

Abstract
The hepadnavirus reverse transcriptase (RT) has the unique ability to initiate viral DNA synthesis using RT itself as a protein primer. Protein priming requires complex interactions between the N-terminal TP (terminal protein) domain, where the primer (a specific Y residue) resides, and the central RT domain, which harbors the polymerase active site. While it normally utilizes the cis-linked TP to prime DNA synthesis (cis-priming), we found that the duck hepatitis B virus (DHBV) RT domain, in the context of the full-length RT protein or a mini-RT construct containing only truncated TP and RT domains, could additionally use a separate TP or RT domain in trans as a primer (trans-priming). trans interaction could also be demonstrated by the inhibitory effect (trans-inhibition) on cis-priming by TP and RT domain sequences provided in trans. Protein priming was further shown to induce RT conformational changes that resulted in TP-RT domain dissociation, altered priming site selection, and a gain of sensitivity to a pyrophosphate analog inhibitor. trans-priming, trans-inhibition, and trans-complementation, which requires separate TP and RT domains to reconstitute a functional RT protein, were employed to define the sequences in the TP and RT domains that could mediate physical or functional inter- and intradomain interactions. These results provide new insights into TP-RT domain interactions and conformational dynamics during protein priming and suggest novel means to inhibit protein priming by targeting these interactions and the associated conformational transitions.
INTRODUCTION
The hepatitis B virus (HBV) is a major human pathogen that chronically infects over 350 million people worldwide (15, 32). Chronic HBV infection is a major cause of end-stage liver diseases, including cirrhosis and hepatocellular carcinoma, resulting in a million fatalities annually. HBV is a member of the Hepadnaviridae family, which also includes related viruses that infect other mammalian and avian species (39). In particular, the duck hepatitis B virus (DHBV) has been a widely used model to study many different aspects of HBV replication and pathogenesis. All hepadnaviruses, as pararetroviruses, replicate a short (ca. 3-kb), partially double-stranded (DS), relaxed circular DNA genome via packaging and reverse transcription of a pregenomic RNA (pgRNA) by a virally encoded reverse transcriptase (RT) (37, 39, 41).
The hepadnavirus RT is a multifunctional protein with unique structural and functional properties (20, 21). Like its retroviral counterparts, RT catalyzes the synthesis of the DS viral DNA, first the minus strand from the pgRNA template and then the plus strand from the minus-strand DNA template (10, 12, 35, 47). Also in common with retroviral RTs, the hepadnavirus RT has an RNase H activity that degrades the pgRNA template during the synthesis of the viral minus-strand DNA (10, 11, 35). Thus, the central and C-terminal regions of RT harbor, respectively, the RT and RNase H domains that are homologous to retroviral RTs. Uniquely, however, the hepadnavirus RT has a so-called N-terminal, terminal protein (TP) domain, which is conserved among all hepadnaviruses but absent from all other RTs. TP is linked to the RT domain via a flexible spacer region. Furthermore, the hepadnavirus RT is able to initiate minus-strand DNA synthesis using itself as a protein primer, via a complex protein priming mechanism whereby a specific tyrosine residue in the TP domain is used as a primer, resulting in a covalent linkage between the 5′ end of the viral minus-strand DNA and the RT protein via a phosphotyrosyl bond (4, 28, 43, 49, 53, 56). Furthermore, protein priming requires RT recognition of a specific viral RNA, an RNA stem-loop structure located on the 5′ end of pgRNA called ε (14, 20, 23, 33, 34, 36, 44, 46, 48, 50). In particular, the sequence from an internal bulge of ε serves as the specific template for protein priming to direct the synthesis of a short (3- to 4-nucleotide [nt]) minus-strand DNA oligomer that is covalently linked to RT. Based on distinct sequence and structural requirements, protein priming in vitro by the DHBV RT has been subdivided into two sequential stages, i.e., the first stage of priming initiation resulting in the formation of the phosphotyrosyl bond between the primer Y residue (Y96 in DHBV) and the first nucleotide (dGMP) of the minus-strand DNA and the second stage of DNA polymerization involving the addition of the next 2 to 3 nt to the initiating dGMP via conventional DNA phosphodiester linkages (31, 51).
Extensive genetic and biochemical studies have shown that both the TP and RT domains of the hepadnavirus RT protein are required to interact with ε and to carry out protein priming whereas the spacer and the C-terminal RNase H are dispensable (17, 27, 34, 50, 51). For protein priming to occur, the TP and RT domains must interact precisely so that the primer Y residing in TP is properly positioned into the DNA polymerase active site in the RT domain. Furthermore, these two domains must simultaneously engage the ε RNA so that its internal bulge template sequence is properly positioned next to the TP primer as well as the RT active site for phosphotyrosyl bond formation (i.e., initiation of protein priming or TP deoxynucleotidylation). In order for RT to adopt a conformation competent for interaction with ε, specific host factors, including a cellular chaperone complex consisting of the heat shock protein 90 (Hsp90), Hsp70, and other cochaperones, are recruited to associate with RT and facilitate the establishment of the ε-binding competent state (18, 19, 22, 25). Furthermore, upon specific RT and ε interaction, conformational changes are triggered in both the RT protein and the ε RNA of the resulting ribonucleoprotein (RNP) complex through an induced-fit mechanism and are thought to activate the RT enzymatic activity and the ε template function (6, 40, 42, 45).
We and others have reconstituted DHBV RT-ε interaction and protein priming using purified, bacterially expressed RT, its cognate ε RNA, and the eukaryotic chaperone proteins (7, 17, 24, 40). Detailed analyses of the DHBV RT requirements for protein priming also led us to the construction of a severely truncated RT protein, MiniRT2, which lacks part of the TP, the spacer, part of the RT, and the entire RNase H domain and retains the ability to carry out authentic, ε-dependent protein priming in vitro but is no longer dependent on the host chaperone proteins for ε binding and protein priming (31, 52). In addition, separately expressed TP and RT domains, containing the minimal TP and RT domain sequences as defined in MiniRT2, can interact in trans, i.e., intermolecularly, to reconstitute a functional RT protein and carry out protein priming (7, 27, 29, 31).
Recent work using the simplified in vitro DHBV priming systems, including MiniRT2, has demonstrated that hepadnavirus RT is remarkably flexible and dynamic in structure and function. For example, we have shown that the divalent metal ions, Mn2+ versus Mg2+, can induce significantly different RT conformations that dramatically affect the RT protein priming functions, including catalytic efficiency, template and nucleotide selectivity, and the transition from priming initiation to DNA polymerization (31). In addition, sensitivity to inhibition by a pyrophosphate analog, phosphonoformic acid (PFA), can be induced during the polymerization but not the initiation stage of protein priming in the presence of Mn2+, whereas PFA shows no effect on protein priming (either initiation or polymerization) in the presence of Mg2+ and inhibits viral DNA synthesis only following protein priming (31, 49). Furthermore, we and others have recently discovered that DHBV RT protein can utilize so-called cryptic priming sites, i.e., Y as well as S/T residues, other than Y96, in both the TP and RT domains to initiate DNA synthesis (5, 8). As with authentic priming at Y96, priming at the cryptic sites requires both the TP and RT (in particular, its polymerase active site) domains and the ε RNA and is stimulated by Mn2+ relative to Mg2+.
In the present study, we found that MiniRT2, and the full-length DHBV RT, was able to initiate protein priming on a separate TP or RT domain provided in trans (trans-priming), in addition to carrying out protein priming from the cis-linked TP (cis-priming). Further studies showed that TP and RT domain sequences provided in trans could also exert an inhibitory effect on cis-priming (trans-inhibition). Taking advantage of these trans-priming and trans-inhibition effects, as well as the trans-complementation priming assay, we have defined the TP and RT domain sequences that were necessary for primer function, priming inhibition, or priming reconstitution through inter- and intradomain interactions in cis (i.e., intramolecular) or in trans (i.e., intermolecular).
MATERIALS AND METHODS
Plasmids.
pGEX-MiniRT2 and pQE-MiniRT2 express the truncated DHBV MiniRT2 protein that is fused to the glutathione S-transferase (GST) and the six-histidine (6×His) tag, respectively (17). pSUMO-MiniRT2 expresses the same DHBV MiniRT2 protein with an N-terminal SUMO tag as well as the 6×His tag. It was created by subcloning the MiniRT2 coding sequence downstream of the SUMO coding sequence in pSUMO-T7-Amp (Lifesensors) with an additional triple-Flag tag inserted between SUMO and the MiniRT2 sequences. pGEX-MiniRT2-YMHA was derived from pGEX-MiniRT2 and contains two amino acid substitutions in the RT active site (changing the conserved YMDD motif into YMHA) (10, 51) that abolish the polymerase activity of RT. pQE-TP and pQE-RT express the 6×His-tagged, truncated TP (residues 75 to 220 [75-220]) and RT (residues 349 to 575 [349-575]) domains, respectively, and were derived from pQE-MiniRT2 by removing the RT and TP coding sequences, respectively (31). Similarly, pGEX-TP and pGEX-RT were derived from pGEX-MiniRT2 and express the GST-tagged, truncated DHBV TP and DHBV RT domains, respectively (30). The pQE-TP-Y96F was derived from pQE-TP by changing the tyrosine residue at position 96 to phenylalanine (8, 56). Additional truncated pGEX-TP and -RT fragments were generated by PCR. The PCR-amplified TP or RT fragments were then cloned into the pGEX vector fused in frame downstream of the GST coding sequence. pHP expresses the full-length DHBV RT under the phage SP6 promoter in vitro (56). All mutations were verified by DNA sequencing.
Protein expression and purification.
The DHBV full-length RT protein and SUMO-MiniRT2 were expressed in vitro using a coupled in vitro transcription and translation reaction kit, the TnT rabbit reticulocyte lysate (RRL) system (Promega), according to the manufacturer's instructions. The amount of translated SUMO-MiniRT2 (3×Flag tagged) was estimated to be 5 to 10 ng/μl translation reaction mixture, based on Western blotting using the anti-Flag antibody (Sigma) and SUMO-MiniRT2 standards purified from bacteria. The translation yield of the full-length RT protein was similar to that of SUMO-MiniRT2. GST-MiniRT2, -RT, and -TP were expressed in the BL21(DE3)-CodonPlus-RIL cells and purified using the glutathione resin, and SUMO-MiniRT2 was expressed using the same cells and purified using Ni+ affinity resins (17, 19). His-MiniRT2, -RT, and -TP were expressed in M15(pREP4) cells and purified using Ni+ affinity resins under native conditions (Qiagen) (17). His-RT, His-TP, and SUMO-MiniRT2 proteins were also purified under denaturing conditions and refolded as described previously (8, 52).
In vitro protein priming.
The protein priming reaction was carried out as previously described (8). Briefly, 1 pmol of purified RT or 2.5 μl of the in vitro translation reaction mixture was mixed with the DHBV mini-ε RNA (6 pmol) (17), 1× EDTA-free protease inhibitor cocktail (Roche), 0.5 μl [α-32P]dGTP (3,000 Ci/mmol and 10 mCi/ml) or another labeled deoxynucleoside triphosphate (dNTP) as indicated per 10-μl reaction mixture, and TMnNK (10 mM Tris-HCl, pH 8.0, 1 mM MnCl2, 15 mM NaCl, 20 mM KCl) or TMgNK (same as TMnNK, except containing 2 mM MgCl2 instead of 1 mM MnCl2) buffer. NP-40 (0.2%, vol/vol) was added to stimulate protein priming (52). For the trans-complementation assay, equimolar amounts (1 pmol each) of TP and RT domains were used. For the trans-priming assay, 1 pmol of MiniRT2 was used. In addition, 1 pmol (unless indicated otherwise) of the RT or TP domain fragments was also used. To test the inhibitory effect of the different TP and RT fragments on protein priming, TP and RT fragments (32 pmol unless otherwise indicated) were mixed with 1 pmol of purified MiniRT2 or 2.5 μl of the full-length RT or MiniRT2 translated in RRL, for 15 min at room temperature. The ε RNA was then added, and the reaction mixtures were incubated for an additional 15 min at room temperature before adding the TMnNK or TMgNK buffer and [α-32P]dGTP. Alternatively, 1 pmol of purified MiniRT2 was mixed first with the ε RNA and incubated for 15 min at room temperature. For the full-length RT and MiniRT2 translated in RRL, the ε RNA was added during translation to allow the RNA binding to RT. TP and RT fragments (32 pmol unless indicated otherwise) were then added to the RNP complex, and the samples were further incubated for 15 min at room temperature before adding the TMnNK or TMgNK buffer and [α-32P]dGTP. The protein priming reaction was carried out at 30°C for 2 h. The reaction products were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and quantified by phosphorimaging.
Phosphoamino acid analysis.
Phosphoamino acid analysis was carried out as described previously (8). Briefly, following SDS-PAGE, the gel was cut into two pieces. Both pieces were fixed with a 10% acetic acid and isopropanol mixture for 1 h with two changes. The gel pieces were rinsed with distilled water twice. One piece was then treated with 3 M KOH at 55°C for 14 h, and the other was mock treated with water. After KOH or water treatment, the gel pieces were treated with a 10% acetic acid and isopropanol mixture and then with water. Subsequently, the gel pieces were dried and protein priming signals were quantified by phosphorimaging.
RESULTS
Expression and purification of DHBV RT proteins and domains.
To facilitate expression and purification, we produced the DHBV MiniRT2 protein and the individual TP (75–220) and RT (349–575) domains derived from MiniRT2 (Fig. 1A) with the GST or 6×His tag and purified them by glutathione or Ni+ affinity methods as described before (17, 19, 30). As an additional attempt to enhance solubility, MiniRT2 was also fused to the SUMO tag. Under native purification conditions, the TP and RT domains and SUMO-MiniRT2 were copurified with GroEL and DnaK, two bacterial chaperone proteins known to bind the DHBV and HBV RT proteins (Fig. 1B, lanes 1, 8, and 18) (17, 19). The SUMO-MiniRT2 that was purified under denaturing conditions did not contain the bacterial chaperone proteins and was >95% pure (Fig. 1B, lane 19), with only some degradation products as minor contaminants. Additional TP and RT domain constructs (other than 75–220 and 349–575) derived from further truncations were also made as GST fusions and similarly purified using glutathione resins (Fig. 1) and will be described below.
Fig 1.

Construction of DHBV RT proteins and individual TP and RT domains. (A) Schematic diagrams of the RT protein and individual TP and RT domain constructs. The top diagram depicts the full-length DHBV RT protein, with the primer Y residue (Y96) in the TP domain and the 511YMDD514 active site in the RT domain denoted. The boundaries (in amino acid positions) of the truncated MiniRT2 (second diagram) and the TP (left) and RT (right) domain constructs (below the MiniRT2 diagram) are indicated. (B) Purification of RT proteins and domains. The DHBV SUMO-MiniRT2 protein and the GST-tagged TP and RT domain constructs were expressed in Escherichia coli and purified by affinity methods as described in Materials and Methods. The GST-tagged TP and RT domains were purified under native conditions (lanes 1 to 17). SUMO-MiniRT2 (His tagged) was purified under either native (N, lane 18) or denaturing (and refolding [R], lane 19) conditions. The purified MiniRT2 protein and domains were analyzed by SDS-PAGE and Coomassie blue staining. The intact protein or domain species are denoted by the stars to the left of the corresponding bands. The copurifying bacterial chaperone proteins, DnaK and GroEL, are also indicated. The absence of GroEL in TP/110–220 (lane 7) is denoted by the dashed box. The protein molecular mass markers are indicated on the left in kDa. The bracket denotes the degradation products from the GST-tagged TP and RT domain constructs or SUMO-MiniRT2, consisting mostly of the GST or SUMO tag plus variable amounts of the TP or RT sequences remaining attached to the tag.
TP and RT domains served as primers in trans as well as in cis.
It is known that the separate TP and RT domains can trans-complement each other to carry out protein priming using the authentic Y96 site in TP and, to a much lesser degree, the cryptic priming sites in both the TP and RT domains (7, 8, 29, 31) (Fig. 2, lanes 1 and 2). When the priming reaction was carried out using MiniRT2 in the presence of an extra RT or TP domain, in trans, we found, surprisingly, that the separate RT and TP domains were also apparently used as protein primers for initiating DNA synthesis (Fig. 2) by MiniRT2. Furthermore, the ability of the TP and RT domains to serve as primers used by MiniRT2 was independent of the nature of the tag on the RT or TP domain. Thus, 6×His-tagged (Fig. 2, lanes 1 to 4, 6, 9, 13, and 17) or GST-tagged (lanes 7 and 10) TP and 6×His-tagged RT (lanes 12 and 16) all served as primers. As MiniRT2 was able to utilize the separate TP (and apparently separate RT, but see below) in trans as well as its own cis-linked TP as a primer, we called priming from the separate TP by MiniRT2 trans-priming to differentiate it from cis-priming, whereby MiniRT2 uses its own cis-linked TP as the primer. As with cis-priming, trans-priming was also detected from TP-Y96F, which lacks the authentic priming site, indicating that trans-priming was occurring at the cryptic site(s) (Fig. 2, lanes 14 and 18). Both refolded and natively purified TP domain served as a trans-primer (Fig. 2, lanes 3, 4, 6, 9, 13, and 17). The ability of MiniRT2 to initiate protein priming on the separate RT and TP domains in trans was also independent of the nature of its own tag. Thus, SUMO-tagged (Fig. 2, lanes 3, 4, and 8 to 10), 6×His-tagged (lanes 12 to 14), and GST-tagged (lanes 5 to 7) MiniRT2s were all able to carry out priming using the TP and RT domains in trans. Both refolded and natively purified SUMO-MiniRT2 was able to carry out trans-priming (Fig. 2, lane 4 versus lane 3). Equimolar amounts of the MiniRT2 proteins and the individual TP or RT domains (1 pmol each) were used in all the priming reactions, except that 4 pmol of His-TP or His-RT was used in the reactions shown in lanes 11 to 18 in Fig. 2, due to the lower priming activity of His-MiniRT2 than of SUMO-MiniRT2 or GST-MiniRT2.
Fig 2.

trans-priming from the TP and RT domains by purified MiniRT2. Purified MiniRT2, TP, and RT domains were mixed together as indicated for protein priming in the presence of [α-32P]dGTP and the DHBV ε RNA. The RT proteins used were SUMO-MiniRT2 refolded (lane 4) or natively purified (lanes 3 and 8 to 10), natively purified GST-MiniRT2 (lanes 5 to 7), His-MiniRT2 (lanes 11 to 14), and His-MiniRT2-Y96F (lanes 15 to 18). The individual domains used were His-tagged and natively purified (lanes 1, 3, 13, and 17) or refolded (lanes 2, 4, 6, and 9) TP or TP-Y96F (lanes 14 and 18), natively purified GST-TP (lanes 7 and 10), and His-tagged RT domain natively purified (lanes 1, 12, and 16) or refolded (lane 2). All priming reactions were conducted in TMnNK. Priming reaction products were resolved by SDS-PAGE and detected by autoradiography. The labeled RT proteins and domains, as a result of cis- or trans-priming, are indicated. All reactions used 1 pmol of proteins/domains, except those shown in lanes 11 to 18, which used 4 pmol of His-TP or His-RT.
When the RT or TP domain was mixed with MiniRT2-Y96F, the mutant MiniRT2 was able to initiate protein priming in cis (i.e., using cryptic sites) (Fig. 2, lanes 15 to 18), as shown previously (8), but also in trans on the RT (lane 16) and the wild-type (WT)- and Y96F-TP (lanes 17 and 18) domain, indicating that Y96 was not essential for trans-priming, as was true for cis-priming and priming via trans-complementation (i.e., priming in the presence of separate TP and RT domains but no MiniRT2 or full-length RT; see below) (8). On the other hand, a mutant (YMHA) RT domain that lacks a functional polymerase active site was not able to serve as a primer when mixed with MiniRT2 (see Fig. S1A, lane 3, in the supplemental material), indicating that the apparent trans-priming observed on the WT RT domain (Fig. 2, lanes 12 and 16; see also Fig. S1A, lane 2) was in fact priming in cis (i.e., the RT domain acting on itself) and that the RT domain was unable to serve as a primer in trans to be used by another RT domain (also see more results below using additional RT domain constructs), in agreement with a recent report (5). As the RT domain by itself, in the absence of a functional TP, is unable to carry out protein priming on either the TP or the RT domain (8), this result indicated that the RT domain in trans was able to access TP in MiniRT2.
The above reactions were all carried out in the presence of Mn2+ due to the higher priming activity with this metal cofactor (31). Since RT presumably utilizes Mg2+ rather than Mn2+ for protein priming under in vivo conditions, we also tested the ability of MiniRT2 to carry out protein priming on the separate TP domain in trans, in the presence of Mg2+. The result showed that the GST- or SUMO-tagged MiniRT2 protein was able to carry out protein priming on TP in trans in the presence of Mg2+ (see Fig. S1B in the supplemental material, lanes 2 and 4), though much less efficiently than with Mn2+. Furthermore, the full-length DHBV RT protein was also able to initiate priming on TP in trans, although excess TP (40 pmol of GST-TP) was needed to obtained a clear trans-priming signal (see Fig. S1C, lane 2). In summary, the RT domain clearly could use a TP domain in trans, as well as a cis-linked TP, as a protein primer to initiate DNA synthesis.
The nucleotide selectivity in trans-priming was similar to that during cis-priming.
The initiating nucleotide (dGMP) in DHBV protein priming is strictly dependent on (i.e., templated by) the last nucleotide of the ε RNA bulge (CMP) when Mg2+ is used as the metal cofactor (48, 49, 51). We have found previously that when Mn2+, instead of Mg2+, is used, the preferred initiating nucleotide for protein priming is still dGMP, but at a lower level, dAMP and TMP (and, to a still much smaller extent, dCMP) can also be used as the initiating nucleotide, at both the authentic Y96 TP site and the cryptic priming sites in the TP and RT domains (8, 31). This indicates that under the Mn2+ condition, the RT protein still uses the correct ε template site for priming. To test if trans-priming discovered here also used the same ε template site, we determined the nucleotide selectivity of trans-priming in comparison to cis-priming. As shown in Fig. S2 in the supplemental material, the dNTP selectivity in trans-priming (labeling of His-TP by SUMO-MiniRT2) was the same as that in cis-priming (labeling of SUMO-MiniRT2 itself), with the following nucleotide selectivity in both cases: dGTP ≫ dATP = TTP ≫ dCTP. Thus, trans-priming appeared to use the same ε RNA template site as did cis-priming.
Protein priming in cis led to a change in priming site selection in trans.
Since MiniRT2 was able to initiate protein priming from a separate TP domain, we reasoned that protein priming in cis might have triggered a conformational change in the TP and/or RT domain that dissociated the two domains linked in cis and allowed the RT domain in MiniRT2 to initiate DNA synthesis from the separate TP domain in trans. As one potential indication for the putative conformational change occurring in the RT domain following cis-priming, we determined the priming site selected on the separate TP domain by MiniRT2. Recently, we showed that KOH treatment of the primed RT or TP domain can be used to distinguish the protein-nucleotide (DNA) linkage involving either serine/threonine (sensitive to KOH cleavage) or tyrosine (resistant to KOH) from the TP or RT domain. Using this method, we showed previously that during trans-complementation with the RT and TP domains, the RT domain was able to initiate protein priming using S/T on TP as an alternative priming site(s), in addition to the authentic Y96 priming site (8). Therefore, we treated the trans-primed TP (WT or Y96F) domain with 3 M KOH following resolution of the labeled proteins by SDS-PAGE to determine the nature of the phosphoester linkage between the labeled nucleotide (dGMP) and TP (Fig. 3). The priming signal from the WT TP with Mg2+ served as an internal control since priming in the presence of Mg2+ is almost exclusively (>95%) from the authentic Y96 site and was not affected by KOH (Fig. 3, lane 5), whereas the priming signal from TP-Y96F produced by the RT domain with Mn2+ served as a control for priming exclusively from the cryptic S/T site but not the Y site, which was completely eliminated by the KOH treatment (lane 8) (8). With trans-complementation, the priming signal on the RT domain was decreased only slightly, suggesting that priming on the RT domain predominantly occurred at Y (Fig. 3, lanes 6 and 8, top versus bottom panels), as reported recently by us and others (5, 8). Similarly, the cryptic priming site(s) on the RT domain used during trans-priming was also predominantly Y (Fig. 3, lane 2). When the WT TP domain was trans-complemented with the RT domain, the priming signal on the TP domain was decreased by 3- to 4-fold by the KOH treatment, indicating that priming initiation in the presence of Mn2+ occurred (mostly) at the cryptic S/T site(s) as well as at the authentic Y96 site (Fig. 3, lane 6, top versus bottom panels), as we reported recently (8). In contrast, the priming signal from WT or mutant (Y96F) TP domains trans-primed with MiniRT2 was not reduced upon KOH treatment (Fig. 3, lanes 3 and 4, top versus bottom panels; also lanes 11 and 12), indicating that the RT domain from MiniRT2 initiated protein priming on TP using mostly, if not exclusively, Y residues (i.e., Y96 plus at least one other Y on TP). Thus, differently from the usage of S/T cryptic priming sites on TP during trans-complementation and cis-priming (8), trans-priming on TP carried out by MiniRT2 utilized predominantly or exclusively a Y residue(s).
Fig 3.
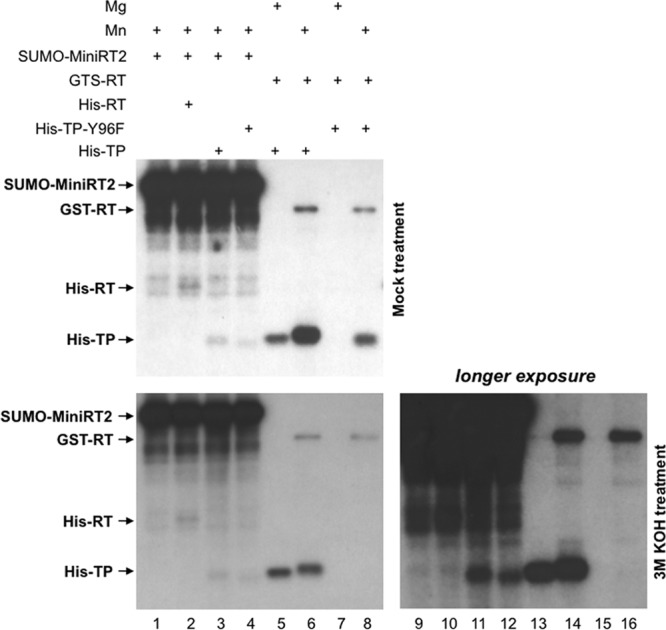
Determination of priming site usage in cis- and trans-priming by alkaline treatment of the priming products. Priming reactions were carried out in TMnNK (lanes 1 to 4, 6, and 8) or TMgNK (lanes 5 and 7), using SUMO-MiniRT2 (lanes 1 to 4), GST-RT (lanes 5 to 8), His-RT (lane 2), His-TP (lanes 3, 5, and 6), or His-TP-Y96F (lanes 4 and 8). The reaction products were resolved by SDS-PAGE. The gel was then cut into two parts; one part, containing one set of the priming reaction products, was mock treated (top panel), and the other part, containing a second set of the same priming reaction products, was treated with 3 M KOH (bottom panels). The priming products were then detected by autoradiography. Lanes 9 to 16 represent a longer exposure of lanes 1 to 8 of the bottom panel.
PFA sensitivity was induced following initiation of protein priming at Y96.
The pyrophosphate analog PFA is reported to block hepadnavirus DNA synthesis but only after protein priming, as it fails to inhibit protein priming either at the initiation or at the polymerization stage (31, 49). However, we recently showed that PFA could inhibit the polymerization stage of protein priming when Mn2+, instead of Mg2+, was used as the polymerase cofactor, though it still failed to inhibit the initiation stage of priming even with Mn2+ (31). These results suggest that RT undergoes a conformational change immediately after initiation of protein priming in the presence of Mn2+ that renders it sensitive to PFA inhibition, whereas with Mg2+, this PFA-sensitive RT conformation is not adopted until after the polymerization stage of priming. Thus, PFA can be a useful tool to probe the RT conformational change during protein priming (and subsequent viral DNA synthesis). Since priming in cis apparently induced a conformational change in the RT (and possibly TP) domain of MiniRT2 as evidenced by the altered priming site selection on the independent TP domain, we were next interested in determining the effect of PFA on priming initiation on TP in trans by MiniRT2.
Therefore, we carried out the trans-priming reaction using MiniRT2 and different concentrations of WT or mutant (Y96F) TP, in the presence of Mn2+, with or without PFA. As reported before (31), PFA did not inhibit priming initiation in cis (i.e., the labeling of MiniRT2 itself) (Fig. 4A, lane 2 versus lane 1). On the other hand, PFA inhibited (by ca. 2-fold) priming initiation on the TP domain in trans by MiniRT2 (lanes 6 to 8 versus lanes 3 to 5). Interestingly, a degradation product from SUMO-MiniRT2 (marked by the arrowhead in Fig. 4A) that migrated above His-TP and presumably corresponded to SUMO fused to the TP domain (with the RT domain being degraded) also was labeled, probably through trans-priming by the intact SUMO-MiniRT2. trans-priming from this endogenous degradation product (i.e., SUMO-TP) was sensitive to PFA inhibition, too (lanes 2, 6, 7, and 8). It was also apparent that the addition of increasing amounts of His-TP diminished trans-priming initiation from this putative degradation product (lanes 3 to 5), probably through competition for access to the RT domain in SUMO-MiniRT2. Also, the slightly decreased cis-priming signal of MiniRT2 in the presence of increasing amounts of TP (lanes 3 to 5 versus lane 1) might be attributed to the inhibitory effect exerted by the excess TP domain in trans (see below). Thus, the PFA inhibition of priming initiation on the TP domain in trans by MiniRT2 was consistent with the notion that cis-priming, which likely occurred before trans-priming (Fig. 4B below; see also the Discussion), induced the PFA-sensitive conformation in MiniRT2, which subsequently acted on the TP in trans to initiate another round of priming.
Fig 4.

Sensitivity of cis- and trans-priming to inhibition by PFA. (A) Priming reactions were performed in TMnNK using SUMO-MiniRT2 (1 pmol) alone (lanes 1 and 2) or together with increasing amounts of His-TP (1 pmol, lanes 3 and 6; 2 pmol, lanes 4 and 7; 4 pmol, lanes 5 and 8). The pyrophosphate analog PFA (at a 1 mM final concentration) was added to the priming reaction mixtures shown in lanes 2 and 6 to 8. The arrowhead denotes a degradation product from SUMO-MiniRT2 that apparently was able to serve as a primer in trans and most likely consisted of the SUMO tag plus the TP domain remaining attached (i.e., SUMO-TP). (B) trans-complementation priming reactions were performed in TMnNK using GST-RT (lanes 1 to 4) and His-TP (lanes 1 and 2) or His-TP-Y96F (lanes 3 and 4). PFA (at a 1 mM final concentration) was added to the reaction mixtures shown in lanes 3 and 4. Priming products were resolved by SDS-PAGE and detected by autoradiography. Priming signals were quantified using phosphorimaging and are represented at the graphs shown at the bottom, with those in the absence of PFA set at 100. The means and standard errors are shown. Statistical significance was calculated using the one-tailed, unpaired Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significant.
We next tested the effect of PFA on protein priming during trans-complementation where separate TP and RT domains reconstitute a priming active polymerase. As shown in Fig. 4B, PFA did not inhibit priming initiation from the TP domain, whether WT or Y96F. However, PFA was able to inhibit priming initiation on the RT domain strongly (by 6-fold) when the WT TP and RT domains were used in trans-complementation (Fig. 4B, lane 3 versus lane 1). In contrast, when the mutant Y96F TP was used, PFA had only a modest effect (2-fold inhibition) on priming initiation from the RT domain (lane 2 versus lane 4). As reported earlier (8), the mutation at TP (Y96F) also decreased priming from the RT domain (lane 2 versus lane 1), suggesting that priming at Y96 could stimulate RT catalytic activity. Together, these results suggested that it was priming initiation specifically at Y96 that induced the putative RT conformational state that was more catalytically active but also more sensitive to PFA inhibition, whereas priming at the cryptic sites was not able or less able to do so. This result also suggested that during trans-complementation, priming at the RT domain probably occurred only after that at TP.
An inactive RT domain blocked the interaction between its cis-linked TP and another independent RT domain in trans.
The results above indicated that the RT domain in MiniRT2 could initiate protein priming on a separate TP domain in trans through intermolecular interaction. As intramolecular TP-RT interaction (i.e., in cis within MiniRT2) is expected to dominate over the intermolecular (in trans) TP-RT interaction, we hypothesized that in order for trans-priming to occur, protein priming in cis might induce a conformational change, as already suggested above based on the alteration in priming site selection and the induction of PFA sensitivity following cis-priming, which would also weaken the intramolecular TP-RT interaction in MiniRT2 (i.e., at least partially dissociate the RT from the TP domain) so that the RT domain in MiniRT2 could access, in trans, a separate TP domain to initiate priming there. In support of this hypothesis, we found that the priming sites on TP were apparently inaccessible to an RT domain in trans when the TP was linked in cis to an RT domain that is catalytically inactive. Thus, when MiniRT2-YMHA, which contains a mutant RT domain with no catalytic activity and is thus unable to carry out protein priming from either the TP or the RT domain (Fig. 5A, lane 1), was complemented in trans with a functional RT domain (either 6×His or GST tagged), little to no priming from the mutant MiniRT2 was observed (lanes 2 to 5), even though the TP domain, as contained in the mutant MiniRT2, tagged with either 6×His (lane 5) or GST (lane 4), was able to serve readily as a functional protein primer when provided as an isolated domain by itself (without any RT domain sequences) to the same RT domain in trans in the same reaction (also Fig. 2 to 4), as reported before (31). Furthermore, the small amounts of apparent degradation products (indicated by the bracket) from GST-MiniRT2-YMHA, invisible on the stained gel (Fig. 5B) but comigrating or migrating just above GST-TP upon labeling by protein priming (Fig. 5A, lanes 2 to 5), were nevertheless labeled at even higher levels than was the intact MiniRT2-YMHA protein, suggesting that the degradation products, containing the TP domain but with little RT domain sequence remaining attached, served as a much more efficient primer than did MiniRT2-YMHA for the active RT domain in trans. These results thus further supported the interpretation that the TP domain was sequestered by the inactive RT domain in MiniRT2-YMHA and was inaccessible to the active RT domain in trans, as a result of the strong cis TP-RT interaction dominant over TP-RT interactions in trans. Furthermore, these results combined suggested that protein priming in cis indeed dissociated or weakened intramolecular TP-RT domain interactions.
Fig 5.

Failure of an active RT domain in trans to rescue the catalytically inactive MiniRT2-YMHA mutant. (A) The priming reactions were performed in the presence of TMnNK. Proteins or domains used were GST-MiniRT2-YMHA (lanes 1 to 5), GST-MiniRT2 (lane 6), GST-RT (lanes 2, 4, and 7), His-RT (lanes 3 and 5), GST-TP (lanes 4 and 7), and His-TP (lane 5). The bracket denotes degradation products from GST-MiniRT2-YMHA that were able to serve as primers in trans. (B) SDS-PAGE and Coomassie blue staining of natively purified GST-MiniRT2 (lane 1) and GST-MiniRT2-YMHA (lane 2). The intact MiniRT2 fusion protein and two copurifying bacterial chaperone proteins, DnaK and GroEL, are indicated. The bracket denotes degradation products from GST-MiniRT2-YMHA (representing mostly just the GST tag itself).
The inactive RT domain in trans inhibited protein priming in a dose-dependent manner.
Since the inactive RT domain that was cis-linked to a functional TP domain in the MiniRT2-YMHA mutant blocked the function of the TP to serve as a primer by an active RT domain in trans, it was possible that an inactive RT domain (with the same YMHA catalytic mutations as those in MiniRT2-YMHA), when added in trans, could also be employed to inhibit protein priming via nonproductive interaction with TP and thus block the interactions between TP and an active RT domain. We tested this notion under two different conditions of protein priming, either through trans-complementation reconstituted with functional TP and RT domains or through cis-priming with MiniRT2. As the above results suggested that TP-RT interaction in cis would dominate over the same interaction in trans (Fig. 5), increasing amounts of the mutant RT-YMHA domain were added to the priming reactions. The mutant (YMHA) RT domain was indeed able to inhibit, dose dependently, protein priming on both the TP and RT domains in the trans-complementation reaction (Fig. 6, lanes 1 to 5). Furthermore, the mutant RT domain also inhibited cis-priming by MiniRT2 in a dose-dependent manner (lanes 6 to 10), suggesting that the mutant RT domain, acting in trans, could indeed block the intramolecular (i.e., in cis) TP-RT domain interactions, especially when provided in excess.
Fig 6.
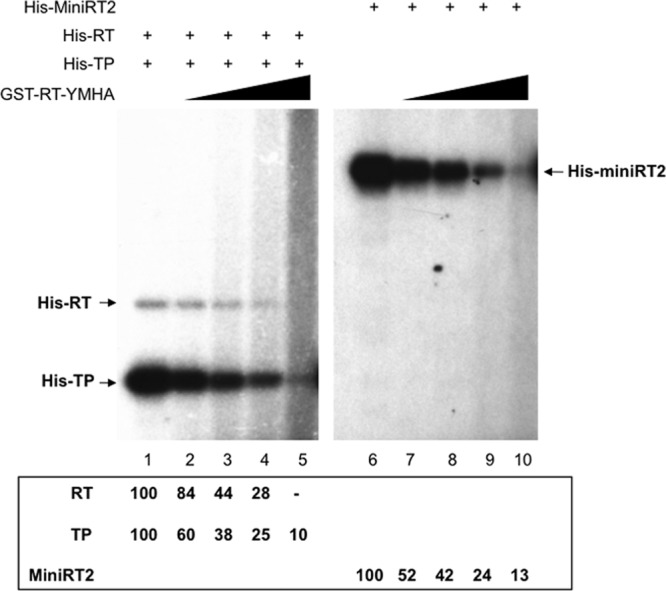
Inhibition of protein priming by the RT-YMHA mutant in trans. trans-complementation priming reactions were performed in TMnNK using His-RT and His-TP (lanes 1 to 5), and cis-priming was performed using His-MiniRT2 (lanes 6 to 10). RT-YMHA at a 1-, 2-, 4-, or 8-fold molar excess (i.e., 1, 2, 4, or 8 pmol) was added to the trans-complementation (lanes 2 to 5) or cis-priming (lanes 7 to 10) reactions. The priming signals of the RT and TP domains or MiniRT2 in the presence of RT-YMHA are indicated at the bottom as percentages of those in the absence of RT-YMHA (lanes 1 and 6).
Inhibition of protein priming by sequences derived from the RT domain.
The strong inhibitory effect of the RT-YMHA mutant on protein priming prompted us to determine in more detail the sequences in the RT domain that could inhibit protein priming when provided in trans. To this end, a series of RT domain fragments derived from C- and N-terminal truncations were constructed as GST fusions (Fig. 1A). All the fragments could be expressed in bacteria and purified at levels similar to or higher than that of the initial RT (349–575) domain construct derived from MiniRT2, with RT/366–555 producing the largest amount of purified proteins among all the RT fragments. As with the initial RT domain construct (17), all the newly derived RT fragments purified were associated with the bacterial chaperones DnaK and GroEL (Fig. 1B).
To test the inhibitory effects of the RT fragments on protein priming, we added an excess amount (32 pmol per 10-μl reaction mixture, or 3.2 μM) of these fragments to SUMO-MiniRT2 (1 pmol), as trans-inhibition by the mutant RT (YMHA) domain was more effective when it was present in excess over the target of inhibition (either MiniRT2 or the TP plus RT domain in trans-complementation) (Fig. 6). Priming reactions were conducted with Mg2+, which is presumably the relevant ion for RT function in vivo. The mutant RT (YMHA) domain again strongly inhibited protein priming by SUMO-MiniRT2 (Fig. 7A, lane 2 versus lane 1). The other RT domain fragments also showed a strong (though not as strong as the RT-YMHA mutant) inhibitory effect (lanes 3 to 11). Surprisingly, even 349–575, the initial RT domain construct derived from MiniRT2 that is active in reconstituting a functional RT protein with TP in trans-complementation (Fig. 2 to 6) (8, 30, 31), also blocked MiniRT2 priming, though less effectively than did its mutant (RT-YMHA) counterpart (Fig. 7A, lane 3 versus lane 2). Also, RT/349–555 and RT/366–555, which were as active as or more active than the starting RT fragment (349–575) in trans-complementation (see Fig. 9 below), still inhibited priming (lanes 4 and 8). The RT fragments also inhibited priming when used at smaller amounts (16 pmol), albeit the inhibition was less efficient, as anticipated (Fig. 7B). These results suggested that multiple inhibitory sequences might be distributed across the RT domain.
Fig 7.
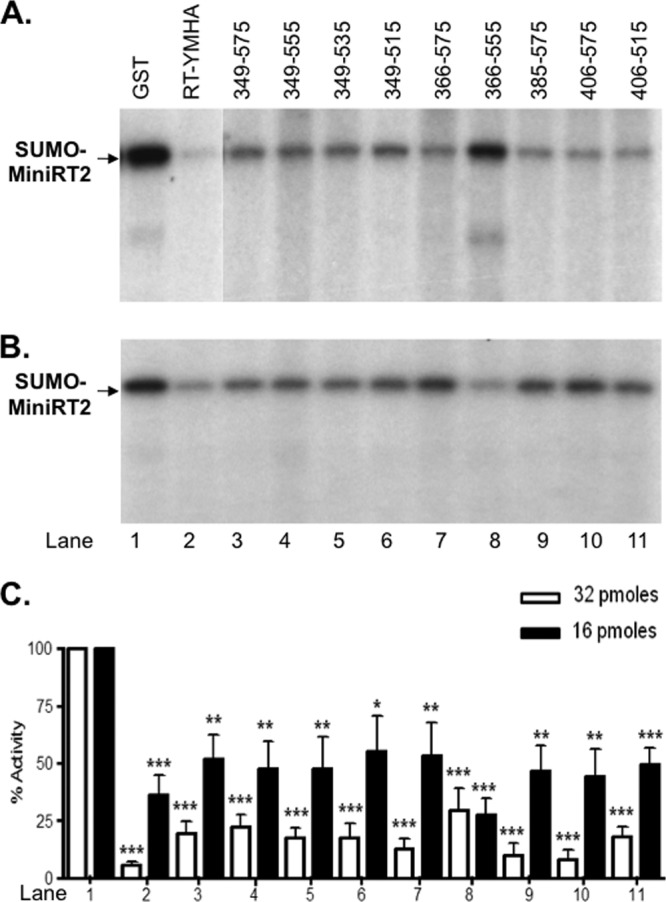
Inhibition of MiniRT2 priming by RT domain fragments. Protein priming reactions were conducted in TMgNK using natively purified SUMO-MiniRT2 (1 pmol), in the presence of the indicated (GST-tagged) RT domain fragments (lanes 3 to 11) (32 pmol in panel A and 16 pmol in panel B). GST (lane 1) and the RT-YMHA mutant (lane 2) (32 pmol in panel A and 16 pmol in panel B) were used as negative and positive controls for trans-inhibition, respectively. The ε RNA was preincubated with MiniRT2 before the RT fragments were added. The protein priming signals are indicated in panel C as percentages of those without inhibition (i.e., in the presence of GST; lane 1). The means and standard errors are shown in the bar graph. Statistical significance was calculated using the one-tailed, unpaired Student t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig 9.

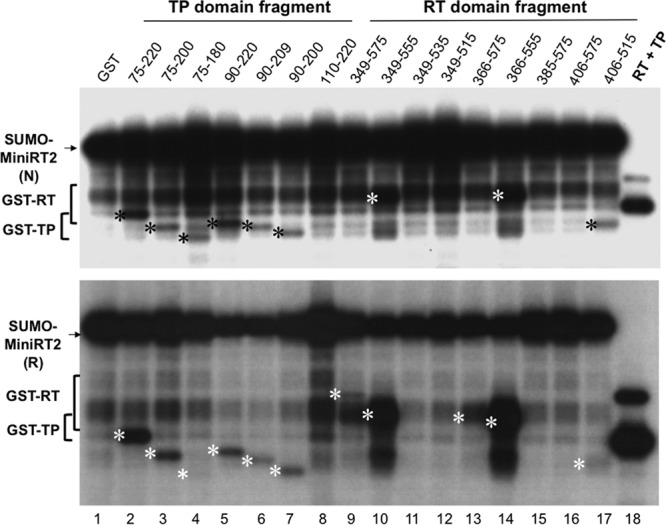
trans-complementation priming reaction using TP and RT domain fragments. (A) trans-complementation priming reactions were performed using the starting GST-RT (RT) plus the starting GST-TP domain (TP, lanes 1 and 16) or the newly constructed TP fragments (lanes 2 to 7), using the starting GST-TP plus the newly constructed RT domain fragments (lanes 8 to 15), or using TP/90–209 plus RT/366–555 (lane 17) in TMnNK (top panel) or TMgNK (bottom panel). The protein priming signals on the TP (top and bottom panels) and RT (top panel) domain fragments are indicated at the bottom of the images, as percentages of the priming signals using the starting TP and RT domain constructs (lanes 1 and 16). The question marks denote that the RT domain signals from these constructs were difficult to quantify due to their comigration with the TP signal (see also panel B). (B) trans-complementation priming reactions were performed using GST-RT/349–575 (lane 1), GST-RT/349–555 (lane 2), or GST-RT/366–555 (lane 3) plus His-TP (lanes 1 to 3) in TMnNK, to show more clearly the priming signals on GST-RT/349–555 and GST-RT/366–555, which were not well resolved from the strong and closely migrating GST-TP signals in panel A (lanes 8 and 12). The protein priming signals on the RT and TP domain fragments are indicated at the bottom of the image, as percentages of the priming signals using the starting (His-) TP and (GST-) RT domain constructs (lane 1).
Given the inhibitory effect of the RT fragments on priming by MiniRT2, we were next interested in determining their potential effect on protein priming by the full-length RT protein. Therefore, priming active full-length DHBV RT protein was expressed by in vitro translation in RRL as described previously (16, 49), and the RT fragments (32 pmol each) were mixed with the translated full-length RT (ca. 0.1 pmol). Also, to test the potential influence of the ε RNA on the inhibitory effect of the TP and RT fragments, we added ε either during translation (i.e., to allow RT-ε RNP formation before the addition of the RT fragments) or after preincubating the translated full-length RT with the RT fragments. Priming reactions were then performed in the presence of Mg2+. When the RT fragments were added before ε, none of the RT fragments, including the RT-YMHA mutant, inhibited protein priming by the full-length RT (see Fig. S3A in the supplemental material). In contrast, when the RT fragments were added to the preformed full-length RT-ε RNP complex, some inhibitory effect (2- to 3-fold reduction) on priming was observed in most cases (see Fig. S3B), except for 366-555 (see Fig. S3B, lane 8) and 406-515 (lane 11), which showed only a minor effect (a less-than-2-fold reduction). These results suggested that some of the interacting sites in the full-length RT protein may become more accessible upon RNP complex formation and thus could be more readily targeted by the inhibitory sequences added in trans.
In general, the inhibitory effect of the RT domain fragments on the full-length RT was less than that on SUMO-MiniRT2, suggesting that factors in RRL may be affecting the inhibitory effect of the fragments on priming or that the full-length RT was less sensitive to inhibition than was MiniRT2. To differentiate between these possibilities, we translated SUMO-MiniRT2 as well as the full-length RT in RRL and tested the effects of the RT fragments on priming by the in vitro-translated MiniRT2. SUMO-MiniRT2, even when translated in RRL, was also more sensitive to the inhibitory effect of the RT domain fragments than was the full-length RT expressed in the same system (data not shown), thus indicating that the truncated MiniRT2 was indeed more amenable than the full-length RT protein to trans-inhibition. This could be explained by the more-extensive intramolecular (i.e., cis) interactions in the full-length RT than in MiniRT2, which would be contributed by the C-terminal RT and RNase H domain sequences absent from MiniRT2 and might make it more difficult for the separate TP or RT domain fragments to insert into the full-length RT in trans. We also attempted to determine if TP domain sequences could exert the trans-inhibitory effects as well. Although some inhibitory effects were observed with some of the TP domain fragments (Fig. 1 and 4A; data not shown), the effects were much weaker than those with the RT domain fragments and were more variable, precluding a definitive analysis using the current system.
Although it is well established that the TP and RT domains together are required for specific interaction with the ε RNA, it was possible that the excess RT fragments added in trans might bind ε nonspecifically and thus make it unavailable to support protein priming by MiniRT2 or the full-length RT in the priming reaction. However, this was made unlikely by the fact that addition of a large excess of tRNA (100-fold excess over the ε RNA and 20-fold excess over the TP or RT fragments) to the priming reaction mixtures did not alleviate the inhibitory effect of these fragments on protein priming (data not shown). The observation that adding the inhibitory RT fragments to the preformed full-length RT-ε or MiniRT2-ε complex was as effective or even more effective in inhibiting protein priming than was adding these fragments before the ε RNA (Fig. 7; see also Fig. S3 in the supplemental material; also data not shown) also helped exclude this nonspecific RNA binding effect. If the inhibitory RT fragments had simply inhibited priming by sequestering the ε RNA away from MiniRT2 or the full-length RT, little inhibition of the preformed RT-ε RNP complex would have been expected.
There was also a concern that some of the RT domain fragments might not remain soluble during the priming reaction and could have aggregated and caused precipitation of the MiniRT2 or full-length RT protein, accounting for their inhibitory effects on priming. This was unlikely because all the RT domain fragments were purified under native conditions routinely at 0.3 to 1.2 mg/ml, levels which were 2- to 10-fold above those used in the priming reactions. To formally exclude this possibility, SUMO-MiniRT2 as well as all the RT domain fragments remaining in solution after the priming reactions was visualized by SDS-PAGE and silver staining. The amount of SUMO-MiniRT2 remaining in solution was constant whether it was incubated with the GST-RT domain fragments or GST alone, and the GST-RT fragments remained in solution during the priming reactions (see Fig. S4 in the supplemental material).
Mapping of protein primer sequences in the TP and RT domains during trans-priming.
Given the ability of the TP fragment 75 to 220 to serve as primer in the trans-priming reaction (Fig. 2 to 5; see also Fig. S1 and S2 in the supplemental material), we were interested in determining the potential of additional TP truncation fragments, as well as the series of RT domain fragments described above, to serve as a protein primer in trans. As with the RT domain series described above, a series of TP domain fragments derived from C- and N-terminal truncations were constructed as GST fusions (Fig. 1A). All the fragments could be expressed in bacteria and purified at levels similar to or higher than that of the initial TP (75–220) fragment derived from MiniRT2, with TP/90–200 producing the largest amount of purified proteins among all the TP fragments. As with the initial TP (17) and all the RT domain constructs (Fig. 1B), all the newly derived TP fragments purified were associated with the bacterial chaperone DnaK and, with the exception of TP/110–220, also with another bacterial chaperone, GroEL (Fig. 1B).
While the trans-inhibitory effect of the RT domain fragments was dominant when added in excess to MiniRT2 or the full-length RT (Fig. 6 and 7; see also Fig. S3 in the supplemental material), trans-priming was readily detectable when an equimolar amount (1 pmol) or a slightly larger amount (4 pmol) of the TP or RT domain (relative to MiniRT2) was used (Fig. 2 to 4; see also Fig. S1 and S2). Therefore, a slight excess of the newly constructed TP or RT domain fragments (4 pmol each) was mixed with SUMO-MiniRT2 (1 pmol) and priming reactions were carried out. Similar to the starting TP construct (75–220), all new TP constructs (Fig. 8, lanes 2 to 7) except one (110–220) (lane 8) were able to serve as a trans primer used by MiniRT2. With the RT domain, in addition to the starting construct (349–575) (lane 9), three other constructs, 349–555, 366–575, and 366–555 (lanes 10, 13, and 14, respectively), also apparently served as primers for trans-priming. The priming signals from 349–575 (lane 9) and 366–575 (lane 13) were more difficult to visualize in Fig. 8 due to their weak labeling and their comigration with MiniRT2 degradation products, but these two RT fragments clearly showed trans-priming signals in other experiments (e.g., see Fig. S1A, lane 2; also Fig. 3, lane 2, and data not shown). As described below (see Fig. 9), these three RT domain constructs, like the starting RT construct (349–575), also retained the ability to reconstitute priming (i.e., catalytic activity) in the trans-complementation assay and thus, like 349–575, probably carried out priming really in cis: i.e., the same RT domain, upon interaction with the TP domain in MiniRT2, initiated priming from itself (see Fig. S1A). In contrast, the shortest RT domain construct, 406–515, could still serve as a primer even though it completely lacked any RT activity in trans-complementation (see Fig. 9, below) and thus was incapable of self-priming in cis, indicating that it truly served as a primer in trans.
Fig 8.
trans-priming from TP and RT domain fragments by MiniRT2. The indicated TP (lanes 2 to 8) or RT (lanes 9 to 17) domain fragments (at 4 pmol each) or GST (4 pmol, as a negative control; lane 1) was added to SUMO-MiniRT2, natively purified (N, top) or refolded (R, bottom), and priming reactions were conducted in TMnNK. The trans-complementation priming reaction conducted with GST-TP (75 to 220) and GST-RT (349 to 575) served as a positive control (lane 18). The asterisks to the left of the labeled bands denote the trans-priming signals from the corresponding TP or RT domain fragments. Note that the RT domain priming signal shown in lanes 9, 10, 13, and 14 probably represented RT domain self-priming (i.e., in cis) instead of true trans-priming, as in Fig. S1A in the supplemental material (see the text for details).
Mapping of minimal TP and RT domain sequences required to reconstitute protein priming through trans-complementation.
The availability of the various TP and RT domain fragments also provided the opportunity to map further the minimal TP and RT domain sequences required to reconstitute a priming active RT protein through trans-complementation and to compare the TP and RT domain sequence requirements for trans-inhibition (Fig. 7; see Fig. S3 in the supplemental material) and trans-priming (Fig. 8) with those for trans-complementation. Therefore, we performed a trans-complementation priming assay using the newly constructed (further truncated) TP and RT fragments, under either Mn2+ (Fig. 9A, top, and 9B) or Mg2+ (Fig. 9A, bottom) conditions. Except for the stronger priming signals overall and the clearer RT domain (cryptic sites) priming signals with Mn2+, as reported recently (8, 31), the results obtained with Mn2+ and those obtained with Mg2+ were identical in terms of functional mapping.
The priming activity of the new TP and RT fragments was normalized to that obtained with the starting TP and RT domain constructs (75–220 and 349–575, respectively) (Fig. 9A, lanes 1 and 16). TP/75–200 retained low priming activity (ca. 1/3) (lane 2), whereas the further C-terminal deletion (75–180) lost all priming activity (lane 3), which placed the TP C-terminal boundary essential for protein priming between position 180 and 200. On the other hand, TP/90–220 (lane 4) was almost as active as the starting TP while 110–220 lost all priming activity (lane 7), placing the N-terminal boundary of TP between position 90 and 110. Further truncations showed that 90–209 retained full priming activity (lane 5) while 90–200 retained 10% activity (lane 6). Thus, residues from position 90 to 200 of TP were determined to be the minimal TP domain sequences essential for priming and the TP sequences from position 200 to 209 contributed significantly to TP priming function, although they were not absolutely required. With the RT domain, the sequence from position 366 to 555 (lane 12) was shown to be the minimal region that retained efficient priming function (even better than that of the starting construct 349–575). Also, extending this minimal sequence either N or C terminally (349–555, lane 8, or 366–575, lane 11) or both (349–575, lane 1) decreased the RT priming function, suggesting that these additional sequences (349–366 and 555–575) might interfere with 366–555 function under these in vitro conditions (see Discussion). None of the other RT fragments showed any priming activity (lanes 9, 10, and 13 to 15), suggesting that sequences after position 366 and before position 555 were essential for priming in vitro. When TP/90–209 and RT/366–555 were combined, they also reconstituted high priming activity in vitro (lane 17).
One cryptic priming site in the RT domain was mapped to Y561 (5), but our previous results also indicated the existence of additional cryptic sites in the RT domain (8). The strong priming function of 349–555 and 366–555 provided the opportunity to determine the presence of a cryptic RT domain priming site(s) N terminal to Y561. Indeed, both 349–555 and 366–555 showed a clear priming signal, albeit less than that of 349–575, suggesting that an additional priming site(s) was indeed present N terminal to position 555 in the RT domain. The (GST-tagged) RT domain priming signals were not clearly separated from the strong (GST-tagged) TP domain signal (Fig. 9A, lanes 8 and 12), but they were detected clearly when trans-complemented with the shorter, 6×His-tagged TP domain (Fig. 9B). As with trans-complementation using GST-TP (Fig. 9A), trans-complementation using His-TP also showed that 355–566 was the most active RT fragment, followed by 349–555 and 349–575 (Fig. 9B).
DISCUSSION
Protein-primed initiation of reverse transcription in hepadnaviruses is a highly dynamic process that requires precise interactions between the TP and RT domains of the RT protein and between these domains and the specific viral RNA signal ε. In the present study, we have discovered that the DHBV RT domain, with its cis-linked TP in the context of a truncated RT protein called MiniRT2 and, to a lesser extent, the full-length RT protein, was able to use a separate TP domain provided in trans to prime DNA synthesis. We call this trans-priming, to differentiate it from cis-priming, in which the RT domain uses its cis-linked TP domain as a primer, as well as from trans-complementation priming, whereby the separate TP and RT domains come together to reconstitute a functional protein. We have exploited the trans-priming system to show that the RT (and possibly TP) domain underwent a conformational change upon protein priming in cis, which dissociated, or at least weakened, the intramolecular (i.e., cis) TP-RT domain interactions to allow the RT domain to initiate protein priming on a separate TP in trans (Fig. 10A). The altered RT conformation following cis-priming could also be demonstrated as a change in priming site selection on TP in trans and a gain of susceptibility to inhibition by the pyrophosphate analog PFA. In addition to trans-priming, intermolecular (i.e., trans) interactions between the TP and RT domains were also indicated by trans-inhibition, whereby sequences derived from the RT domain were able to inhibit cis-priming. Comparison of the sequence requirements for trans-priming, trans-inhibition, and trans-complementation priming allowed further mapping of the minimal TP and RT sequences essential not only for productive priming (requiring functional TP-RT interactions as well as TP/RT-ε interactions) but also for the capacity to serve as a protein primer per se (minimally requiring productive TP-RT domain interactions) and of those for trans-inhibition (requiring physical TP-RT interactions that are not necessarily productive for priming) (Fig. 10B).
Fig 10.

Proposed RT conformational dynamics and TP and RT domain interactions in protein priming. (A) RT conformational changes following initiation of protein priming. The authentic primer site (Y96 in TP) and cryptic priming sites (S/T or Y in TP) are indicated. For clarity, the cryptic priming sites in the RT domain are omitted. The resistance (left) of cis-priming and sensitivity (right) of trans-priming to PFA inhibition are also indicated. The dissociation of cis-linked TP and RT domains upon cis-priming is depicted as an opening of the protein structure. The proposed conformational change in the RT domain is depicted as a change in the shape and shading of the RT domain. (B) Definition of TP and RT domain sequences required for trans-inhibition, trans-priming, and trans-complementation. The top two diagrams depict the domain structures of the full-length DHBV RT and MiniRT2, as explained in Fig. 1A. The dashed lines on the third diagram indicate that multiple sequences in the RT domain could function to inhibit priming in trans. The light shading in the fourth diagram signifies the fact that the longer RT domain construct (containing position 366 to 555) failed to serve as a primer, in trans, to be used by another RT domain but could prime from itself in cis (when provided with a functional TP domain). The thinner line in the fifth diagram (denoting residues 200 to 209) signifies that these TP sequences, while not essential for trans-complementation, nevertheless contribute substantially to the reaction. See the text for details.
The fact that RT proteins with cis-linked TP and RT domains could utilize a separate TP domain in trans to prime DNA synthesis indicates that the RT domain in these proteins is able to interact, intermolecularly, with TP in trans, as well as intramolecularly with their cis-linked TP domain. The difference between primer site selection on TP during cis-priming and that during trans-priming (8) suggests that the RT domain adopts a conformation during trans-priming that is different from that during cis-priming (Fig. 10A). Two additional lines of evidence argue that trans-priming occurred after cis-priming, which further supports the putative RT conformational change occurring after cis-priming (Fig. 10A). First, the intramolecular interactions between the cis-linked TP and RT domains have to be at least partially disrupted or weakened to allow the intermolecular TP-RT interactions required for trans-priming, as indicated by the inaccessibility of a TP domain that is linked in cis to a catalytically inactive RT domain (i.e., as in MiniRT2-YMHA) to a functional RT domain in trans. The result is consistent with the previous finding that two full-length RT mutants containing either the Y96F mutation (in TP) or the YMHA mutation (in the RT domain) could not complement each other for protein priming (55, 56) and supports the notion that the RT protein functions as a monomer (55). This apparent sequestration of the cis-linked TP, together with the ability of an active RT domain with a cis-linked TP to use a separate TP in trans as a primer, supports the notion that some conformational change in the RT and/or TP domain occurs following cis-priming, which dissociates, at least partially, the intermolecular TP-RT interactions and allows the RT domain to interact with a separate TP for trans-priming. Second, in contrast to cis-priming initiation, trans-priming initiation was found to be sensitive to inhibition by PFA. As we have previously shown that PFA, under the Mn2+ priming conditions as used here, also inhibits the second stage of cis-priming (polymerization) following initiation (31), these results suggest that the RT conformation during trans-priming initiation is similar to that during polymerization of cis-priming and that both trans-priming and polymerization occur following cis-priming initiation, which induces the PFA-sensitive RT conformational state. Furthermore, the efficient induction of the PFA-sensitive RT conformation following cis-priming required the authentic Y96 site, which on the one hand stimulates the enzymatic activity of the RT (8) and on the other enhances PFA sensitivity. A recent crystal structure of PFA in complex with a DNA polymerase indeed shows that PFA sensitivity is determined by a specific polymerase conformation rather than specific side chains (54), affirming the utility of PFA as a sensitive probe for polymerase conformational changes.
The TP-RT domain interactions in cis, while strong, were evidently dynamic and could be disrupted by excess RT domain fragments provided in trans, which were shown to inhibit cis-priming in a dose-dependent manner. Efforts to localize the inhibitory sequences within the RT domain showed that multiple sequences within it could function, in trans, to inhibit cis-priming by MiniRT2 and, to a lesser extent, by the full-length RT protein, through intermolecular (trans) RT-TP and RT-RT domain interactions (Fig. 10B). Even catalytically active RT domain fragments, including the starting construct 349–575 and the newly constructed 349–555, 366–555, and 366–575 (see also below), could inhibit cis-priming. In these cases, simple competition by the RT domain in trans to snatch (deprive) TP from its cis-linked RT domain in MiniRT2 or the full-length RT may not account entirely for the inhibitory effect, as the catalytically active RT domain fragments in trans, having interacted with TP, would be able to carry out priming via trans-complementation (see below). Even considering that priming via trans-complementation may be less efficient than cis-priming, the rather strong inhibitory effects of the RT domain fragments on protein priming were likely mediated in part via trans RT-RT domain interactions, as well as RT-TP domain interactions. As the RT domain itself has to maintain multiple intradomain interactions (e.g., between its palm and finger subdomains) in order to establish an enzymatically active conformation, it is likely that the extra RT domain in trans can disrupt these intradomain (and intramolecular or cis) interactions by establishing intermolecular (trans) interactions between their subdomains and those in the RT domain in MiniRT2 (and, less effectively, in the full-length RT), leading to inhibition of MiniRT2 (and full-length RT) cis-priming.
Efforts to map the TP and RT sequences that were required for them to serve as a protein primer by MiniRT2 localized the minimal TP sequence sufficient for trans-priming to between positions 90 and 180 (Fig. 10B). The fact that TP sequences after position 180 are required for ε binding and protein priming during cis-priming (and trans-complementation priming [see below]) indicates that the TP domain in trans may not need to contribute, at least fully, to ε binding in order to serve as a primer per se in trans-priming, in contrast to the requirements for TP in trans-complementation (see below). Conceivably, the RT domain within MiniRT2 (or the full-length RT), upon priming in cis and dissociation from its cis-linked TP, could maintain enzymatic activity and initiate another round of protein priming from another TP in trans. This lack of requirement for the trans TP domain to contribute to ε binding increases the possibility, as we suggested recently (8), that a host protein may be used by the viral RT as a primer and thus modified by covalent nucleotide or DNA attachment (26). The TP fragment 110–220, which lacks the authentic Y96 priming site as well as the previously identified cryptic priming site S93 (8), was not able to serve as a primer in trans. This may suggest that no additional cryptic priming sites reside within 110–220. However, the inability of 110–220 to serve as a primer could also be due to its lack of appropriate interactions with the RT domain in MiniRT2 to position it in the RT active site to allow priming (see below also).
Regarding the sequences from the RT domain, we found that, with one notable exception, only those that retain catalytic activity (349–575, 349–555, 366–555, and 366–575), as determined in trans-complementation (see below), could serve as a primer in the trans-priming assay (Fig. 10B). RT-YMHA, and most of the other RT fragments that were inactive in trans-complementation, also did not show any signal in the trans-priming assay. These results are consistent with the notion that the cryptic priming sites in the RT domain can be accessed only by the polymerase active site of the same RT domain (i.e., in cis), as suggested recently (5). Those RT domain fragments that retained the catalytic capacity were thus able to interact with TP contained in MiniRT2 to carry out priming on themselves. On the other hand, the shortest RT fragment, 406–515, which did not show any trans-complementation activity, could serve as a trans-primer for MiniRT2 (Fig. 10B). This result indicates that at least one cryptic priming site is located within this RT region. Furthermore, as this cryptic site is contained in all the other (longer) RT domain constructs tested, which nevertheless failed to serve as trans primers, it was apparently inaccessible to another RT domain in trans in the context of the longer RT constructs, probably because it was sequestered by intradomain (and intramolecular) interactions present in these longer RT constructs.
Regarding trans-complementation, we further narrowed down the minimal TP sequence to a 110-amino-acid segment from 90 to 200, although TP sequences from position 200 to 209 also substantially contribute to trans-complementation (Fig. 10B). Consistent with the suggestion that the TP sequences from position 90 to 200 may constitute a functional, minimal “core” TP domain is the fact that this segment produced the highest yield of purified protein among all TP fragments tested, indicative of its improved folding characteristics, at least in bacteria. Our mapping results here are also consistent with previous reports identifying critical roles of sequences near the so-called T3 motif (176–183) in ε binding and protein priming (3, 9, 38, 40) and the dispensability of TP sequence 1–90 for cis-priming shown recently (5). That the N-terminal TP boundary is only six amino acid residues away from the authentic Y96 priming site (and three residues away from the cryptic S93 priming site) indicates remarkably little sequence requirement N terminal to the priming site for ε binding or positioning of the primer residue in the RT active site. TP/110–220 may not be able to serve as a primer simply because it no longer contains the authentic Y96 and the cryptic S93 priming sites (8). However, no priming from the RT domain (cryptic priming sites) was detected in the trans-complementation assay using this TP fragment either. Therefore, the TP sequences from position 90 to 110, in addition to harboring the priming sites, may also be required for ε binding or stimulation of RT catalytic activity. Interestingly, this TP fragment was the only one among all the TP fragments tested that lost association with the bacterial chaperone GroEL, suggesting that at least some aspect of its folding characteristic is different from those of the other TP fragments.
The minimal RT domain sequence required for trans-complementation was also further narrowed down to position 366 to 555 (Fig. 10B). It was previously reported that a C-terminal truncation at position 559, in the context of the full-length RT protein translated in RRL, abolished protein priming (though not ε binding) (50), suggesting that either factors in the RRL or the rest of the RT protein sequences (position 1 to 75 of TP and/or the spacer) could influence the C-terminal boundary of the RT domain required for protein priming. Indeed, in several cases, the additional RT domain sequences either N terminal or C terminal to the 366–555 “core” RT domain apparently decreased the priming activity of the RT “core” domain (comparing 349–575 with 349–555, 349–555 with 366–555, and 366–575 with 366–555), suggesting that these additional (“extraneous” in this assay context) sequences might interfere with the core RT domain folding and function, especially in the absence of the eukaryotic chaperones that are known to be required for the folding of the full-length RT protein (19, 22, 24, 25, 40). We have previously proposed that sequences C terminal to MiniRT2 (i.e., after position 575), though obviously required for other aspects of viral replication, act as “autoinhibitory” sequences as far as protein priming is concerned and that their inhibitory effect is counteracted normally by the host chaperones (52). From the further mapping results presented here, it appears that the autoinhibitory sequences extend further (N terminally) to position 555. As with the proposed minimal core TP (90–200) domain above, the putative core RT domain (366–555) also produced the highest yield of purified protein among all the RT fragments tested, further indicative of its improved folding property. Efforts are under way to link, in cis, the minimal TP and RT domain sequences required for protein priming, defined here using trans-complementation, to verify if these same minimal sequences are indeed sufficient for normal (cis) priming.
A comparison of the TP and RT domain sequence requirements defined in the three different assays here, trans-inhibition, trans-priming, and trans-complementation, thus helps to dissect different kinds of inter- and intradomain interactions that may occur during protein priming (Fig. 10B). For example, trans-inhibition may be the least demanding in that mere physical interactions between the TP or RT fragments in trans and the target RT protein would suffice. There would be no need for the primer site(s) or its proper positioning in the catalytic active site of the RT domain, and no need for contribution to ε binding. For the TP or RT fragments to serve as a primer in the trans-priming assay, they must, at a minimum, harbor a priming site and properly position it in the active site of the RT domain. On the other hand, trans-complementation would have the most stringent requirements of the three assays here: besides the obvious requirements for the priming site (in the TP or RT domain) and the catalytic active site (the RT domain), productive TP-RT domain interactions must be maintained to properly position the primer residue in the active site of the RT domain and allow proper association with the ε RNA (including induced-fit conformational changes [see the introduction]) and possibly allosteric activation of the RT active site by TP (8).
The multiple intra- and interdomain interactions in the hepadnavirus RT protein during protein-primed initiation of reverse transcription and the associated dynamic conformational changes represent excellent but yet-to-be exploited opportunities for developing new generations of anti-HBV agents. Some small sequence motifs have been localized within the DHBV TP and RT domain, termed T3 (176–183) and RT1 (381–416), respectively, which are thought to be molecular contact sites involved in protein priming, and peptides derived from these motifs could indeed block protein priming (2, 9). Our results here imply the existence of multiple contact points in the RT domain that can be accessed in trans and may thus be targeted by novel antiviral peptides or small molecules. In this regard, peptides derived from different domains of the human immunodeficiency virus type 1 (HIV-1) RT protein have been shown to inhibit the HIV-1 RT activity by blocking either its dimerization or structural maturation following dimerization (1, 13). The trans-priming and trans-inhibition systems developed here should not only help to elucidate further the complex TP and RT domain interactions and the structural dynamics during hepadnavirus protein priming but also facilitate screening of peptide- and small-molecule-based anti-HBV agents to block the TP and RT domain interactions and the conformational dynamics essential for protein priming.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Public Health Service grant (R01 AI043453 to J.H.) from the National Institutes of Health.
We thank the Biostatistics Core, Penn State Hershey Cancer Institute, for assistance with statistical analyses.
Footnotes
Published ahead of print 18 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Agopian A, et al. 2009. A new generation of peptide-based inhibitors targeting HIV-1 reverse transcriptase conformational flexibility. J. Biol. Chem. 284:254–264 [DOI] [PubMed] [Google Scholar]
- 2. Badtke MP, Cao F, Tavis JE. 2006. Combining genetic and biochemical approaches to identify functional molecular contact points. Biol. Proced. Online 8:77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Badtke MP, Khan I, Cao F, Hu J, Tavis JE. 2009. An interdomain RNA binding site on the hepadnaviral polymerase that is essential for reverse transcription. Virology 390:130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartenschlager R, Schaller H. 1988. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked protein) believed to prime reverse transcription. EMBO J. 7:4185–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beck J, Nassal M. 2011. A Tyr residue in the reverse transcriptase domain can mimic the protein-priming Tyr residue in the terminal protein domain of a hepadnavirus P protein. J. Virol. 85:7742–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beck J, Nassal M. 1998. Formation of a functional hepatitis B virus replication initiation complex involves a major structural alteration in the RNA template. Mol. Cell. Biol. 18:6265–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beck J, Nassal M. 2001. Reconstitution of a functional duck hepatitis B virus replication initiation complex from separate reverse transcriptase domains expressed in Escherichia coli. J. Virol. 75:7410–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boregowda RK, Lin L, Zhu Q, Tian F, Hu J. 2011. Cryptic protein priming sites in two different domains of duck hepatitis B virus reverse transcriptase for initiating DNA synthesis in vitro. J. Virol. 85:7754–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cao F, et al. 2005. Identification of an essential molecular contact point on the duck hepatitis B virus reverse transcriptase. J. Virol. 79:10164–10170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang LJ, Hirsch RC, Ganem D, Varmus HE. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y, Marion PL. 1996. Amino acids essential for RNase H activity of hepadnaviruses are also required for efficient elongation of minus-strand viral DNA. J. Virol. 70:6151–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen Y, Robinson WS, Marion PL. 1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol. 68:5232–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Divita G, Restle T, Goody RS, Chermann JC, Baillon JG. 1994. Inhibition of human immunodeficiency virus type 1 reverse transcriptase dimerization using synthetic peptides derived from the connection domain. J. Biol. Chem. 269:13080–13083 [PubMed] [Google Scholar]
- 14. Fallows DA, Goff SP. 1995. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69:3067–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ganem D, Prince AM. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118–1129 [DOI] [PubMed] [Google Scholar]
- 16. Hu J. 2004. Studying DHBV polymerase by in vitro transcription and translation. Methods Mol. Med. 95:259–269 [DOI] [PubMed] [Google Scholar]
- 17. Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol. 74:11447–11455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu J, Boyer M. 2006. Hepatitis B virus reverse transcriptase and epsilon RNA sequences required for specific interaction in vitro. J. Virol. 80:2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 78:13122–13131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu J, Lin L. 2009. RNA-protein interactions in hepadnavirus reverse transcription. Front. Biosci. 14:1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu J, Seeger C. 1996. Expression and characterization of hepadnavirus reverse transcriptases. Methods Enzymol. 275:195–208 [DOI] [PubMed] [Google Scholar]
- 22. Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 93:1060–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu J, Seeger C. 1997. RNA signals that control DNA replication in hepadnaviruses. Semin. Virol. 8:205–211 [Google Scholar]
- 24. Hu J, Toft D, Anselmo D, Wang X. 2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol. 76:269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 16:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Itzen A, Blankenfeldt W, Goody RS. 2011. Adenylylation: renaissance of a forgotten post-translational modification. Trends Biochem. Sci. 36:221–228 [DOI] [PubMed] [Google Scholar]
- 27. Lanford RE, Kim YH, Lee H, Notvall L, Beames B. 1999. Mapping of the hepatitis B virus reverse transcriptase TP and RT domains by transcomplementation for nucleotide priming and by protein-protein interaction. J. Virol. 73:1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lanford RE, Notvall L, Beames B. 1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 69:4431–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lanford RE, Notvall L, Lee H, Beames B. 1997. Transcomplementation of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcriptase. J. Virol. 71:2996–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin L, Hu J. 2008. Inhibition of hepadnavirus reverse transcriptase-epsilon RNA interaction by porphyrin compounds. J. Virol. 82:2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin L, Wan F, Hu J. 2008. Functional and structural dynamics of hepadnavirus reverse transcriptase during protein-primed initiation of reverse transcription: effects of metal ions. J. Virol. 82:5703–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGlynn KA, London WT. 2011. The global epidemiology of hepatocellular carcinoma: present and future. Clin. Liver Dis. 15:223–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nassal M, Rieger A. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pollack JR, Ganem D. 1994. Site-specific RNA binding by a hepatitis B virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol. 68:5579–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radziwill G, Tucker W, Schaller H. 1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol. 64:613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rieger A, Nassal M. 1996. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 70:585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seeger C, Hu J. 1997. Why are hepadnaviruses DNA and not RNA viruses? Trends Microbiol. 5:447–450 [DOI] [PubMed] [Google Scholar]
- 38. Seeger C, Leber EH, Wiens LK, Hu J. 1996. Mutagenesis of a hepatitis B virus reverse transcriptase yields temperature-sensitive virus. Virology 222:430–439 [DOI] [PubMed] [Google Scholar]
- 39. Seeger C, Zoulim F, Mason WS. 2007. Hepadnaviruses, p 2977–3030 In Knipe DM, et al. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 40. Stahl M, Beck J, Nassal M. 2007. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J. Virol. 81:13354–13364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415 [DOI] [PubMed] [Google Scholar]
- 42. Tavis JE, Ganem D. 1996. Evidence for activation of the hepatitis B virus polymerase by binding of its RNA template. J. Virol. 70:5741–5750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tavis JE, Ganem D. 1993. Expression of functional hepatitis B virus polymerase in yeast reveals it to be the sole viral protein required for correct initiation of reverse transcription. Proc. Natl. Acad. Sci. U. S. A. 90:4107–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tavis JE, Ganem D. 1995. RNA sequences controlling the initiation and transfer of duck hepatitis B virus minus-strand DNA. J. Virol. 69:4283–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tavis JE, Massey B, Gong Y. 1998. The duck hepatitis B virus polymerase is activated by its RNA packaging signal, epsilon. J. Virol. 72:5789–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toh H, Hayashida H, Miyata T. 1983. Sequence homology between retroviral reverse transcriptase and putative polymerases of hepatitis B virus and cauliflower mosaic virus. Nature 305:827–829 [DOI] [PubMed] [Google Scholar]
- 48. Wang GH, Seeger C. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670 [DOI] [PubMed] [Google Scholar]
- 50. Wang GH, Zoulim F, Leber EH, Kitson J, Seeger C. 1994. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol. 68:8437–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang X, Hu J. 2002. Distinct requirement for two stages of protein-primed initiation of reverse transcription in hepadnaviruses. J. Virol. 76:5857–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang X, Qian X, Guo HC, Hu J. 2003. Heat shock protein 90-independent activation of truncated hepadnavirus reverse transcriptase. J. Virol. 77:4471–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weber M, et al. 1994. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 68:2994–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zahn KE, Tchesnokov EP, Gotte M, Doublie S. 2011. Phosphonoformic acid inhibits viral replication by trapping the closed form of the DNA polymerase. J. Biol. Chem. 286:25246–25255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang Z, Tavis JE. 2006. The duck hepatitis B virus reverse transcriptase functions as a full-length monomer. J. Biol. Chem. 281:35794–35801 [DOI] [PubMed] [Google Scholar]
- 56. Zoulim F, Seeger C. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.