

Abstract
Herpes simplex virus 2 (HSV-2) is the primary cause of genital herpes, which is one of the most common sexually transmitted viral infections worldwide and a major cofactor for human immunodeficiency virus infection. The lack of an effective vaccine or treatment and the emergence of drug-resistant strains highlight the need for developing new antivirals for HSV-2. Here, we demonstrate that a low-molecular-weight peptide isolated against 3-O-sulfated heparan sulfate (3-OS HS) can efficiently block HSV-2 infection. Treatment with the peptide inhibited viral entry and cell-to-cell spread both in vitro and in vivo using a mouse model of genital HSV-2 infection. Quite interestingly, the peptide showed a preferential binding to HSV-2-infected cells, with more than 200% increased binding compared to uninfected cells. Our additional results show that heparan sulfate expression is upregulated by 25% upon HSV-2 infection, which is a significant new finding that could be exploited for designing new diagnostic tests and treatment strategies against HSV-2-infected cells. In addition, our results also raise the possibility that 3-OS HS modifications within HS may be upregulated even more to accommodate for a significantly higher increase in the peptide binding to the infected cells.
INTRODUCTION
Herpes simplex virus 2 (HSV-2) is responsible for approximately two-thirds of the sexually transmitted mucocutaneous lesions commonly known as genital herpes in the United States, with a gender, age, and racial disparity in its seropositivity prevalence that ranges from 3.4% in Caucasian males and 18.2% in black males in about their early 20s to 31.4% in Caucasian women and 66.5% in black women in about their late 40s (40). HSV-2 enters susceptible cells by first attaching to heparan sulfate (HS) linear polysaccharide side chains of cell surface heparan sulfate proteoglycans (HSPGs) (48). Attachment is mediated by the viral envelope glycoproteins gB and gC (19, 48). The interaction between HSV attachment glycoproteins and HS most probably relies on the noncovalent associations between the positively charged amino acid residues of the HS binding sites of viral glycoproteins and the negatively charged sulfate and carboxylate groups of HS chains (10). The absence of this interaction does not prevent entry but simply slows the process (18). This is followed by a postattachment receptor-mediated docking of HSV virions to one of the gD receptors: herpesvirus entry mediator (HVEM), nectin-1, or nectin-2 (17, 23, 27, 45). The binding of gD to its receptor initiates the penetration process involving gB, gH, and gL and possibly the gB or gH receptor, or both (6, 33, 34, 35).
Recently, the role of HS in HSV infection has been shown to be more than just providing attachment sites for the virus. Some rare modifications in HS can create an HSV-1 gD receptor (31). It is also involved in negatively regulating virus-induced cell-to-cell fusion and mediating HSV movement along plasma membrane protrusions, a process that is termed surfing (30, 32). Moreover, HS has been shown to serve as coreceptor for many viruses, including human immunodeficiency virus (HIV), human cytomegalovirus (HCMV), and varicella-zoster virus (VZV) (13, 14, 20, 44).
The biosynthesis of HS and subsequent modifications are performed by multiple enzymes. Combinatorial expression of these enzymes gives rise to the structural diversity of HS chains to modulate a wide variety of tissue-specific functions (16). One of the most critical modifications in HS structure, 3-O-sulfation, is catalyzed by a family of enzymes called 3-O-sulfotransferases (3-OSTs) (24). Seven members have been identified in this family, where each isoform has the ability to recognize a distinct saccharide sequence around the modification site and thus generate its own unique 3-O-sulfated motifs. Some of the unique motifs have been shown to have strong regulatory functions, while others remain poorly understood (25, 38). 3-O-Sulfated HS (3-OS HS) modified by the 3-OST isoforms, except for 3-OST isoform 1 (3-OST-1), has been shown to function as a gD receptor for HSV-1. However, 3-OS HS fails to provide gD receptor activity for HSV-2 entry into HSV-resistant cells that lack a gD receptor (31, 36, 41, 49, 50). It probably only mediates HSV-2 attachment to cells (37). In this regard, it is not very clear whether blocking 3-OS HS can block HSV-2 infection in vivo, and likewise, very limited information on the expression of 3-OS HS in vitro and in vivo is available.
Cationic peptides targeting HS on the surface of the host cell provide an attractive approach for the development of antiherpetic agents. Some recently identified peptides include human apolipoprotein E-derived peptide (apoEdp), rabbit neutrophil peptide-1 (NP-1), lactoferrin (LF), indolicidin (a tryptophan-rich peptide from bovine neutrophils), and brevinin-1 (a peptide found in frog skin) (2, 3, 4, 8, 39, 51). We recently used a phage display random peptide library screening to isolate a 12-mer cationic peptide (G2 peptide) that binds 3-OS HS and blocks HSV-1 infection (43). Very interestingly, we show here that despite the inability of 3-OS HS to allow HSV-2 entry, G2 peptide blocks HSV-2 infection and cell-to-cell fusion mediated by the virus. We also provide new details on the mechanism of antiviral activity of G2 peptide in vitro and demonstrate its efficacy in vivo using a mouse model of genital herpes infection. Additionally, our study demonstrates that HSV-2-infected cells show a significantly higher binding of G2 peptide than uninfected cells. We also observed an increase in HS expression and propose an even higher increase in 3-OS HS modifications, which may explain the 200% increase in the peptide binding to infected cells. The enhanced HS expression and preferential binding of G2 are novel pieces of information with significant long-term implications for anti-HSV-2 prognosis and future drug designs for therapy.
MATERIALS AND METHODS
Cell culture and viruses.
African green monkey kidney (Vero) cells were provided by P. G. Spear (Northwestern University, Chicago, IL). Human cervical (HeLa) cells were provided by B. S. Prabhakar (University of Illinois at Chicago, Chicago, IL). The human corneal epithelial (HCE) cell line (RCB1834 HCE-T) was provided by Kozaburo Hayashi (National Eye Institute, Bethesda, MD) (5). Vero and HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate at 37°C in an atmosphere of 5% CO2. HCE cells were maintained in minimum essential medium (MEM; Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate at 37°C in an atmosphere of 5% CO2.
Three strains of HSV-2 were used: HSV-2 strain 333 and the galactosidase enzyme-inducing (gJ negative [gJ−]) HSV-2 strain 333 (both provided by P. G. Spear, Northwestern University) and green fluorescent protein (GFP)-tagged HSV-2 derived from HSV-2 strain 333 with an EF-1alpha promoter expressing GFP inserted between UL26 and UL27 (provided by J. Vieira, University of Washington, Seattle, WA). High-titer stocks of all of the strains used were prepared by infecting Vero cells at a low multiplicity of infection (MOI; 0.01) and harvesting the cells when the cytopathic effect was 90 to 100%. Cell-bound virus particles were released by sonication. Purified stocks of all HSV-2 strains were prepared by purification on a sucrose gradient (47).
Isolation and synthesis of HS antagonist peptide.
Phage-displayed combinatorial 12-mer peptide libraries were screened for phage populations exposing peptides that are able to bind purified HS and 3-OS HS that has been modified by 3-OST-3 as described previously (43). G2 and a control peptide, Cp (RVCGSIGKEVLG), were synthesized using the 9-fluorenylmethoxy carbonyl synthesis method at the University of Illinois Research Resources Center Proteomics Laboratory, with the purity of the synthesized peptide measured by mass spectrometry/high-performance liquid chromatography to be more than 90% (29). Working stocks of the peptides were prepared by dissolving in 1× phosphate-buffered saline (PBS) and filter sterilization through a 0.2-μm-pore-size low-protein-binding syringe filter (Millipore Corp., Billerica, MA), followed by protein estimation.
Heparinase assay.
Vero cell monolayers were pretreated with 10 mIU of heparinase III (provided by J. Liu, University of North Carolina, Chapel Hill, NC) in reduced-serum medium (Opti-MEM; Invitrogen, Carlsbad, CA) or with medium alone for 2 h at 37°C. Cells were then washed three times with warm 1× PBS and then incubated with fluorescein isothiocyanate (FITC)-conjugated G2 peptide at room temperature for 30 min. Detection of bound fluorescent G2 peptide was performed by fluorescence microscopy and flow cytometry. Images were captured using a Nikon Eclipse T2000 microscope equipped with a low-light-sensitive charge-coupled-device (CCD) camera (Photometrics Cascade II) under the control of imaging software (Metamorph; Molecular Devices, Sunnyvale, CA). For flow cytometry, cells were detached from culture plates using enzyme-free cell dissociation buffer (Life Technologies, Grand Island, NY), and cells treated with heparanase III only were used as a background control.
Viral entry assays.
The effect of G2 peptide on HSV-2 entry was assessed using a standard entry assay as described previously (36). Briefly, confluent cultures of HeLa cells in a 96-well plate were incubated for 1 h with a serial dilution of G2 peptide. Cells were then infected with the β-galactosidase-expressing recombinant HSV-2(333) gJ− at a high MOI of 20 PFU/cell for 6 h. Enzymatic activity was then assayed using the galactosidase enzyme substrate o-nitrophenyl-β-d-galactopyranoside (ONPG; Pierce, Rockford, IL), and the optical density (OD) for the reaction was measured at 410 nm using a microplate reader (Spectra Max 190; Molecular Devices, Sunnyvale, CA).
Fluorescence microscopy was also utilized to assess HSV-2 entry after G2 peptide treatment. Confluent cultures of Vero cells were not treated, treated with control nonspecific peptide, or treated with G2 peptide at 1 mg/ml for 1 h at 37°C. Cells were then infected with GFP-expressing HSV-2 (GFP–HSV-2) at an MOI of 10 for 2 h at 37°C, washed, and incubated for another 12 h at 37°C. Imaging was performed using a 10× objective on a Zeiss Axiovert 200 fluorescence microscope.
Western blotting for viral protein VP16.
Confluent cultures of HeLa cells were either untreated or treated with 2 mg/ml G2 peptide for 30 min at 37°C. Cells were then infected with HSV-2(333) at an MOI of 10 for 4 h at 37°C. After 4 h, cells were washed 3 times with 1× PBS and once with citrate buffer (pH 3) to remove any bound virus particles. The Western blot assay was performed according to protocols described previously (21). Briefly, whole-cell lysates were denatured in NuPAGE LDS sample buffer (NP0007; Invitrogen, Carlsbad, CA) and heated to 86°C for 8 min before gel loading. Equal amounts of protein were subjected to 4 to 12% SDS-PAGE and electroblotted onto a nitrocellulose membrane. Nonspecific binding was blocked using 5% nonfat milk in Tris-buffered saline (TBS) for 2 h at 37°C. The membranes were then incubated with primary mouse monoclonal antibodies to VP16 (1:500; sc-7545; Santa Cruz Biotech, Santa Cruz, CA) overnight at 4°C. The blots were rinsed 5 times with 0.1% TTBS (0.1% Tween 20 in TBS) for 5 min, followed by incubation for 1 h at room temperature with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (IgG; 1:25,000; 115-035-062; Jackson ImmunoResearch Laboratories, West Grove, PA). Protein bands were detected using SuperSignal West Femto maximum-sensitivity substrate (34096; Pierce, Rockford, IL) and visualized using an ImageQuant LAS 4000 imager (GE Healthcare Life Sciences, Piscataway, NJ). Protein bands were quantified using ImageQuant TL image analysis software (version 7).
Plaque reduction assay.
Plaque reduction assays were performed in a pretreatment approach to test for the antiviral properties of the G2 peptide. Confluent monolayers of Vero cells in 24-well plates were pretreated with serial dilutions of the G2 peptide or with PBS for 1 h at 37°C. Cells were then infected with HSV-2(333) or green fluorescent protein (GFP)-tagged HSV-2 to yield 20 to 30 plaques per well for 2 h to allow virus adsorption. After 2 h, cells were washed three times with 1× PBS to remove unbound viral particles and then overlaid with 1% methylcellulose in DMEM supplemented with 0.05% human pooled IgG (Sigma, St. Louis, MO) to neutralize any unbound virions. Forty-eight to 72 h later, cells infected with HSV-2(333) were fixed with 100% methanol and stained with 0.1% crystal violet, and the plaques were enumerated using a microscope or, alternatively, by direct visualization. In the case of plaques formed by GFP-tagged HSV-2, plaques were visualized using fluorescence microscopy (Zeiss Axioscope microscope equipped with a digital low-light CCD camera under the control of the imaging software Axiovision) at a ×100 magnification, and plaque size was measured using Axiovision software (version 4).
Viability and cytotoxicity assays.
Fifty-percent-confluent Vero and HCE cells in 24-well plates were either untreated or treated with a serial dilution of G2 peptide in complete medium for 24 h at 37°C. Cells were then either visualized under the bright field directly for HCE cells or stained with the Hoechst 33342 live-cell nuclear stain in case of Vero cells to count viable cells' nuclei using a direct cell-counting method.
Cell-to-cell fusion assay.
Standard cell-to-cell fusion assay was used as previously described (43). Cells were split into two populations. Target cells were transfected with plasmid expressing nectin-1 as a gD receptor (1.0 μg) and the luciferase gene (0.5 μg). Effector cells were transfected with plasmids expressing HSV-2 glycoproteins gD, gB, gH, and gL and T7 RNA polymerase (0.5 μg each). At 24 h posttransfection, target cells were either treated with serial dilutions of G2 peptide for 1 h at 37°C or left untreated. Target cells were then washed 3 times with 1× PBS and were mixed with effector cells in a 1:1 ratio and replated in 24-well dishes. Luciferase activity was measured after 18 h. As a negative control, target cells were mixed with effector cells that lack HSV-2 gB.
Flow cytometry.
HS cell surface expression was detected after HSV-2(333) infection. Vero cells were either uninfected or infected with HSV-2(333) (MOI, 10) for 2 h. Cells were then washed with PBS, harvested, and incubated with mouse anti-human HS monoclonal antibody 10E4 (1:50; US Biological, Swampscott, MA) in 1% bovine serum albumin (BSA) in 1× PBS for 30 min at 4°C. After that, cells were washed and incubated for 30 min at 4°C with FITC-conjugated anti-mouse IgM (1:100; F9259; Sigma). Cells stained only with FITC-conjugated anti-mouse IgM were used as background controls.
G2 peptide binding assay.
Vero cells were either uninfected or infected with HSV-2(333) at an MOI of 10 for 2 h at 37°C. Cells were then washed and treated with 1 mg/ml FITC-conjugated G2 peptide (G2-FITC) for 30 min at 37°C. G2-FITC binding to Vero cells was examined by fluorescence microscopy at ×100 magnification. Images were analyzed using the NIH imageJ, version 1.41, software histogram analysis function. G2-FITC binding to uninfected or HSV-2-infected cells was also analyzed by flow cytometry, where untreated cells were used as a background control.
In vivo study to examine G2 peptide inhibitory effect in a mouse model of herpesvirus genital infection.
Four female BALB/c mice were pretreated for 1 h with either PBS (n = 2) or 25 μl G2-peptide (2 mg/ml) (n = 2). Mice were then infected with 1 × 103 PFU β-galactosidase-expressing HSV-2 (gJ−). At 24 h after infection, mice were sacrificed by euthanasia. Genital organs were harvested, vaginal tubes were longitudinally opened and fixed in 2% paraformaldehyde plus 0.02% Nonidet for 12 h, and then tissues were incubated for 24 h in X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) in ferricyanide buffer to stain infected cell colonies dark blue. Tissue samples were compressed to a glass slide and imaged using a Zeiss Axioscope microscope equipped with a digital camera at ×100 magnification. Mice were housed in the animal facility at the University of Illinois at Chicago Medical School. Experimental protocols involving animals were performed in accordance with the guideline of the University of Illinois at Chicago.
RESULTS
Hydrophilic properties of G2 peptide and distribution of negative charges on 3-O-sulfated HS.
G2 peptide contains mostly positively charged amino acids in the sequence MPRRRRIRRRQK with a molecular mass of 1.71 kDa. Our analysis shows that its estimated charge at pH 7.00 is approximately +8 and the ratio of its hydrophilic residues to the total number of residues is 75% (Fig. 1A). We also found that G2 peptide is soluble in PBS and reduced-serum Opti-MEM without any significant pH shift up to 20 mg/ml (11.6 mM). The putative cellular target of G2 peptide is 3-O-sulfated HS (3-OS HS), which is a polysaccharide with repeating units of a disaccharide (Fig. 1B) (43). A complex distribution of negative charges originates from multiple modifications, including sulfations, during the biosynthesis of 3-OS HS (16) (Fig. 1, broken arrows), many of which may be important for G2's ability to bind 3-OS HS.
Fig 1.
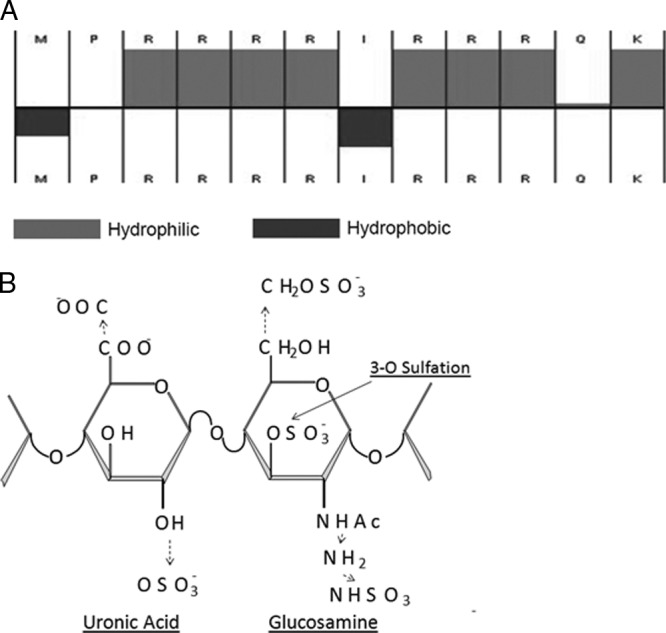
G2 peptide structure. (A) Arrangement of the hydrophilic (gray) and hydrophobic (black) amino acid residues of the G2 peptide as calculated by Innovagen peptide property calculator (Hopp & Woods), accessible from http://www.innovagen.se/custom-peptide-synthesis/peptide-property-calculator/peptide-property-calculator.asp (as of August 2011). (B) 3-O-Sulfated HS. The disaccharide structure representative of 3-O-sulfated HS is shown. The solid arrow shows the 3-O position of the glucosamine residue where sulfation is essential for HSV-1 gD binding. The broken arrows show the additional modifications, one or more of which may also be present as part of the gD-binding 3-OS HS.
G2 peptide binds to cell surface HS.
In order to examine the affinity of G2 peptide to cell surface HS, it was conjugated with FITC to generate a fluorescent peptide (G2-FITC). Cells were pretreated with heparinase III (10 IU), which selectively cleaves HS chains from cellular surfaces and from extracellular matrices (22). G2-FITC binding to heparinase-treated cells was determined by fluorescence microscopy (Fig. 2A) and by flow cytometry (Fig. 2B) and compared with the G2-FITC binding observed with untreated cells. Removal of HS from the cell surface resulted in significant loss of G2-FITC binding to Vero cells, and thus, fewer fluorescence signals were detected for treated cells than untreated cells (Fig. 2). Vero cells treated with heparinase III only were used as a background control. Similar results were obtained with HeLa cells (data not shown).
Fig 2.
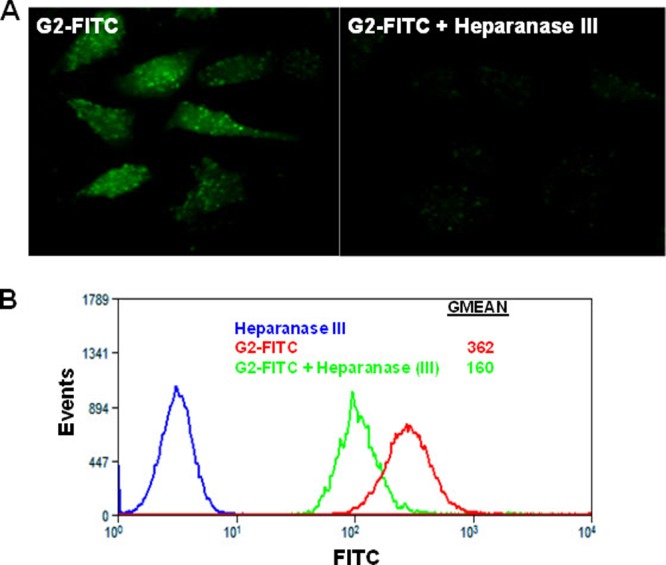
G2 peptide binds specifically to HS. (A) FITC-conjugated G2 peptide (FITC-G2) binding to Vero cells with or without heparanase III treatment imaged by fluorescence microscopy. Vero cells were grown in chamber slides and either treated or not treated with heparanase III (10 mIU) for 2 h at 37°C. This was followed by incubation at 37°C for 30 min with FITC-G2 peptide. (B) FITC-G2 peptide binding to Vero cells with or without heparanase III treatment quantified by flow cytometry. Vero cells were treated as described for panel A and processed for flow cytometry. Vero cells treated with heparanase III only were used as a background control. Results are representative of three independent experiments.
G2 peptide inhibits HSV-2 entry into host cells.
After verifying that G2 peptide binds cell surface HS, the effect of the peptide on HSV-2 entry into HeLa cells was examined. A standard HSV entry assay that utilizes a recombinant virus, HSV-2(333) gJ−, which expresses a reporter β-galactosidase upon viral entry, was used (1). G2 peptide treatment resulted in an inhibition of HSV-2 entry into HeLa cells compared to untreated cells. The concentration of G2 peptide that showed its maximum inhibitory effect was determined to be about 2.5 mg/ml (Fig. 3A). It was interesting that the peptide concentrations higher than 2.5 mg/ml were a little less effective. To demonstrate that the antiviral effect of the G2 peptide on HSV-2 entry was not cell type specific, HSV-2 entry was analyzed by fluorescence microscopy using Vero cells. Vero cells treated with 1 mg/ml G2-FITC peptide showed less of a fluorescence signal than untreated or control peptide-treated cells (Fig. 3B). For further confirmation, the effect of G2 peptide treatment on HSV-2 entry was analyzed by examining viral protein expression by Western blot analysis. HeLa cells were either untreated or pretreated with 2 mg/ml G2 peptide, followed by HSV-2 infection. G2 peptide treatment resulted in about an 80% reduction in VP16 levels, indicating that less virus was able to enter these cells than untreated HeLa cells (Fig. 4). The positive control represents a control sample for Western blotting. Together, these results suggest that G2 peptide inhibits HSV-2 entry into host cells.
Fig 3.

G2 peptide inhibits HSV-2 entry into HeLa cells. (A) Confluent cultures of HeLa cells in a 96-well plate were either left untreated or treated with serial dilutions of G2 peptide for 1 h at 37°C. Cells were then infected with the recombinant β-galactosidase-expressing HSV-2 strain 333 (gJ−) at an MOI of 20. After 6 h, the soluble substrate ONPG was added and enzymatic activity was measured. Results are representative of three independent experiments. (B) The effect of entry-blocking activity of G2 peptide was examined by fluorescence microscopy. Vero cells were not treated (No G2), treated with control nonspecific peptide (Cont. peptide), or treated with G2 peptide (G2) at 1 mg/ml for 1 h. Cells were then infected with GFP-expressing HSV-2 (GFP–HSV-2) at an MOI of 10 for 2 h, washed, and incubated for another 12 h. Imaging was performed using a ×10 objective on a Zeiss Axiovert 200 fluorescence microscope. Representative images from one experiment performed in triplicate are shown. Images of G2 peptide-treated cells under both the fluorescent light and the bright field are shown.
Fig 4.
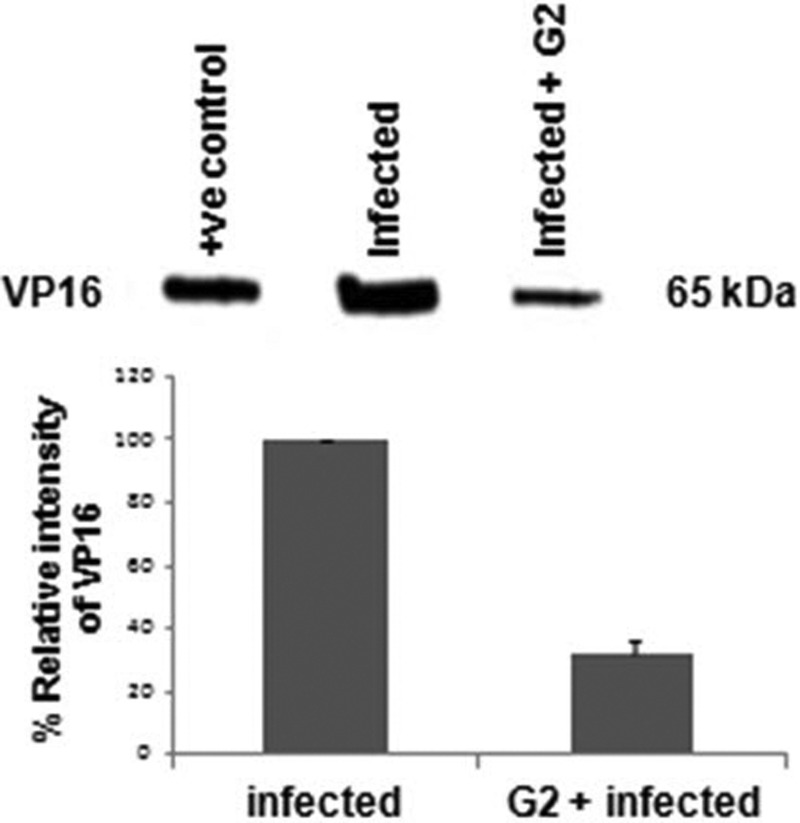
G2 peptide pretreatment reduces VP16 protein translocation into cells. As a marker for HSV-2 entry, the VP16 level was measured by Western blotting using lysates from HeLa cells untreated or treated with G2 peptide (2 mg/ml) for 30 min, followed by HSV-2 infection for 4 h. A representative Western blot is shown. The positive control represents lysates from cells transiently expressing VP16. The results are expressed as mean ± 1 SD values from two independent experiments.
In order to determine the concentration of G2 peptide that produces 50% of its maximum inhibitory effect, or the IC50 value, plaque reduction assays were performed using two different HSV-2 strains: HSV-2 strain 333 (gJ−) and GFP-tagged HSV-2 (GFP–HSV-2). G2 peptide pretreatment of Vero cells resulted in smaller plaques, as well as fewer plaques, than those for untreated cells (Fig. 5A). On the basis of the inhibition levels of both plaque size and number after G2 peptide treatment, the IC50 for G2 peptide is estimated to be approximately 1 mg/ml. It was also interesting to note that the best inhibition was observed at 2.5 mg/ml, and increasing G2 peptide concentration further did not produce better results. Representative images of plaques formed after HSV-2(333) gJ− and GFP–HSV-2 infection are shown in Fig. 5B.
Fig 5.

The IC50 of G2 peptide is approximately 1 mg/ml by plaque reduction assay. Vero cells were either untreated or treated with serial dilutions of G2 peptide for 1 h at 37°C. Cells were then infected at 20 to 30 PFU/well with either HSV-2(333) gJ− or GFP-tagged HSV-2 (GFP–HSV-2) for 2 h at 37°C. After 2 h, cells were washed and overlaid with 1% methylcellulose in DMEM supplemented with 0.05% human pooled IgG for 48 to 72 h. (A) HSV-2 plaque number and size. Plaque number was counted under the microscope. Plaque size was measured using the Axiovision software, version 4, program. Results are representative of three independent experiments. (B) Representative images of HSV-2(333) gJ− plaques after crystal violet staining (top) and GFP–HSV-2 plaques under fluorescent excitation light (bottom). Imaging was performed using a Zeiss Axioscope microscope equipped with a digital low-light CCD camera under the control of the imaging software Axiovision at a ×100 magnification.
G2 peptide is nontoxic at concentrations of 2.5 mg/ml or lower.
In order to examine the effect of G2 peptide on cell viability, direct cell-counting methods were utilized to estimate the fraction of viable cells among treated cells. The morphology of HCE cells treated with serial dilutions of G2 peptide showed that G2 peptide has cytotoxic effects at concentrations above 2.5 mg/ml. Bright-field microscopy images of HCE cells treated with serial dilutions of G2 peptide are shown in Fig. 6A. Additionally, viability of G2 peptide-treated Vero cells was evaluated by counting fluorescent nuclei stained with live-cell nuclear stain Hoechst 33342. The number of live-cell fluorescent nuclei of G2 peptide-treated Vero cells indicates that G2 cytotoxic properties are exhibited at concentrations above 2.5 mg/ml (Fig. 6B). Taken together, G2 peptide does not affect cell viability at concentrations below 2.5 mg/ml. Therefore, the determined value of the G2 peptide IC50 (1 mg/ml) has no effect on cell viability.
Fig 6.

G2 peptide affects cell viability at concentrations above 2.5 mg/ml in a dose-dependent manner. (A) HCE cells were either untreated or treated with serial dilutions of G2 peptide in MEM complete medium for 24 h at 37°C in 5% CO2. The morphological appearance of treated HCE cells was observed and compared to that of untreated cells at ×100 magnification. (B) Viability of Vero cells that were either untreated or treated with serial dilutions of G2 peptide in DMEM complete medium for 24 h at 37°C in 5% CO2 was examined by counting fluorescent nuclei of viable cells after staining the cells with Hoechst 33342 live-cell nuclear stain. Representative images of each condition are shown, as is the relative number of viable cells. Results are representative of three independent experiments.
Pretreatment with G2 peptide affects cell-to-cell fusion.
G2 peptide was isolated on the basis of its ability to bind 3-OS HS (43). Although 3-OS HS is identified as an HSV-1 entry receptor, studies have shown that it does not function as a receptor for HSV-2 (36). However, since G2 peptide pretreatment resulted in significant inhibition of HSV-2 entry and plaque formation in various HSV-2-susceptible cell lines, we sought to investigate whether G2 peptide is capable of blocking HSV-2-induced cell-to-cell fusion. Membrane fusion represents a critical step during HSV entry and an important event during virus spread (28, 42).
HSV-2-induced cell-to-cell fusion can be studied by cocultivating two cell populations: target and effector cell populations. Target cells express gD receptor and the luciferase reporter gene under the control of the T7 promoter. Effector cells express HSV-2 glycoproteins that are absolutely required for virus fusion (gB, gD, gH, and gL) plus T7 polymerase (33). In order to quantify cell-to-cell fusion, luciferase reporter gene activity is determined at 18 h postmixing of the two populations. As a negative control, target cells are mixed with effector cells that lack HSV-2 gB, where cell-to-cell fusion is expected to be reduced dramatically (28). Chinese hamster ovary (CHO-K1) cells were used in the cell-cell fusion assay and were transfected with nectin-1 plasmid as a gD receptor. Target CHO-K1 cells were treated with serial dilutions of G2 peptide for 1 h at 37°C and then washed and mixed with the effector cell population. Interestingly, G2 peptide treatment inhibited HSV-2-induced cell-to-cell fusion in a dose-dependent manner (Fig. 7). This result indicates that G2 peptide acts on HSV-2 entry by blocking membrane fusion.
Fig 7.
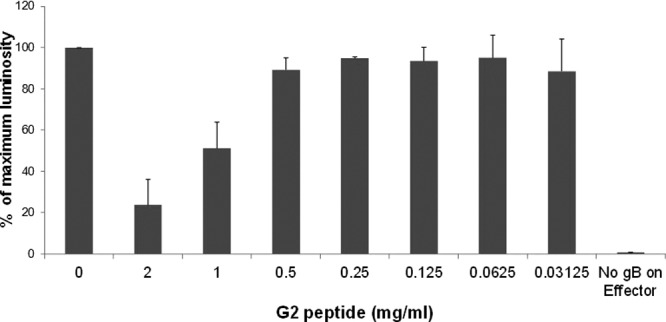
Pretreatment of target cells with G2 peptide affects cell-to-cell fusion in a dose-dependent manner. Target CHO-K1 cells expressing HSV-2 gD receptor nectin-1 and the luciferase gene under the control of the T7 promoter were preincubated with a serial dilution of G2 peptide or left untreated for 1 h at 37°C. Target cells were then mixed with an effector cell population that expresses HSV-1 fusion glycoproteins plus T7 polymerase. As a negative control for the cell fusion assay, untreated target cells were mixed with effector cells that lack HSV-2 gB. Luciferase reporter gene activity was determined to quantify cell-to-cell fusion. Results are presented as mean ± 1 SD of 3 independent experiments.
G2 peptide preferentially binds to infected cells.
In order to investigate the binding of G2 peptide to HSV-2-infected cells, G2-FITC attachment to Vero cells that were preinfected with HSV-2 was compared to the attachment of G2-FITC to uninfected Vero cells. According to the results, G2-FITC peptide showed more binding to HSV-2-infected cells than to uninfected cells. G2-FITC attachment was evaluated by fluorescence microscopy, which demonstrates about a 400 times increase, as well as flow cytometric analysis, which shows about a 214 times increase (Fig. 8). This result raises the possibility that HS expression is enhanced upon HSV-2 infection. It also suggests that G2 peptide has the capability of being used in targeted therapy, as it preferentially binds to HSV-2-infected cells.
Fig 8.

G2 peptide is preferentially bound by an infected cell population. Vero cells were either uninfected or infected with HSV-2 at an MOI of 10 for 2 h at 37°C. Cells were then treated with 1 mg/ml FITC-conjugated G2 peptide (G2-FITC) for 30 min at 37°C. (A) G2-FITC binding was examined by fluorescence microscopy at ×100 magnification. Images were analyzed using the imageJ histogram analysis function. (B) G2-FITC binding by uninfected or HSV-2-infected cells was further quantified using flow cytometry analysis. Results are representative of six independent experiments.
HSV-2 infection results in an increase in HS expression on the surface of infected cells.
Next, we sought to examine the possibility that HSV-2 infection affects HS expression on the cell surface. Vero cells were infected with HSV-2(333) at an MOI of 10 for 2 h. HS expression on the cell surface was then examined using flow cytometry and compared to the levels of HS expression on uninfected cells. HSV-2 infection resulted in a 25% increase in HS cell surface levels compared to the HS levels on uninfected cells (Fig. 9). The increase in HS cell surface expression after HSV-2 infection may explain, at least partially, the higher affinity of G2 peptide to HSV-2-infected cells than to uninfected cells, as was shown in Fig. 8.
Fig 9.
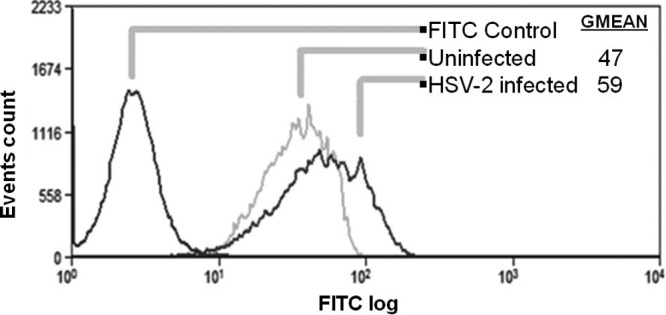
HSV-2 infection results in an increase in HS expression on the surface of infected Vero cells. The effect of HSV-2 infection on HS expression was analyzed using flow cytometric analysis of HS expression probed by FITC-conjugated anti-HS antibody specific to epitope 10E4. Vero cells were infected with HSV-2 strain 333 at an MOI of 10 for 2 h. Vero cells incubated with a FITC-conjugated secondary antibody (isotypic control) were used as a background control. Results are representative of three independent experiments.
Intravaginal G2 peptide pretreatment reduces the number of HSV-2-infected cell colonies.
A mouse genital herpes model was utilized to investigate whether G2 peptide's antiviral action can lead to protection in vivo. Mice were pretreated with either PBS or 25 μl G2 peptide (2 mg/ml) for 1 h. Mice were then inoculated with β-galactosidase-expressing HSV-2 (gJ−) and sacrificed at 24 h postinfection. Vaginal tissues were dissected and stained with X-Gal, and the number of genital lesions was counted. G2 peptide-treated mice showed a significantly lower number of genital lesions than the PBS-treated mice (Fig. 10). Results suggest that G2 peptide is effective as a prophylactic drug against HSV-2. Importantly, this is the first study to show the importance of a 3-OS HS binding peptide in protection against HSV-2 genital infection in vivo.
Fig 10.

Intravaginal G2-peptide pretreatment reduces the number of HSV-2-infected cell colonies. BALB/c female mice were pretreated for 1 h with either PBS or 25 μl G2 peptide (2 mg/ml). Mice were then infected with 1 × 103 PFU β-galactosidase-expressing HSV-2 (gJ−). At 24 h after infection, mice were sacrificed and the vaginal tissue was harvested, fixed, and incubated in X-Gal in ferricyanide buffer to stain infected cell colonies. Tissue samples were compressed onto a glass slide and imaged using a Zeiss Axioscope microscope equipped with a digital camera at ×100 magnification. (A) Vaginal tissue from mice of the indicated treatments; (B) HSV-2 (gJ−) vaginal lesions found in the vaginas from mice for the treatments noted; (C) quantitation of the vaginal lesions.
DISCUSSION
Our study has shown that G2 peptide, a 12-mer cationic peptide, exhibits antiviral activity against HSV-2. It inhibits HSV-2 entry, cell-to-cell fusion, and virus-induced plaque formation. Even more interestingly, G2 peptide demonstrates a significantly enhanced targeting of HSV-2-infected cells. As a possible explanation for that, we also demonstrate that HSV-2-infected cells show an increase in HS expression. This and virtually all other results shown here are novel and highly significant. They strongly raise the possibility that anti-3-OS HS agents can inhibit HSV-2 infection in vivo and therefore provide significant protection from the development of disease symptoms. In addition, our data suggest that G2 peptide can be used to perform two independent roles in anti-HSV-2 therapy. It could be used as a microbicide against the infection, and in parallel, it can also be exploited for preferential delivery of other antiviral drugs to infected cells.
The effect of G2 peptide is not only at the level of virus attachment; in addition, it also affects membrane fusion during entry. The evidence comes from our observation that G2 peptide blocks cell-to-cell membrane fusion induced by HSV-2 glycoproteins (Fig. 7). In addition, G2 peptide treatment significantly reduced the number of plaques formed by HSV-2 and it also resulted in plaques smaller than those for untreated cells (Fig. 5). Plaque assays were performed in the presence of neutralizing antibodies, where plaque sizes depended exclusively on cell-to-cell spread of the virus, suggesting that G2 peptide reduces the efficiency of virus spread, most likely by blocking the membrane fusion required for spread. Interestingly, the concentration that most effectively blocks HSV-2 entry and plaque formation is 2.5 mg/ml (Fig. 3 and 5); increasing the concentration further shows less of an inhibitory effect. The rebound in percent reduction in plaque sizes and numbers noticed at 5 mg/ml and 10 mg/ml might be attributable to transmembrane channel opening, since G2 peptide has structural similarity to cell-penetrating arginine-rich peptides (44). The higher concentrations might result in the disruption of the host cell membrane integrity and a receptor-independent entry, and similarly, spread of the virions may be less inhibited. Studies will be performed in the future to further investigate this possibility.
Since G2 peptide was isolated against 3-OS HS, the suggested mechanism of activity is that G2 peptide binds to modified 3-OS HS on the cell surface, preventing virus attachment and eventually the fusion of the virions with cells, thus reducing virus entry. Consistent with that, heparanase treatment of cells reduced the ability of G2 peptide to bind cultured cells (Fig. 2). The effect on membrane fusion was indicated by the observation that G2 peptide treatment of target cells inhibited HSV-2-induced cell-to-cell fusion (Fig. 7). So, the intriguing question is that if 3-OS HS is not a receptor for HSV-2, then what is the mechanism by which G2 peptide blocks entry and fusion? One possible explanation is that HSV-2 gD may contain a partial 3-OS HS binding domain, blocking of which may block the membrane fusion altogether. The putative 3-OS HS binding domain of HSV-1 gD overlaps the HVEM and/or nectin-1 binding domain, and that is why blocking the 3-OS HS binding domain blocks entry via all the receptors (11, 43). Therefore, the suggested mechanism of activity of the G2 peptide is a 2-fold effect by inhibiting virus attachment as well as membrane fusion during virus entry.
A related interesting finding is that G2 peptide binding by HSV-2-infected cells was significantly higher than that by uninfected cells. The dramatic, 200% increase in G2 peptide binding to HSV-2-infected cells (Fig. 8) cannot be explained solely in light of the increased HS availability on the surface of HSV-2-infected cells. Our interesting results indicate that HS cell surface expression also increased, but only by 25% (Fig. 9). We propose that the infection-dependent increase in HS is accompanied by hyper 3-OS HS modifications, and that may be a reason why the 3-OS HS binding G2 peptide shows a 200% increase in binding to the infected cells, while the total HS is increased by only 25%. It has been reported that modifications within HS are altered in response to certain inflammatory and stress signals mediated by inflammatory cytokines (12). HSV infection is considered a viral inflammatory condition, with proinflammatory cytokines and chemokines released from HSV-infected cells during different phases of the infection. It is possible that the cytokines enhance the enzymatic activities of 6 or more 3-O-sulfotransferases (25, 50), which in turn can induce hyper 3-O-sulfation in newly synthesized HS moieties during HSV-2 infection. Currently, the precise functions of 3-O-sulfotransferases are not very clear, and it is possible that they respond to stress by increasing the 3-O-sulfation in HS. Further studies must be done to identify the structural changes induced in HS upon HSV-2 infection.
The therapeutic margin of the G2 peptide is relatively narrow, as its IC50 equals 1 mg/ml, while its 50% cytotoxic concentration (CC50) is about 10 mg/ml. Nevertheless, the binding of G2 peptide by infected cells is enhanced and is more than 200% higher than that by noninfected cells. This opens the door for targeted therapeutic potential and considerably widens the therapeutic margin of the G2 peptide. In addition, the higher affinity of G2 peptide to HSV-2-infected cells than to uninfected cells advances the therapeutic applications of G2 peptide for use as a delivery molecule, in addition to its use for its antiviral effect. It also paves the way for the discovery of new HS binding small molecules that can be used for preferential targeting of infected cells, which will avoid healthy cells and thereby reduce any associated cytotoxicity to healthy cells (7, 9, 15, 26, 46).
To our understanding, our animal study provides a first-time proof of the importance of blocking of 3-OS HS during HSV-2 infection in vivo. In this regard, administering G2 peptide to female BALB/c mice before HSV-2 challenge significantly reduced the number of vaginal herpetic lesions compared to that in untreated mice (Fig. 10). The peptide was well tolerated, and the animals did not exhibit any adverse reaction to its application before or after the infection. However, extensive toxicology studies and more work need to be done to determine whether a preventive application of the peptide can block the virus from establishing latency, and likewise, the efficacy of the peptide needs to be examined under therapeutic conditions for treating an existing infection.
In summary, our results demonstrate that the cationic G2 peptide inhibits HSV-2 infection in cell culture as well as in vivo using the mouse model of genital herpes. G2 peptide binds to 3-OS HS, disrupting virus attachment and entry into cells. G2 peptide protects mice from HSV infection and resulted in fewer herpetic genital lesions compared to the number in untreated animals. These studies show the promise that G2 peptide or similar 3-OS HS binding agents can be developed into effective anti-HSV agents. Similarly, the 200% increase in G2 peptide binding to HSV-2-infected cells can be exploited in the future for diagnosis and targeted delivery of antiviral agents to the infected cells. The possibility that 3-O-sulfation of HS itself may increase upon HSV-2 infection will almost certainly spark new studies on HS modifications and how they may be regulated in response to infection and stress.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants AI057860 and AI081869 (both to D. Shukla) and core grant EY01792.
We thank Patricia G. Spear (Northwestern University, Chicago, IL), Bellur S. Prabhakar (University of Illinois at Chicago, Chicago, IL), Kozaburo Hayashi (National Eye Institute, Bethesda, MD), and Jeffrey Vieira (University of Washington, Seattle, WA) for providing cell lines, viruses, and plasmids used in this study.
Footnotes
Published ahead of print 4 April 2012
REFERENCES
- 1. Akhtar J, et al. 2008. HVEM and nectin-1 are the major mediators of herpes simplex virus 1 (HSV-1) entry into human conjunctival epithelium. Invest. Ophthalmol. Vis. Sci. 49:4026–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Albiol Matanic VC, Castilla V. 2004. Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int. J. Antimicrob. Agents 23:382–389 [DOI] [PubMed] [Google Scholar]
- 3. Andersen JH, Jenssen H, Gutteberg TJ. 2003. Lactoferrin and lactoferricin inhibit herpes simplex 1 and 2 infection and exhibit synergy when combined with acyclovir. Antiviral Res. 58:209–215 [DOI] [PubMed] [Google Scholar]
- 4. Andersen JH, Jenssen H, Sandvik K, Gutteberg TJ. 2004. Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J. Med. Virol. 74:262–271 [DOI] [PubMed] [Google Scholar]
- 5. Araki-Sasaki K, et al. 1995. An SV40-immortalized human corneal epithelial cell line and its characterization. Invest. Ophthalmol. Vis. Sci. 36:614–621 [PubMed] [Google Scholar]
- 6. Arii J, et al. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862 [DOI] [PubMed] [Google Scholar]
- 7. Bernstein DI, Ireland J, Bourne N. 2000. Pathogenesis of acyclovir-resistant herpes simplex type 2 isolates in animal models of genital herpes: models for antiviral evaluations. Antiviral Res. 47:159–169 [DOI] [PubMed] [Google Scholar]
- 8. Bhattacharjee PS, Neumann DM, Hill JM. 2009. A human apolipoprotein E mimetic peptide effectively inhibits HSV-1 TK-positive and TK-negative acute epithelial keratitis in rabbits. Curr. Eye Res. 34:99–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brady RC, Bernstein DI. 2004. Treatment of herpes simplex virus infections. Antiviral Res. 61:73–81 [DOI] [PubMed] [Google Scholar]
- 10. Cardin AD, Weintraub HJ. 1989. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9:21–32 [DOI] [PubMed] [Google Scholar]
- 11. Carfí A, et al. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 8:169–179 [DOI] [PubMed] [Google Scholar]
- 12. Carter NM, Ali S, Kirby JA. 2003. Endothelial inflammation: the role of differential expression of N-deacetylase/N-sulphotransferase enzymes in alteration of the immunological properties of heparan sulphate. J. Cell Sci. 116:3591–3600 [DOI] [PubMed] [Google Scholar]
- 13. Compton T, Nowlin DM, Cooper NR. 1993. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 193:834–841 [DOI] [PubMed] [Google Scholar]
- 14. de Jong MA, de Witte L, Taylor ME, Geijtenbeek TB. 2010. Herpes simplex virus type 2 enhances HIV-1 susceptibility by affecting Langerhans cell function. J. Immunol. 185:1633–1641 [DOI] [PubMed] [Google Scholar]
- 15. Eizuru Y. 2003. Development of new antivirals for herpesviruses. Antivir. Chem. Chemother. 14:299–308 [DOI] [PubMed] [Google Scholar]
- 16. Esko JD, Lindahl U. 2001. Molecular diversity of heparan sulfate. J. Clin. Invest. 108:169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 18. Gruenheid S, Gatzke L, Meadows H, Tufaro F. 1993. Herpes simplex virus infection and propagation in a mouse L cell mutant lacking heparan sulfate proteoglycans. J. Virol. 67:93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65:1090–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jacquet A, et al. 1998. The varicella zoster virus glycoprotein B (gB) plays a role in virus binding to cell surface heparan sulfate proteoglycans. Virus Res. 53:197–207 [DOI] [PubMed] [Google Scholar]
- 21. Karasneh GA, Ali M, Shukla D. 2011. An important role for syndecan-1 in herpes simplex virus type-1 induced cell-to-cell fusion and virus spread. PLoS One 6:e25252 doi:10.1371/journal.pone.0025252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kimura C, Oike M. 2008. Heparan sulfate proteoglycan is essential to thrombin-induced calcium transients and nitric oxide production in aortic endothelial cells. Thromb. Haemost. 100:483–488 [PubMed] [Google Scholar]
- 23. Krummenacher C, et al. 1999. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity, while the third domain is involved in oligomerization of HveC. J. Virol. 73:8127–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu J, Shworak NW, Fritze LM, Edelberg JM, Rosenberg RD. 1996. Purification of heparan sulfate D-glucosaminyl 3-O-sulfotransferase. J. Biol. Chem. 271:27072–27082 [DOI] [PubMed] [Google Scholar]
- 25. Liu J, et al. 1999. Expression of heparan sulfate D-glucosaminyl 3-O-sulfotransferase isoforms reveals novel substrate specificities. J. Biol. Chem. 274:5185–5192 [DOI] [PubMed] [Google Scholar]
- 26. Martens MG, Fife KH, Leone PA, Dix LP, Brennan CA. 2009. Once daily valacyclovir for reducing viral shedding in subjects newly diagnosed with genital herpes. Infect. Dis. Obstet. Gynecol. 2009:105376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 28. Muggeridge MI. 2000. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J. Gen. Virol. 81:2017–2027 [DOI] [PubMed] [Google Scholar]
- 29. Nilsson BL, Soellner MB, Raines RT. 2005. Chemical synthesis of proteins. Annu. Rev. Biophys. Biomol. Structure 34:91–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Donnell CD, Shukla D. 2009. A novel function of heparan sulfate in the regulation of cell-cell fusion. J. Biol. Chem. 284:29654–29665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Donnell CD, Tiwari V, Oh MJ, Shukla D. 2006. A role for heparan sulfate 3-O-sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology 346:452–459 [DOI] [PubMed] [Google Scholar]
- 32. Oh MJ, Akhtar J, Desai P, Shukla D. 2010. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 391:176–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 34. Satoh T, et al. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scanlan PM, Tiwari V, Bommireddy S, Shukla D. 2003. Cellular expression of gH confers resistance to herpes simplex virus type-1 entry. Virology 312:14–24 [DOI] [PubMed] [Google Scholar]
- 36. Shukla D, et al. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22 [DOI] [PubMed] [Google Scholar]
- 37. Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shworak NW, et al. 1999. Multiple isoforms of heparan sulfate d-glucosaminyl 3-O-sulfotransferase. Isolation, characterization, and expression of human cDNAs and identification of distinct genomic loci. J. Biol. Chem. 274:5170–5184 [DOI] [PubMed] [Google Scholar]
- 39. Sinha S, Cheshenko N, Lehrer RI, Herold BC. 2003. NP-1, a rabbit alpha-defensin, prevents the entry and intercellular spread of herpes simplex virus type 2. Antimicrob. Agents Chemother. 47:494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stanberry L, et al. 1999. New developments in the epidemiology, natural history and management of genital herpes. Antiviral Res. 42:1–14 [DOI] [PubMed] [Google Scholar]
- 41. Tiwari V, O'Donnell CD, Oh MJ, Valyi-Nagy T, Shukla D. 2005. A role for 3-O-sulfotransferase isoform-4 in assisting HSV-1 entry and spread. Biochem. Biophys. Res. Commun. 338:930–937 [DOI] [PubMed] [Google Scholar]
- 42. Tiwari V, ten Dam GB, Yue BY, van Kuppevelt TH, Shukla D. 2007. Role of 3-O-sulfated heparan sulfate in virus-induced polykaryocyte formation. FEBS Lett. 581:4468–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tiwari V, Liu J, Valyi-Nagy T, Shukla D. 2011. Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J. Biol. Chem. 286:25406–25415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tyagi M, Rusnati M, Presta M, Giacca M. 2001. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 276:3254–3261 [DOI] [PubMed] [Google Scholar]
- 45. Warner MS, et al. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189 [DOI] [PubMed] [Google Scholar]
- 46. Weller S, et al. 1993. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin. Pharmacol. Ther. 54:595–605 [DOI] [PubMed] [Google Scholar]
- 47. Wigdahl B, Smith CA, Traglia HM, Rapp F. 1984. Herpes simplex virus latency in isolated human neurons. Proc. Natl. Acad. Sci. U. S. A. 81:6217–6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. WuDunn D, Spear PG. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63:52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xia G, et al. 2002. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus, type 1. J. Biol. Chem. 277:37912–37919 [DOI] [PubMed] [Google Scholar]
- 50. Xu D, et al. 2005. Characterization of heparan sulphate 3-O-sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem. J. 385:451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yasin B, et al. 2000. Evaluation of the inactivation of infectious herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 19:187–194 [DOI] [PubMed] [Google Scholar]