

Abstract
A number of sentinels sense incoming herpes simplex virus (HSV) virions and initiate an immediate innate response. The first line of defense at the cell surface is TLR2 (Toll-like receptor 2), whose signature signaling activity leads to activation of the key transcription factor NF-κB. We report that the HSV pathogen-associated molecular patterns for TLR2 are the virion glycoproteins gH/gL and gB, which constitute the conserved fusion core apparatus across the members of the Herpesviridae family. Specifically, virions devoid singly of one of essential fusion glycoproteins (gD, gB, or gH null), able to attach to cells but defective in fusion/entry, were sufficient to elicit the first wave of NF-κB response to HSV. The most effective were the gD-null virions, positive for gH/gL and gB. A soluble form of gB, truncated upstream of the transmembrane sequence (gB730t-st), was produced in human cells and purified by means of a Strep tag. gH/gL and gB were each able to physically interact with TLR2 in coimmunoprecipitation assays, one independently of the other, yet gHt-st/gL, but not gB730t-st, elicited an NF-κB response. Thus, whereas both HSV gH/gL and gB are ligands to TLR2, only gH/gL is sufficient to initiate a signaling cascade which leads to NF-κB activation.
INTRODUCTION
The cell deploys numerous obstacles to invasion by herpesviruses in general, including herpes simplex virus (HSV). To this end, a number of receptors sense the presence of the HSV virion, or some of its products, and initiate a signaling activity which culminates in the cell innate response (40). Collectively, the receptors are called pattern recognition receptors (PRRs) and include the Toll-like receptors (TLRs), transmembrane proteins located either at the plasma membrane or in endosomes (35, 38, 42). TLRs recognize motifs distinct from those of the host, defined as pathogen-associated molecular patterns (PAMPs), which include virion proteins, viral DNA, or double-stranded viral RNAs. The TLRs important for HSV-1 recognition are TLR2 (27, 28), which is located in the plasma membrane and is therefore the first line of defense; TLR9, which is able to recognize double-stranded DNA in endosomes—mainly in plasmacytoid cells (32); and TLR3 (22, 61), which is also present in endosomes and is able to sense viral RNA. Pathogen recognition by TLRs triggers a series of signaling events characterized by the recruitment of adaptors and activation of endpoint proteins. This signature TLR2 signaling leads to the activation of the key transcription factor NF-κB. These dimers are maintained in the cytoplasm in inactive form by inhibitors (IκB proteins). In the canonical pathway of activation, the phosphorylated IKK (IκB kinase), a trimer made of IKKα, IKKβ, and the regulatory subunit IKKγ, also named NEMO, phosphorylates and promotes subsequent ubiquitination and proteasomal degradation of IκBα. This leads to the release of the RelA-p50 dimer, which is translocated to the nucleus, where it regulates the transcription of NF-κB-responsive genes, in particular, cytokines, including beta interferon, inflammatory cytokines, and chemokines (24, 53).
The cell response to HSV can be differentiated in two temporal waves. The first, immediate wave is exemplified by UV-inactivated human cytomegalovirus (HCMV) and HSV; it activates NF-κB and other lines of defense within a few minutes after infection (8, 34, 38, 60). UV-inactivated virions are able to enter cells but fail to express any viral gene, implying that one or more virion components induce NF-κB activation. A second and more sustained wave of NF-κB activation occurs at later times and requires viral gene products (21, 41, 44, 54, 55). Because the NF-κB response is, at least in part, detrimental to the virus, HSV has evolved strategies to counteract it or even take advantage of it. Thus, vhs (virion host shutoff) and the immediate-early proteins ICP0 and ICP27 inhibit TLR2-dependent NF-κB activation (13, 23, 25, 39, 57, 59).
In beta- and gammaherpesviruses, the virion envelope components which activate NF-κB signaling are HCMV envelope glycoproteins gH and gB (8) or Epstein-Barr virus (EBV) and Kaposi's sarcoma herpesvirus (KSHV) gp350 and K1, respectively (14, 16). gp350 and K1 mediate EBV and KSHV attachment to cells; gH/gL and gB represent the conserved fusion core apparatus in all human herpesviruses. In HSV, the tegument protein UL37 activates NF-κB signaling through the TRAF6 adaptor protein (30). A glycoprotein-dependent and TLR-2-independent innate response has been reported in dendritic cells (43). The issue of whether HSV envelope glycoproteins activate NF-κB has been addressed in two series of studies. An insightful study found that gD was able to block Fas-mediated apoptosis (34). Monocytic U937 cells were rendered resistant to apoptosis following infection with UV-inactivated virions or exposure to cells expressing gD or to a soluble form of gD. gD-mediated NF-κB signaling was seen in cells expressing HVEM (herpesvirus entry mediator) as a gD receptor (49). A second study made use of HSV virions devoid of one of the glycoproteins essential for HSV entry/fusion (33), namely, gD, the heterodimer gH/gL, or gB. gD serves as a receptor binding glycoprotein, gH/gL likely serves as an intermediate between gD and gB in promoting virus fusion/entry, and gB serves as a fusion glycoprotein (10, 11, 12). Virus particles lacking one of these proteins are entry/fusion defective although still capable of attachment to cells. In a microarray and quantitative reverse transcription-PCR study, binding of virions was sufficient to stimulate an immediate cell gene expression response. In particular, gD was confirmed as implicated in NF-κB activation and several pathways were found to be induced by entry/fusion-defective virions (33).
The aims of this study were to identify the HSV TLR2 PAMPs and to define whether the HSV glycoproteins involved in entry/fusion can activate TLR-2-dependent NF-κB signaling. We report that entry-defective virions devoid of one of the essential glycoproteins, in particular, gD-null virions, elicited an NF-κB response; soluble forms of gH/gL, but not of gB, were sufficient to elicit NF-κB activation; and both gH/gL and gB physically interacted with TLR2 independently of one another.
MATERIALS AND METHODS
Cells and viruses.
293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). Wild-type HSV-1(F) and the ICP0− mutant R7910 were described previously (17, 31). The gD−/+, gH−/+, and gB−/+ mutant viruses were described previously (9, 18, 29).
Plasmids.
The mammalian expression plasmids encoding gH, gL, and gB in the MTS vector and gD and HVEM (pBEC) in pcDNA3.1, all under the control of the cytomegalovirus promoter, were described previously (6, 36). Empty vector pcDNA3.1 was from Invitrogen. Plasmids encoding TLR2-flag and pNF-κB-luc were a gift from E. Kurt-Jones and were described previously (57).
Antibodies (Abs).
Monoclonal Ab (MAb) H1817 to gB recognizes a conformation-independent N-terminal epitope; MAb H233 to gB recognizes a conformation-dependent epitope, neutralizes infectivity, and was purchased from the Goodwin Institute. MAb 52S to gH recognizes a conformation-dependent epitope and neutralizes virion infectivity (50). Polyclonal Ab (PAb) to gHt-st/gL was derived by standard techniques by PRIMM (Milan, Italy) (20). PE (phycoerythrin)-conjugated Strep-Tactin was from IBA GmbH (Göttingen, Germany). Anti-flag M2 and anti-β-tubulin MAbs were from Sigma-Aldrich, and anti-Iκ-Bα MAb was from Cell Signaling.
Soluble glycoproteins.
gB730t-st was truncated upstream of the transmembrane sequences and tagged with a Strep tag as follows. The starting plasmid, named gBC (4), encodes HSV-1(F) gB (amino acids [aa] 1 to 867) fused to the C-terminal half of the enhanced green fluorescent protein cloned into the pcDNA3.1 vector. The plasmid was mutagenized by means of oligonucleotides 5′-ACGCCGACGCCAACGCCGCCAGATCTGCGGGCCTGGGCGCGTTCTTCG-3′ and 5′-GAAGAACGCGCCCAGGCCCGCAGATCTGGCGGCGTTGGCGTCGGCGT-3′ (the BglII site is underlined) in order to introduce a BglII site at aa 730 of gB, upstream of the transmembrane sequence. The mutant plasmid was digested with BglII-HindIII to remove the transmembrane sequence and cytoplasmic tail of gB. We ligated an oligonucleotide generated by PCR by means of oligonucleotides 5′-GGAAGATCTAGCGGAGGTGGACATCATCACCATCACCATAGCGGAGGTGGAAGCGCTTGGAGCCACCCGCAGTTCGAGAAAGGTGGAGGTTCCGGAGGT-3′ and 5′-AAAAGCTTTCATTTTTCGAACTGCGGGTGGCTCCACGATCCACCTCCCGATCCACCTCCGGAACCTCCACCTTTCTCGAACTGCGGGTGGCTCCAAGCG-3′ (BglII and HindIII sites are underlined) into the BglII-HindIII-digested plasmid, thus generating pgB730t-st. The PCR-generated oligonucleotide inserts a C-terminal One-STrEP tag consisting of two tandem Strep tags and an intervening Ser-Gly linker. 293T cells were transfected with pgB730t-st by means of Arrest-in (Thermo Scientific). Twelve hours after transfection, cells were overlaid with CD293, a chemically defined, protein- and serum-free medium (Invitrogen). Medium was harvested 3 to 4 days later, replaced with fresh medium, and harvested 3 to 4 days later. The two media were pooled and then concentrated by ultrafiltration. gB730t-st was purified by means of Strep-Tactin columns according to the IBA protocol at the IBA facility (IBA GmbH, Göttingen, Germany). Soluble gHt-st/gL was produced in insect cells as described previously (20). Soluble gD290-299 was described previously (20).
ELISA.
gB730t-st reactivity to MAbs H1817 and H233 was measured by enzyme-linked immunosorbent assay (ELISA). gB730t-st or fetuin (negative control) was immobilized on 96-well trays at a concentration of 8 nM in bicarbonate buffer for 16 h at 4°C. Unspecific binding sites were blocked with 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 2 h at 37°C. Serial dilutions of MAbs H1817 and H233 (from 1:30 to 1:21,870) were added to the wells in PBS containing 1% BSA and incubated for 1 h at 37°C. Excess Abs were removed, and the bound Abs were reacted with anti-mouse Abs conjugated to peroxidase. Binding was detected by incubation with o-phenylenediamine (Sigma-Aldrich) at 0.5 mg/ml, and the optical density at 490 nm was read.
Binding of gB730t-st to cells.
293T cells were washed once with PBS and once with PBS containing 10% FBS and then allowed to react with 0.8 μM gB730t-st in 100 μl of DMEM containing 5% FBS and 30 mM HEPES for 1 h at 4°C. Cells were washed three times with PBS and reacted with Strep-Tactin–PE, a PE-conjugated MAb directed to the Strep tag engineered at the C terminus of truncated gB, in PBS containing 5% FBS for 1 h at 4°C. Control cells were incubated only with Strep-Tactin–PE. Cells were analyzed with a FACScalibur cytometer (BD). For each sample, a minimum of 20,000 cells were acquired in list mode as described previously (20). Heparinase treatment of cells was done as described previously (58).
NF-κB activity.
293T cells were transfected by means of Arrest-in or Lipofectamine 2000 (Invitrogen) with a plasmid encoding firefly luciferase under the control of an NF-κB-regulated promoter and Renilla luciferase at a firefly/Renilla luciferase ratio of 130:1 plus TLR2-flag or with the pcDNA 3.1 empty vector, as indicated. When indicated, pBEC encoding HVEM was added. After transfection, cells were incubated in pre-exhausted medium for 2 days before use. Cells were exposed to the indicated viruses (UV irradiated or not) for the indicated times or stimulated with lipopolysaccharide (LPS; catalog number L2630; Sigma-Aldrich, Milan, Italy) at 100 ng/ml, gDt290-299 at 1.5 μM, gB730t-st at 0.75 μM, or increasing concentrations of gHt-st/gL for 4 h. When indicated, gHt-st/gL were heat inactivated for 15 min at 100°C or preincubated with MAb 52S for 1 h at 37°C before addition to cells. Luciferase activity was quantified by means of the Dual-Glo luciferase reporter assay system (Promega).
Coimmunoprecipitation experiments.
293T cells were transfected by means of Arrest-In with a TLR2-flag-encoding plasmid (1.5 μg DNA/10-cm2 dish) plus plasmids encoding full-length gH (1 μg) and gL (0,5 μg) or full-length gB (1 μg). Negative-control cells were transfected with the empty vector or with a plasmid encoding a flag-tagged irrelevant protein (TOM1, target of Myb protein 1 [a gift from A. Calistri]). Eighteen hours after transfection, cells were lysed in PBS buffer containing 1% NP-40, 1% sodium deoxycholate, and protease inhibitors. TLR2 was immunoprecipitated with anti-flag MAb M2 (Sigma-Aldrich). The proteins retained by protein G-Sepharose (Sigma-Aldrich) were separated by PAGE and blotted with PAb to gH/gL (20), MAb H1817 to gB, or MAb M2 to flag. In a reverse coimmunoprecipitation experiment, the glycoproteins were first immunoprecipitated with MAb H1817 or PAb to gH/gL and the retained proteins were probed with anti-flag MAb.
RESULTS
Generation of a Strep-tagged soluble form of gB.
A soluble form of gB (gB730t-st), truncated upstream of the transmembrane sequences, was produced in 293T cells and purified by means of a C-terminal Strep tag. Briefly, the plasmid named gBC (4), encoding HSV-1(F) gB (aa 1 to 867 fused to the C-terminal half of the enhanced green fluorescent protein) in the pcDNA3.1 vector, was mutagenized in order to introduce a BglII site at aa 730. The plasmid was digested with BglII-HindIII to remove the transmembrane sequence and cytoplasmic tail of gB. We ligated a PCR-generated oligonucleotide into the BglII-HindIII-digested plasmid, thus generating pgB730t-st. The PCR-generated oligonucleotide inserts a C-terminal One-STrEP tag consisting of two tandem Strep tags and an intervening Ser-Gly linker. 293T cells were transfected with pgB730t-st by means of Arrest-in. Twelve hours after transfection, cells were overlaid with CD293, a chemically defined protein- and serum-free medium (Invitrogen). Medium was harvested 3 to 4 days later, replaced with fresh medium, and harvested again 3 to 4 days later. The pooled media were concentrated by ultrafiltration. gB730t-st was purified by means of Strep-Tactin columns according to the IBA protocol at the IBA facility (IBA GmbH, Göttingen, Germany). The yield of protein was, at best, around 1 mg/liter of medium. The extent of purification was assessed by silver staining (Fig. 1). The purified protein reacted by Western blotting (WB) with MAb H1817 (Fig. 1).
Fig 1.
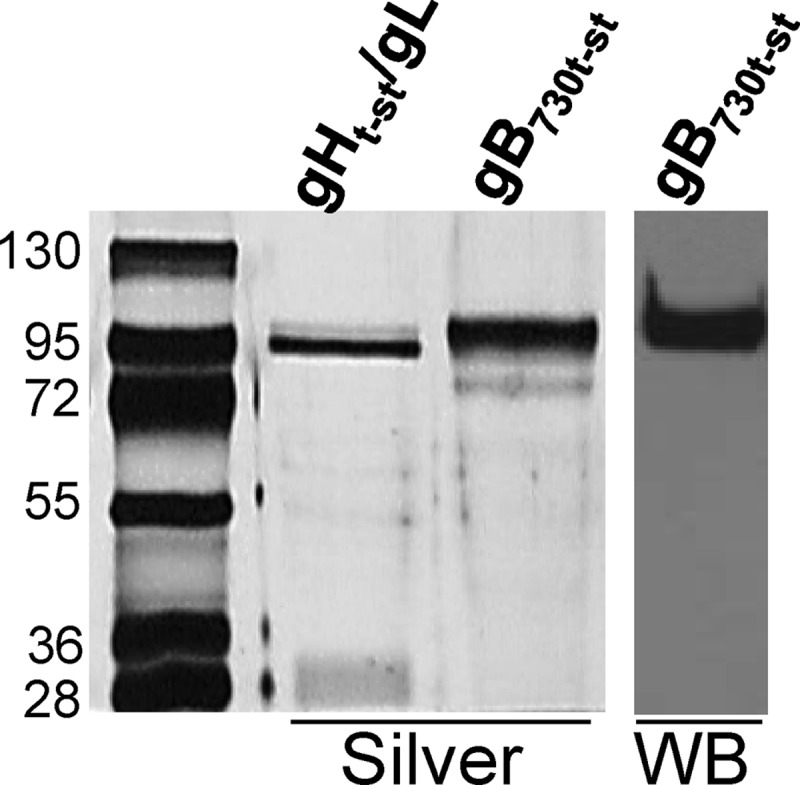
Purity and immunoreactivity of purified gB730t-st. Purified gB730t-st and, for comparison, gHt-st/gL were separated by SDS-PAGE and detected by silver staining (silver) or WB with MAb H1817. The migration positions of molecular mass markers are indicated on the left in kilodaltons.
To test whether gB730t-st represents a biologically active form of the glycoprotein, we measured (i) its reactivity to a neutralizing MAb (H233) (26) in ELISA and (ii) its ability to bind cells by fluorescence-activated cell sorter (FACS). Figure 2A shows the ELISA results. Binding to conformation-independent MAb H1817 was assayed in parallel as a positive control. Binding of the same Abs to fetuin served as a negative control and as the reaction background. gB730t-st bound both MAb H233 and MAb H1817 in a dose-dependent manner. By FACS analysis following incubation with gB730t-st, the vast majority of cells was marked with PE-labeled Strep-Tactin antibody (Ab) (Fig. 2B, bold line); about 60% of them exhibited a strong reactivity (cells within M1). It is known that gB binds cell surface heparan sulfates (51), as well as other receptors (1, 47). To further demonstrate the specificity of gB730t-st binding, cells were treated with heparinase prior to incubation with gB730t-st and Strep-Tactin–PE Ab (Fig. 2B, dashed line). Heparinase treatment caused a dramatic drop in fluorescence, as can also be appreciated by comparing the median fluorescence channels of the samples, obtained by transforming the log scale to a linear scale. The median fluorescence channel of the negative control was 103, that of the sample incubated with gB730t-st. was 271, and that of the heparinase-treated cells was 167. gB730t-st inhibited virus infection. At 1 μM, inhibition was around 30% (data not shown), in agreement with an earlier report (7). Higher concentrations could not be tested. Cumulatively, the results indicate that gB730t-st represents a biologically active form of the glycoprotein.
Fig 2.

Binding of gB730t-st to neutralizing and nonneutralizing MAbs and to the 293T cell surface. (A) ELISA reactivity of gB730t-st to neutralizing conformation-dependent (H233) and conformation-independent (H1817) MAbs. gB730t-st or fetuin, as a negative control, was immobilized on 96-well plates and allowed to react with increasing dilutions of MAbs. Reactivity was detected by anti-mouse Ab conjugated to peroxidase, followed by o-phenylenediamine and reading of the optical density (O.D.) at 490 nm. Each point represents the average of triplicate measurements. Bars represent standard deviations. (B) Flow cytometric analysis of the binding of gB730t-st. One experiment representative of three is shown. 293T cells were reacted with gB730t-st, followed by Strep-Tactin–PE Ab. The thin line represents the negative control (cells incubated with Strep-Tactin–PE Ab alone), the bold line represents the fluorescence of cells incubated with gB730t-st and Strep-Tactin–PE Ab, and the dashed line represents the fluorescence of cells treated with heparinase prior to staining with gB730t-st and Strep-Tactin–PE Ab. Abscissa, fluorescence intensity; ordinate, relative number of cells. The values shown above the curves represent the percentages of cells positive for staining.
The first wave of NF-κB activation dependent on TLR2 is elicited by entry-defective virions devoid of one of the essential glycoproteins.
To preliminarily confirm the abilities of UV-inactivated virions and the ΔICP0 R7910 mutant (31) to activate NF-κB, we transfected 293T cells—which fail to express endogenous TLR2—with TLR2 or an empty vector plus the NF-κB-luc plasmid, a plasmid encoding an NF-κB-driven luciferase reporter; a Renilla luciferase-encoding plasmid was cotransfected as a transfection control (firefly/Renilla luciferase ratio, 130:1). Results were expressed as units of firefly luciferase relative to Renilla luciferase. Figure 3A shows that the overall NF-κB activity was higher in the presence of TLR2 than in its absence, as expected, given that TLR2 is a sensor of HSV-1 (27). UV-inactivated HSV-1(F) virions, able to penetrate cells but unable to direct expression of viral genes, account for the first wave of the NF-κB response. As expected, the R7910 ΔICP0 mutant exerted a greater effect than the wild-type virus at 4 to 6 h after infection, in agreement with recent findings which indicate that ICP0 counteracts the TLR2 signaling pathway (15, 57).
Fig 3.

NF-κB activation by wild-type HSV-1(F), an ICP0− recombinant (R7910), or virions devoid of gD (gD−/−), gB (gB−/−), or gH/gL (gH−/−). 239T cells were transfected with NF-κB-luc and Renilla luciferase and with TLR2 as indicated. At 18 h after transfection, cells were exposed to the indicated viruses and harvested at the indicated times (hours) for panel A or at 6 h for panel B. UV-inactivated virions, F-UV and R7910-UV. Results are the average of triplicate experiments and are expressed as relative fluorescence units (RFU) × 102 ± the standard deviation. Un, untransfected.
Next, we measured NF-κB activation by virions devoid of gD, gH, or gB in cells negative or positive for TLR2. These viruses lack the indicated genes and are usually grown in complementing cells, generating the corresponding gD−/+, gH−/+, and gB−/+ virions. When grown in noncomplementing cells, they generate gD−/−, gH−/−, and gB−/− virions, which attach to cells but fail to carry out virion-cell fusion and entry. 293T cells were transfected, or not, with TLR2 along with NF-κB-luc- and Renilla luciferase-encoding plasmids. Figure 3B shows that the glycoprotein-null virions elicited reporter NF-κB activation; the greatest effect was observed with the gD−/− virions in TLR2-positive cells. The response to gH-null virions was reported previously in a different cell line (33). Although the virions lacking different glycoproteins exhibited minor differences from one another that were not necessarily significant, these findings point to entry glycoproteins as candidate inducers of TLR-2-dependent NF-κB activation.
Soluble forms of gH/gL, but not of gB, suffice for TLR2-dependent NF-κB activation.
The above findings prompted us to investigate whether soluble forms of the virion glycoproteins suffice to mediate NF-κB activation. gHt-st/gL is a biologically active soluble form, produced in insect cells and carrying a C-terminal Strep tag for purification; it binds conformation-dependent neutralizing MAbs, binds to cell surfaces in a saturable manner, and inhibits virus infection (20). 293T cells negative or positive for TLR-2 expression were exposed to increasing amounts of gHt-st/gL. Figure 4A shows that gHt-st/gL induced NF-κB activation in a TLR2-dependent fashion. NF-κB activation by soluble proteins is marred by the fact that the preparations may contain heat-stable LPS, lipopeptides, or other unspecific activators. Figure 4A shows that heat inactivation of gHt-st/gL reduced TLR2-dependent NF-κB activation by 60 to 70%, suggesting that most of the activity was ascribable to gH/gL. This conclusion was confirmed by the finding that preincubation of gHt-st/gL with MAb 52S reduced NF-κB activation by about 60% (Fig. 4B). With respect to gB730t-st, our preparations of gB730t-st were at much lower concentrations and needed to be concentrated and, overall, larger volumes were employed. At least three independent batches failed to elicit an NF-κB response, in agreement with a previous report (8). A representative result is shown in Fig. 4C, along with NF-κB activation induced by R7910 or by a commercial LPS (100 ng/ml) preparation which elicited a TLR2-dependent response. We note that some batches of gB730t-st elicited an NF-κB response, but this was mostly heat resistant and not decreased by preincubation of the glycoprotein with MAb H233 (data not shown). We conclude that soluble gH/gL, but not soluble gB, was able and sufficient to induce NF-κB activation in a TLR2-dependent fashion.
Fig 4.

NF-κB activation by soluble glycoproteins gHt-st/gL, gB730t-st, and gD290-299. (A) 239T cells were transfected with NF-κB-luc and TLR2 as indicated. At 48 h after transfection, cells were exposed to the indicated amounts of native (N) or heat-denatured (H) gHt-st/gL for 4 h. The results are the average of three experiments with two different batches of gHt-st/gL. (B) 239T cells were transfected with NF-κB-luc and TLR2 as indicated. gH/gL was preincubated for 1 h with MAb 52S (+Ab) prior to administration to cells for 4 h at 48 h after transfection. (C) 293T cells positive or negative for TLR2 were exposed to R7910, gB730t-st, or LPS for 4 h as indicated in panel A. (D) 239T cells were transfected with NF-κB-luc, TLR2, and HVEM as indicated. At 48 h after transfection, cells were exposed to gD290-299 for 4 h. All results are averages of triplicate experiments.
It was reported previously that gD induces NF-κB activation following binding to HVEM, one of its receptors (34, 49). We confirmed that result by transfecting cells with HVEM, exposing them to gD290-299, and measuring NF-κB activity (Fig. 4D). We further noted that gD-HVEM-induced NF-κB activation did not vary whether cells were positive or negative for TLR2 (Fig. 4D). This argues that HSV virions can elicit NF-κB activation through at least two independent signaling pathways, one initiated by gH/gL via TLR2 and one initiated by gD via HVEM and independent of TLR2.
To verify gH/gL-induced NF-κB activation at the endogenous protein level, we detected Iκ-Bα, which undergoes phosphorylation and degradation during the process of NF-κB activation. 293T cells negative or positive for TLR2 were infected with R7910 or exposed to gHt-st/gL or the commercial LPS preparation for 4 h. The lysates were subjected to SDS-PAGE. Iκ-Bα was revealed by WB; a decrease in band intensities denotes NF-κB activation. For relative quantification of the bands by means of ImageJ software, the blots were also reacted with antitubulin Ab (Sigma-Aldrich). Band quantification was expressed in densitometric units (DUs). The ratio of the DUs of the band of interest relative to the DUs of tubulin in the same sample was calculated and expressed as a percentage of the ratio in the uninfected-cell sample. Figure 5A shows a typical WB example. Figure 5B shows the average relative quantification of band intensities from two experiments. In TLR2-positive cells, gHt-st/gL induced a decrease similar to that caused by R7910.
Fig 5.
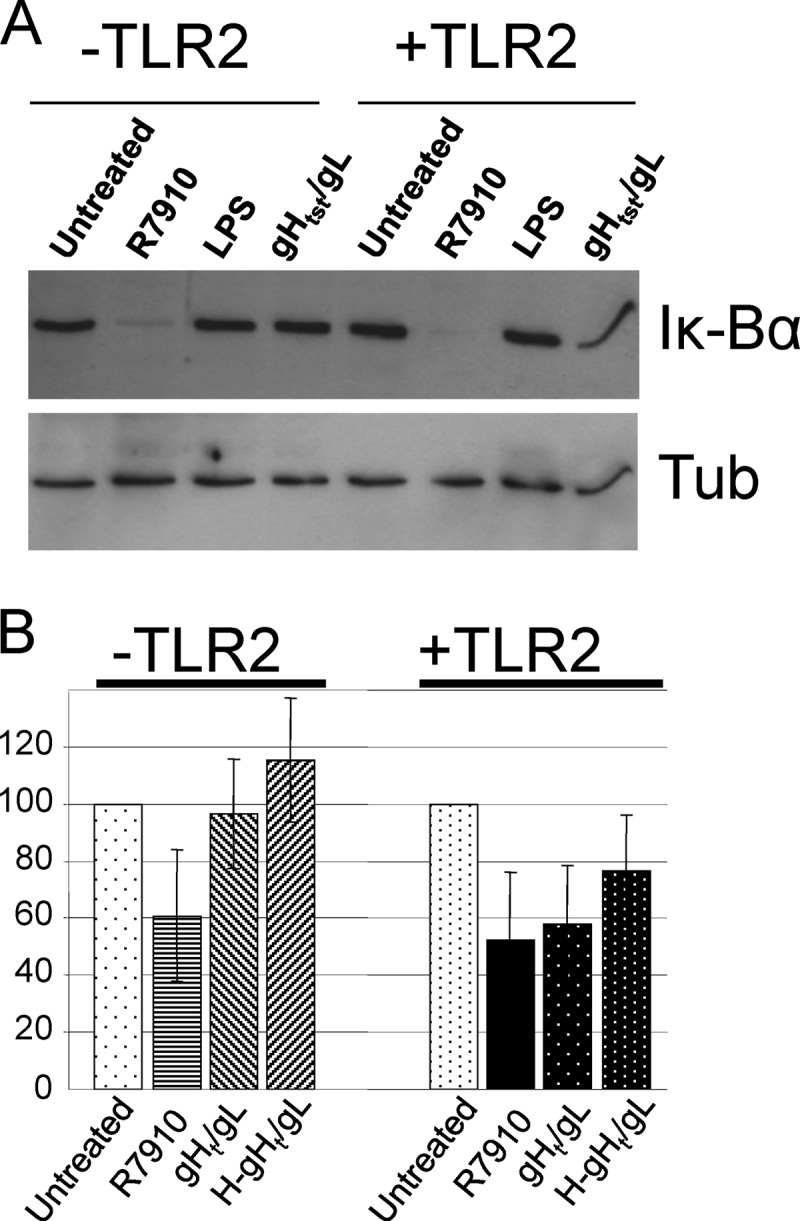
Validation of NF-κB activation by gHt-st/gL through detection of Iκ-Bα degradation. 239T cells were transfected with TLR2 as indicated for 48 h and exposed to R7910 or native or heat-denatured (H) gHt-st/gL for 4 h. Cellular proteins extracted in the presence of protease and phosphatase inhibitors (Sigma-Aldrich) were separated by SDS-PAGE and identified by WB with Abs to Iκ-Bα and tubulin (Tub) (A). For each lane, quantification is expressed as the ratio of Iκ-Bα to tubulin (B).
TLR2 physically interacts with gH/gL and gB.
The greatest extent of NF-κB activation induced by entry/fusion-defective virions or by soluble gH/gL was in a TLR2-dependent fashion. In the case of HCMV, gB and gH/gL induce NF-κB activation by binding TLR2 (8). To investigate whether HSV gH/gL and gB physically interact with TLR2, we transfected 293T cells with TLR2-flag or an irrelevant, flag-tagged protein (TOM1) as a negative control together with full-length gH/gL or gB and carried out coimmunoprecipitation assays. In the first series (Fig. 6A and B), TLR2 was immunoprecipitated with anti-flag MAb (IP flag); the retained proteins were analyzed by WB with PAb to gH/gL or MAb H1817 to gB (panel A). As shown, TLR2-flag was coimmunoprecipitated with gH (lane 3) and gB (lane 6). These glycoproteins were absent (lanes 2 and 5) when immunoprecipitation was carried out from samples which contained TOM1-flag instead of TLR2-flag. The immunoprecipitation of TLR2 was ascertained by blotting the same strips with anti-flag MAb (lanes 3′ and 6′ in panel B). TOM1 is not visible in lanes 2′ and 5′ because it has a much lower Mr than TLR2 and hence migrates faster. To validate the above results, we carried out the reverse coimmunoprecipitation (Fig. 6C). gH and gB were first immunoprecipitated with specific Abs (IP-gH, lane 3; IP-gB, lane 6); coimmunoprecipitated TLR2 was revealed with anti-flag MAb. Figure 6C shows that immunoprecipitation of gH (lane 3) or gB (lane 6) pulled down TLR2-flag. We note that in the gB immunoprecipitations (Fig. 6C, lanes 5 and 6), an unspecific band shows up, with a migration slower than that of TLR2-flag. The lanes marked Lys contain aliquots of the lysates prior to immunoprecipitation and were included to enable identification of the migration position of the proteins of interest.
Fig 6.

gH/gL and gB interact physically with TLR2 in coimmunoprecipitation assays. 293T cells were transfected with TLR2-flag or TOM1-flag and full-length gH/gL or gB, as indicated to the left in italics. (A and B) TLR2-flag or TOM1-flag was immunoprecipitated with anti-flag MAb (IP flag); complexes were harvested with protein G-Sepharose. Coimmunoprecipitated gH or gB was separated by SDS-PAGE and revealed by WB with PAb to gH or MAb H1817 to gB (A). Immunoprecipitated TLR2 was identified with anti-flag MAb (B). (C) 293T cells were cotransfected as indicated to the left in italics. gH or gB was immunoprecipitated with PAb to gH (IP gH) or MAb H1817 to gB (IP gB). Coimmunoprecipitated TLR2, retained by protein A-Sepharose, was separated by SDS-PAGE and identified with anti-flag MAb (TLR2 band). Unsp denotes an unspecific band which showed up only in immunoprecipitations with MAb H1817. In all panels, aliquots of lysates (Lys) were run in parallel for identification of the migration positions of the proteins of interest that were identified by WB.
DISCUSSION
TLR2, a key player in the innate response to HSV (27, 28), is located in the plasma membrane and represents the first line of defense against invading HSV. So far, the HSV virion glycoproteins which interact with TLR2 have remained elusive (16, 35, 40). Here we provide evidence that HSV gH/gL and gB are TLR2 ligands and that soluble gH/gL suffice to elicit TLR2 signaling, which leads to NF-κB activation. First, virions devoid of essential fusion glycoproteins, able to attach to cells but defective in fusion/entry, are sufficient to elicit an immediate NF-κB response to HSV. Of these, the most effective were the gD-null virions, positive for gH/gL and gB. Second, gH/gL and gB physically interact with TLR2 in coimmunoprecipitation assays. Third, soluble truncated forms of gH/gL, but not of gB, were sufficient to elicit an NF-κB response. gD was insufficient and did so only when HVEM was also expressed, in accordance with previous data which indicate that gD can signal to NF-κB through its HVEM receptor (34, 48). The HVEM-mediated gD response was independent of TLR2; hence, its signaling is distinct from that of TLR2, a feature previously unknown. It is unclear why soluble gB failed to elicit an NF-κB response. Failure may be consequent to the postfusion conformation adopted by gB in solution (11, 12), or signaling might require the presentation of envelope-bound gB. Alternatively, this is an intrinsic property of gB, which alone does not suffice to elicit the NF-κB response. The situation in HSV is similar, but not identical, to that in HCMV, where gH/gL and gB are ligands to TLR2 and initiate a signaling cascade (8); it differs from that in EBV and KSHV, in which the NF-κB response is elicited by the attachment glycoprotein gp350 or K1 (14, 16). TLR2 may be endocytosed (56). While a first interaction between the virion glycoproteins and TLR2 will occur at the plasma membrane, it can be envisioned that TLR2 is endocytosed along with HSV, and therefore, the signaling cascade may potentially take place both at the plasma membrane and following endocytosis.
The most surprising features of the present study are that the same glycoproteins which mediate virus entry into the cell also initiate the NF-κB response and furthermore that each of the major entry/fusion glycoproteins, gD, gH/gL, and gB can initiate such a response independently of one another. Some considerations are pertinent here. First, this finding strongly argues for the coevolution of HSV with its human host. Second, the quartet of gD, gH/gL, and gB act in concert to enable virus entry. Complexes of the glycoproteins have been documented (2–5, 19), and the current view of HSV entry envisions that fusion of the virion envelope with cell membranes is the result of a series of interactions that start with the gD encounter with its receptor and are transmitted to gH/gL and ultimately to gB, so that the fusion-active conformation is adopted (10, 12). The simultaneous interaction of gH/gL and gB with a same molecule (TLR2) may favor complex assembly/stabilization and interglycoprotein signaling. Because cells that lack TLR2 can be infected, the effect of TLR2 on entry may not be essential. For example, cells lacking TLR2 might alternatively employ gH/gL and/or gB receptors, like those described recently (20, 47, 52). The finding that soluble gH/gL, but not gB, suffices to elicit an NF-κB response argues that the complex of gH/gL and gB may be the bona fide activator of TLR2 signaling. Third, it has been debated whether the NF-κB response is detrimental or favorable to HSV. HSV has evolved a number of proteins which counteract the innate response, including the tegument protein encoded by vhs and the immediate-early proteins ICP0 and ICP27 (13, 21, 23, 25, 39, 55, 57). These proteins enter into the game after the virion glycoproteins have performed their task, raising the possibility that the immediate response is not totally detrimental to the virus. Beneficial effects of the NF-κB response may include some of the stress response RNAs elicited by NF-κB and/or the ability to promote the synthesis of proteins able to block apoptosis (13, 45, 46). Fourth, an emerging question is why HSV did not evolve its essential glycoproteins so as to escape the TLR2 interaction. An answer may reside in the nature of the PAMPs recognized by TLR2. For example, if the oligosaccharide chains of the virion glycoproteins are part of the molecular patterns, the virus would have no way to escape TLR2 recognition. Nonetheless, implicit in the innate and adaptive responses is the ability of the immune receptors to distinguish between the host and pathogens. Because the oligosaccharide chains of HSV glycoproteins are generated by the cell, a feature distinguishing between the host and the viral PAMPs might be provided by fucosyltransferases which are induced by HSV and are silent in uninfected cells (37) and may add one to four fucose residues for each complex-type N-linked sugar. These glycosyltransferases may potentially confer upon the oligosaccharides of HSV glycoprotein a signature recognized by TLR2.
The innate response represents the first building block of adaptive immunity. A number of efforts have been made in recent years to generate HSV vaccines based on the glycoproteins analyzed here, unfortunately with modest protective and prophylactic results. A better understanding of the innate response to HSV glycoproteins may be key to the design of better HSV subunit vaccines.
ACKNOWLEDGMENTS
We thank IBA GmbH, Göttingen, Germany, for collaborative purification of gHt-st/gL during the TargetHerpes Project and our colleagues R. Eisenberg, G. H. Cohen, E. Kurt-Jones, and A. Calistri for generous gifts of reagents.
This work was supported by investigator grants to G.C.-F. from AIRC (Associazione Italiana per la Ricerca sul Cancro, Milan, Italy), by the Fondazione del Monte di Bologna e Ravenna, by Roberto and Cornelia Pallotti Funds from the Department of Experimental Pathology, by PRIN projects from the Italian Ministry for University and Research, and by University of Bologna RFO (Ricerca Fondamentale Orientata).
Footnotes
Published ahead of print 11 April 2012
REFERENCES
- 1. Arii J, et al. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862 [DOI] [PubMed] [Google Scholar]
- 2. Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atanasiu D, et al. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532–11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avitabile E, Forghieri C, Campadelli-Fiume G. 2009. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J. Virol. 83:10752–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Avitabile E, Lombardi G, Campadelli-Fiume G. 2003. Herpes simplex virus glycoprotein K, but not its syncytial allele, inhibits cell-cell fusion mediated by the four fusogenic glycoproteins, gD, gB, gH, and gL. J. Virol. 77:6836–6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 79:11588–11597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boehme KW, Guerrero M, Compton T. 2006. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 177:7094–7102 [DOI] [PubMed] [Google Scholar]
- 9. Cai WH, Gu B, Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campadelli-Fiume G, et al. 2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 17:313–326 [DOI] [PubMed] [Google Scholar]
- 11. Campadelli-Fiume G, Menotti L, Avitabile E, Gianni T. 2012. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr. Opin. Virol. 2:28–36 [DOI] [PubMed] [Google Scholar]
- 12. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cotter CR, et al. 2011. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J. Virol. 85:12662–12672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. D'Addario M, Ahmad A, Morgan A, Menezes J. 2000. Binding of the Epstein-Barr virus major envelope glycoprotein gp350 results in the upregulation of the TNF-alpha gene expression in monocytic cells via NF-kappaB involving PKC, PI3-K and tyrosine kinases. J. Mol. Biol. 298:765–778 [DOI] [PubMed] [Google Scholar]
- 15. Daubeuf S, et al. 2009. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113:3264–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DE Oliveira DE, Ballon G, Cesarman E. 2010. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 18:248–257 [DOI] [PubMed] [Google Scholar]
- 17. Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357–364 [DOI] [PubMed] [Google Scholar]
- 18. Forrester A, et al. 1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL through the C-terminal profusion. J. Biol. Chem. 284:17370–17382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gianni T, et al. 2010. Herpes simplex virus glycoproteins H/L bind to cells independently of {alpha}V{beta}3 integrin and inhibit virus entry, and their constitutive expression restricts infection. J. Virol. 84:4013–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gregory D, Hargett D, Holmes D, Money E, Bachenheimer SL. 2004. Efficient replication by herpes simplex virus type 1 involves activation of the IkappaB kinase-IkappaB-p65 pathway. J. Virol. 78:13582–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo Y, et al. 2011. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 208:2083–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hargett D, Rice S, Bachenheimer SL. 2006. Herpes simplex virus type 1 ICP27-dependent activation of NF-kappaB. J. Virol. 80:10565–10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawai T, Akira S. 2007. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 13:460–469 [DOI] [PubMed] [Google Scholar]
- 25. Kim JC, et al. 2008. HSV-1 ICP27 suppresses NF-kappaB activity by stabilizing IkappaBalpha. FEBS Lett. 582:2371–2376 [DOI] [PubMed] [Google Scholar]
- 26. Kousoulas KG, Huo B, Pereira L. 1988. Antibody-resistant mutations in cross-reactive and type-specific epitopes of herpes simplex virus 1 glycoprotein B map in separate domains. Virology 166:423–431 [DOI] [PubMed] [Google Scholar]
- 27. Kurt-Jones EA, et al. 2005. The role of Toll-like receptors in herpes simplex infection in neonates. J. Infect. Dis. 191:746–748 [DOI] [PubMed] [Google Scholar]
- 28. Kurt-Jones EA, et al. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. U. S. A. 101:1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu X, Fitzgerald K, Kurt-Jones E, Finberg R, Knipe DM. 2008. Herpesvirus tegument protein activates NF-kappaB signaling through the TRAF6 adaptor protein. Proc. Natl. Acad. Sci. U. S. A. 105:11335–11339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lopez P, Jacob RJ, Roizman B. 2002. Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J. Virol. 76:9355–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. 2003. Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198:513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacLeod IJ, Minson T. 2010. Binding of herpes simplex virus type-1 virions leads to the induction of intracellular signalling in the absence of virus entry. PLoS One 5:e9560 doi:10.1371/journal.pone.0009560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Medici MA, et al. 2003. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: role of nuclear factor kappaB. J. Biol. Chem. 278:36059–36067 [DOI] [PubMed] [Google Scholar]
- 35. Melchjorsen J. 2012. Sensing herpes: more than Toll. Rev. Med. Virol. 22(2):106–121 [DOI] [PubMed] [Google Scholar]
- 36. Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 37. Nordén R, Nystrom K, Olofsson S. 2009. Activation of host antiviral RNA-sensing factors necessary for herpes simplex virus type 1-activated transcription of host cell fucosyltransferase genes FUT3, FUT5, and FUT6 and subsequent expression of sLe(x) in virus-infected cells. Glycobiology 19:776–788 [DOI] [PubMed] [Google Scholar]
- 38. Paladino P, Cummings DT, Noyce RS, Mossman KL. 2006. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J. Immunol. 177:8008–8016 [DOI] [PubMed] [Google Scholar]
- 39. Paladino P, Mossman KL. 2009. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J. Interferon Cytokine Res. 29:599–607 [DOI] [PubMed] [Google Scholar]
- 40. Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11:143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patel A, et al. 1998. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virology 247:212–222 [DOI] [PubMed] [Google Scholar]
- 42. Rasmussen SB, et al. 2009. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene-like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 90:74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reske A, Pollara G, Krummenacher C, Katz DR, Chain BM. 2008. Glycoprotein-dependent and TLR2-independent innate immune recognition of herpes simplex virus-1 by dendritic cells. J. Immunol. 180:7525–7536 [DOI] [PubMed] [Google Scholar]
- 44. Roberts KL, Baines JD. 2011. UL31 of herpes simplex virus 1 is necessary for optimal NF-kappaB activation and expression of viral gene products. J. Virol. 85:4947–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, New York, NY [Google Scholar]
- 46. Santoro MG, Rossi A, Amici C. 2003. NF-kappaB and virus infection: who controls whom. EMBO J. 22:2552–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Satoh T, et al. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sciortino MT, et al. 2008. Involvement of gD/HVEM interaction in NF-kB-dependent inhibition of apoptosis by HSV-1 gD. Biochem. Pharmacol. 76:1522–1532 [DOI] [PubMed] [Google Scholar]
- 49. Sciortino MT, et al. 2008. Involvement of HVEM receptor in activation of nuclear factor kappaB by herpes simplex virus 1 glycoprotein D. Cell Microbiol. 10:2297–2311 [DOI] [PubMed] [Google Scholar]
- 50. Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Suenaga T, et al. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 107:866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun SC. 2011. Non-canonical NF-kappaB signaling pathway. Cell Res. 21:71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taddeo B, Luo TR, Zhang W, Roizman B. 2003. Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc. Natl. Acad. Sci. U. S. A. 100:12408–12413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taddeo B, Zhang W, Lakeman F, Roizman B. 2004. Cells lacking NF-kappaB or in which NF-kappaB is not activated vary with respect to ability to sustain herpes simplex virus 1 replication and are not susceptible to apoptosis induced by a replication-incompetent mutant virus. J. Virol. 78:11615–11621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vabulas RM, et al. 2001. Endocytosed HSP60s use Toll-like receptor 2 (TLR2) and TLR4 to activate the Toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 276:31332–31339 [DOI] [PubMed] [Google Scholar]
- 57. van Lint AL, et al. 2010. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 84:10802–10811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. WuDunn D, Spear PG. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63:52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yao XD, Rosenthal KL. 2011. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J. Gen. Virol. 92:1981–1993 [DOI] [PubMed] [Google Scholar]
- 60. Yurochko AD, et al. 1997. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J. Virol. 71:5051–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang SY, et al. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–1527 [DOI] [PubMed] [Google Scholar]