et al. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Mol. Biol. 15:567–572 [DOI] [PubMed] [Google Scholar]
- 10. Brindley MA, Plemper RK. 2010. Blue native PAGE and biomolecular complementation reveal a tetrameric or higher-order oligomer organization of the physiological measles virus attachment protein H. J. Virol. 84:12174–12184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chua KB, et al. 1999. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 354:1257–1259 [DOI] [PubMed] [Google Scholar]
- 12. Feige MJ, Hendershot LM. 2011. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 23:167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Field HE, Mackenzie JS, Daszak P. 2007. Henipaviruses: emerging paramyxoviruses associated with fruit bats. Curr. Top. Microbiol. Immunol. 315:133–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gurley ES, et al. 2007. Risk of nosocomial transmission of Nipah virus in a Bangladesh hospital. Infect. Control Hosp. Epidemiol. 28:740–742 [DOI] [PubMed] [Google Scholar]
- 15. Iorio RM, Melanson VR, Mahon PJ. 2009. Glycoprotein interactions in paramyxovirus fusion. Future Virol. 4:335–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lam SK, Chua KB. 2002. Nipah virus encephalitis outbreak in Malaysia. Clin. Infect. Dis. 34(Suppl. 2):S48–S51 [DOI] [PubMed] [Google Scholar]
- 17. Lamb RA, Jardetzky TS. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 17:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lamb RA, Paterson RG, Jardetzky TS. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee B, Ataman ZA. 2011. Modes of paramyxovirus fusion: a Henipavirus perspective. Trends Microbiol. 19:389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee JK, et al. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561–16572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levroney EL, et al. 2005. Novel innate immune functions for galectin-1: galectin-1 inhibits cell fusion by Nipah virus envelope glycoproteins and augments dendritic cell secretion of proinflammatory cytokines. J. Immunol. 175:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lo MK, Rota PA. 2008. The emergence of Nipah virus, a highly pathogenic paramyxovirus. J. Clin. Virol. 43:396–400 [DOI] [PubMed] [Google Scholar]
- 23. Luby SP, Gurley ES, Hossain MJ. 2009. Transmission of human infection with Nipah virus. Clin. Infect. Dis. 49:1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luby SP, et al. 2006. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 12:1888–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGinnes LW, Morrison TG. 1994. Modulation of the activities of HN protein of Newcastle disease virus by nonconserved cysteine residues. Virus Res. 34:305–316 [DOI] [PubMed] [Google Scholar]
- 26. McGinnes LW, Morrison TG. 1994. The role of the individual cysteine residues in the formation of the mature, antigenic HN protein of Newcastle disease virus. Virology 200:470–483 [DOI] [PubMed] [Google Scholar]
- 27. Mirza AM, et al. 2011. Triggering of the Newcastle disease virus fusion protein by a chimeric attachment protein that binds to Nipah virus receptors. J. Biol. Chem. 286:17851–17860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montgomery JM, et al. 2008. Risk factors for Nipah virus encephalitis in Bangladesh. Emerg. Infect. Dis. 14:1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Negrete OA, Chu D, Aguilar HC, Lee B. 2007. Single amino acid changes in the Nipah and Hendra virus attachment glycoproteins distinguish ephrinB2 from ephrinB3 usage. J. Virol. 81:10804–10814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Negrete OA, et al. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405 [DOI] [PubMed] [Google Scholar]
- 31. Negrete OA, et al. 2006. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog. 2:e7 doi:10.1371/journal.ppat.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paal T, et al. 2009. Probing the spatial organization of measles virus fusion complexes. J. Virol. 83:10480–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7:e1002058 doi:10.1371/journal.ppat.1002058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Plemper RK, Hammond AL, Cattaneo R. 2000. Characterization of a region of the measles virus hemagglutinin sufficient for its dimerization. J. Virol. 74:6485–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Porotto M, et al. 2011. Spring-loaded model revisited: paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J. Virol. 85:12867–12880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu K, et al. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U. S. A. 105:9953–9958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan P, Leser GP, Demeler B, Lamb RA, Jardetzky TS. 2008. Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378:282–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yuan P, et al. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A. 108:14920–14925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.