

Abstract
The emergence of resistance to existing classes of antiretroviral drugs necessitates finding new HIV-1 targets for drug discovery. The viral capsid (CA) protein represents one such potential new target. CA is sufficient to form mature HIV-1 capsids in vitro, and extensive structure-function and mutational analyses of CA have shown that the proper assembly, morphology, and stability of the mature capsid core are essential for the infectivity of HIV-1 virions. Here we describe the development of an in vitro capsid assembly assay based on the association of CA-NC subunits on immobilized oligonucleotides. This assay was used to screen a compound library, yielding several different families of compounds that inhibited capsid assembly. Optimization of two chemical series, termed the benzodiazepines (BD) and the benzimidazoles (BM), resulted in compounds with potent antiviral activity against wild-type and drug-resistant HIV-1. Nuclear magnetic resonance (NMR) spectroscopic and X-ray crystallographic analyses showed that both series of inhibitors bound to the N-terminal domain of CA. These inhibitors induce the formation of a pocket that overlaps with the binding site for the previously reported CAP inhibitors but is expanded significantly by these new, more potent CA inhibitors. Virus release and electron microscopic (EM) studies showed that the BD compounds prevented virion release, whereas the BM compounds inhibited the formation of the mature capsid. Passage of virus in the presence of the inhibitors selected for resistance mutations that mapped to highly conserved residues surrounding the inhibitor binding pocket, but also to the C-terminal domain of CA. The resistance mutations selected by the two series differed, consistent with differences in their interactions within the pocket, and most also impaired virus replicative capacity. Resistance mutations had two modes of action, either directly impacting inhibitor binding affinity or apparently increasing the overall stability of the viral capsid without affecting inhibitor binding. These studies demonstrate that CA is a viable antiviral target and demonstrate that inhibitors that bind within the same site on CA can have distinct binding modes and mechanisms of action.
INTRODUCTION
The current antiretroviral arsenal against HIV-1 comprises more than 26 FDA-approved drugs from six mechanistic classes that target one of the three viral enzymes or viral entry (5). In spite of this array of drugs and targets and the simplification of therapies, drug resistance can still occur due to lack of adherence, often owing to toxicities associated with the lifelong therapy required for sustained viral suppression (28, 36). Moreover, cross-resistance within mechanistic classes and the emergence of multidrug-resistant isolates can have considerable impact on treatment options and disease outcomes, underscoring the need to discover new classes of HIV inhibitors.
The HIV-1 capsid (CA) protein plays essential roles in viral replication and as such represents an attractive new therapeutic target (11, 18). CA is initially synthesized as the central region of the 55-kDa Gag polyprotein, which is the protein that mediates the assembly and budding of the immature virion. In this context, CA provides key protein-protein interactions required for immature virion assembly (18, 40). During viral maturation, proteolytic cleavage of Gag releases CA, allowing the protein to assemble into the cone-shaped central capsid that surrounds the viral RNA genome and its associated enzymes, reverse transcriptase (RT) and integrase (IN) (34, 35). The capsid is stabilized by multiple weak protein-protein interactions, and CA mutations that impair the assembly and/or stability of the capsid typically inhibit viral replication (10, 17, 40). Thus, HIV-1 CA plays essential roles during the assembly of both the immature virion and the mature viral capsid.
CA is composed of two highly helical domains, the N-terminal domain (CANTD, residues 1 to 146) and the C-terminal domain (CACTD, residues 151 to 231), which are separated by a short flexible linker. Solution nuclear magnetic resonance (NMR) and high-resolution X-ray crystal structures have been reported for both isolated domains (4, 13, 14, 19, 41). Conical HIV-1 capsids belong to a class of geometric structures called fullerene cones, which comprise hexagonal lattices with 12 pentagonal defects that allow the cones to close at both ends. Although individual HIV-1 capsids differ in size and shape, they typically contain ∼250 CA hexagons and have 7 CA pentagons at the wide end and 5 CA pentagons at the narrow end of the cone (15).
The recent availability of high-resolution structures of CA hexagons and pentagons has enabled molecular modeling of the viral capsid (29, 30). The capsid lattice is stabilized by four different types of intermolecular CA-CA interactions: a CANTD/CANTD interaction that creates the hexameric (or pentameric) rings (29, 30), a CANTD/CACTD interaction that forms a “girdle” that reinforces the rings (16, 29), dimeric CACTD/CACTD interactions that link adjacent hexamers across local 2-fold axes (1, 4, 22, 41), and trimeric CACTD/CACTD interactions that link adjacent hexamers across local 3-fold axes. Each of these different interfaces has been characterized structurally, although the interactions that stabilize the CACTD/CACTD trimer are not yet known in atomic detail (4). Moreover, several distinct but related CACTD/CACTD dimers have been observed (1, 4, 22, 41), and it is not yet certain how these different dimers are used to connect the CA hexamers and pentamers within authentic viral capsids (22). Although capsid-like conical assemblies can form in vitro, most conditions that drive CA assembly favor CA hexamerization over pentamerization such that recombinant CA proteins typically assemble into long helical tubes composed exclusively of CA hexamers (4, 26).
CA-binding inhibitors of HIV-1 capsid assembly have been reported, thus providing evidence that CA may be a viable drug target (2, 24, 32, 33, 37, 38). These inhibitors have collectively defined three independent inhibitor binding sites on CA. However, each of these sites ultimately appears to interfere with the formation of the CANTD/CACTD interface, suggesting that this interaction may be an inhibitory “Achilles' heel” for capsid assembly. The small-molecule inhibitor CAP-1 binds to an induced hydrophobic pocket at the base of the CANTD helical bundle (24, 37). The pocket is located at the junction of α-helices 1, 2, 4, and 7 and is normally occupied by the aromatic side chain of Phe-32 in structures of uninhibited CA hexamers, pentamers, and monomers. Binding of CAP-1 distorts the loop between helices 3 and 4, which may inhibit the formation of the CANTD/CACTD interface (16, 24). A second CA inhibitor, termed the CAI peptide, binds to a conserved hydrophobic cleft within the CACTD four-helix bundle and inhibits both Gag and CA assembly in vitro (33, 38). Superposition of the CACTD-CAI complex onto the CANTD/CACTD interface of assembled CA suggested that binding of the peptide would sterically hinder the CANTD/CACTD interaction (16, 29). The peptide may also act allosterically by altering the geometry of CACTD dimers such that propagation of the mature CA lattice is prevented. More recently, a third small-molecule binding site on CA was identified via a high-throughput screen for inhibitors of HIV replication. PF-3450074 and related compounds bind between helix 4 and helix 7 of the CANTD. This latest class of CA inhibitors disrupts the stability of the viral capsid in both the early and late stages of viral replication, again probably by altering CANTD/CACTD interactions (2, 32).
Here we describe two new and highly potent families of CA inhibitors that were identified by a novel high-throughput screening (HTS) assay. Inhibitors from both families bind to the CANTD by further expanding the CAP-binding pocket. Moreover, although these two classes of inhibitors target the same binding pocket, they have distinct binding modes, select for unique patterns of resistance mutations, and have different effects on virion morphology.
MATERIALS AND METHODS
Immobilized capsid assembly assay.
The 5′-biotin-labeled (TG)25 oligonucleotide (Integrated DNA Technology Inc.) was immobilized on Reacti-Bind neutravidin-coated black 384-well plates (Pierce catalog no. 15402) that were washed with 80 μl/well of buffer C {50 mM Tris (pH 8.0), 350 mM NaCl, 10 μM ZnSO4, 0.0025% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 50 μg/ml bovine serum albumin (BSA), 1 mM dithiothreitol (DTT)} prior to the addition of 50 μl/well of a 25 nM solution of oligonucleotide in buffer C plus 5 mg/ml BSA, followed by overnight incubation. Unbound material was removed by two washes with buffer C. Assembly reactions were performed in 60-μl/well reaction mixtures comprising 100 nM 5′-fluorescein-labeled (TG)25 oligonucleotide (Integrated DNA Technology Inc.), 2 μM CA-NC protein, and various concentrations of test compounds diluted in buffer C with a final dimethyl sulfoxide (DMSO) concentration of 1%. Assembly reaction mixtures were incubated for 2 h at room temperature, followed by two washes with buffer C, the addition of 80 μl/well of buffer C plus 0.1% sodium dodecyl sulfate (SDS), and a 15-min incubation prior to quantification of captured fluorescence on a Victor2 plate reader (Perkin-Elmer Life Sciences) equipped with fluorescein excitation and emission filters. The amount of captured fluorescence is proportional to the level of assembly. The concentrations of compound required for 50% inhibition of assembly (IC50s) were generated by fitting inhibition curves from 10-point dilution series to the following equation: % inhibition = (Imaxn × [I]n)/([I]n + IC50n) × 100, where Imax is the maximal percent inhibition, [I] is the corresponding concentration of inhibitor, and the superscript n denotes the Hill coefficient.
Protein expression and purification.
pET-11a vectors were used to express the HIV-1NL4-3 CA-NC (WISP-98-68, Gag residues 133 to 432) carrying the CA G94D mutation, full-length CA (WISP-98-85), CANTD (WISP-96-19, CA residues 1 to 146), and CACTD (WISP-97-07, CA residues 146 to 231) (17, 26). Point mutations were introduced using the QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions.
All proteins were expressed in Escherichia coli BL21(DE3) cells (Novagen). Briefly, LB medium was inoculated with overnight precultures, which were grown at 37°C until mid log-phase (A600, ∼0.6). Protein expression was induced with 0.5 to 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 to 6 h at 30°C. Cells were harvested by centrifugation, and pellets were stored at −80°C until purification. For NMR studies, 15N-labeled proteins were produced using Spectra 9 (15N, 98%) medium (Cambridge Isotope Laboratories Inc.).
Purification of CA-NC (and all mutants) was carried out as follows. Five to 10 g of cell paste was lysed by sonication in 40 ml of buffer A (20 mM Tris [pH 7.5], 1 μM ZnCl2, 10 mM β-mercaptoethanol) supplemented with 0.5 M NaCl and Complete EDTA-free protease inhibitor tablets (Roche). Nucleic acids and cell debris were removed by adding 0.11 volume of 0.2 M ammonium sulfate and an equivalent volume of 10% poly(ethyleneimine) (pH 8.0), stirring the sample for 20 min at 4°C, and centrifuging at 30,000 × g for 20 min. CA-NC protein was recovered from the supernatant by adding 0.35 volume of saturated ammonium sulfate solution, followed by centrifugation at 10,000 × g for 15 min. The pellet was dissolved in 10 ml of buffer A plus 0.1 M NaCl and was dialyzed overnight in buffer A plus 0.05 M NaCl. The sample was cleared by centrifugation and was chromatographed on a 1-ml HiTrap SP HP column (GE Healthcare) preequilibrated with dialysis buffer. CA-NC protein was eluted with buffer A plus 0.5 M NaCl. Fractions containing the protein were pooled, and the concentration was determined by absorbance at 280 nm using the calculated molar extinction coefficient (ε = 40,220 M−1 cm−1).
Purification of CANTD (wild type [WT], all mutants, and 15N labeled) was similar to that of CA-NC, but lysis was performed in buffer B (20 mM morpholineethanesulfonic acid [MES] [pH 6.5], 10 mM β-mercaptoethanol) supplemented with 0.5 M NaCl and Complete EDTA-free protease inhibitor tablets. Nucleic acids and cell debris were removed as described above. CANTD was recovered from the supernatant by the addition of 0.6 volume of saturated ammonium sulfate. The pellet was dissolved in 10 ml of buffer B and was dialyzed in the same buffer using dialysis tubing with a 10,000-molecular-weight cutoff. The sample was clarified by centrifugation and was sequentially passed through HiTrap SP HP and Q HP columns (GE Healthcare) preequilibrated in buffer B. CANTD was recovered in the flowthrough and wash fractions and was concentrated, and the protein concentration was determined by the absorbance at 280 nm using the calculated molar extinction coefficient (ε = 25,320 M−1 cm−1).
Cells.
SupT1 and 293FT cells were obtained from the ATCC (CRL-1942) and Invitrogen (R700-07), respectively. C8166 cells were obtained from J. Sullivan, University of Massachusetts Medical Center. C8166-LTR-Luc cells were produced by stable transfection of C8166 cells with an HIV long terminal repeat (LTR)-luciferase construct followed by selection with 5 μg/ml blasticidin S-HCl through three consecutive limiting dilutions. SupT1 and C8166 cells were maintained in RPMI medium (Wisent) supplemented with 10% fetal bovine serum (FBS; HyClone). C8166-LTR-Luc cells were maintained in the same medium supplemented with 5 μg/ml blasticidin S-HCl. Antibiotic was removed for all antiviral activity assays. 293FT cells were maintained in Dulbecco's modified Eagle medium (DMEM; Wisent) supplemented with 10% FBS (37°C, 5% CO2).
Antiviral activity (EC50) determinations.
Inhibitors were prepared in 10 to 20 mM stocks in 100% DMSO and were serially diluted in RPMI medium plus 10% FBS. C8166-LTR-luciferase cells were infected at a multiplicity of infection (MOI) of 0.005 with HIV-1 NL4-3 for 1.5 h and were seeded at 25,000 cells/well in 96-well black microtiter plates, in wells already containing inhibitors or an equivalent concentration of DMSO (0.5%). Plates were incubated for 3 days (37°C, 5% CO2), and luciferase expression levels were determined by the addition of 50 μl per well of SteadyGlo (Promega) and measurement on the BMG LUMIstar Galaxy luminometer. The inhibitor concentration needed to produce a 50% reduction of viral replication activity (50% effective concentration [EC50]) was determined by nonlinear regression analysis using SAS software (SAS Institute, Cary, NC).
Single-cycle assays for evaluating early- versus late-stage inhibition of HIV-1 replication.
Three constructs for the generation of vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped HIV-1 were made as follows. HIV-1 helper virus was amplified from SODk1CG2 cells (21) and was cloned into pcDNA 3.1. The helper plasmid, which lacks both LTRs as well as functional gp120 and Nef, has all HIV coding sequences expressed from the immediate-early cytomegalovirus (CMV) promoter. Gag-Pol, Vif, and Vpr were derived from NL4-3, while all other sequences were derived from HXB2. The pTV-linker self-inactivating transfer vector (23) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from Lung-Ji Chang. pTV-linker was further modified by inserting the EF-1α promoter and firefly luciferase gene, creating pTV-Luc. The VSV-G expression plasmid was obtained from Ivan Lessard (Boehringer Ingelheim [Canada] Ltd.).
The activity of the CA compounds in the early phase of the replication cycle was evaluated by transducing SupT1 cells with VSV-G-pseudotyped HIV-1 in the presence of test compounds. VSV-G-pseudotyped HIV-1 was prepared by batch transfection of 293FT cells in a T-75 flask with 2.2 μg of VSV-G plasmid, 6.7 μg of HIV-1 helper plasmid, and 9 μg of pTV-luc by using FuGene (Roche Applied Science). The 293FT supernatant was harvested 48 h posttransfection and was centrifuged for 5 min at 2,000 rpm. p24 levels were quantified using an HIV-1 p24 enzyme-linked immunosorbent assay (ELISA) kit (Beckman Coulter). Viral supernatants were stored at −80°C. In a 96-well microtiter plate, 40,000 SupT1 cells (25 μl) were infected with an amount of virus corresponding to 4 ng of p24 (25 μl) in the presence of 50 μl of the test compound (in 1% DMSO). Forty-eight hours postinfection, 25 μl Steady Glo (Promega) was added to each well, and luciferase activity was measured using a TopCount plate reader (Perkin-Elmer).
Inhibitory activity during the late phase of the replication cycle was evaluated by adding test compounds to 293FT cells during the production of VSV-G-pseudotyped HIV-1. Cells were batch transfected (as described above) in the absence of compounds by using FuGene (Roche Applied Science) for 4 to 6 h in a T-75 (75-cm2) flask, after which cells were dislodged by pipetting, and ∼40,000 cells in 50 μl were transferred to a 96-well microtiter plate (Corning Costar) containing an equivalent volume of test compound from 10-point dose-response curves (in 1% DMSO). Forty-eight hours posttransfection, 10 μl viral supernatant was diluted 1:10 in RPMI medium supplemented with 10% FBS in a second 96-well microtiter plate and was then frozen at −80°C for at least 1 h. Following thawing, 10 μl of diluted viral supernatant was used to infect 40,000 SupT1 cells in a final volume of 100 μl. Thus, compounds were diluted 100-fold in order to minimize any potential inhibitory activity in the early phase of the replication cycle. Forty-eight hours postinfection, firefly luciferase activity was measured as described above. The cytotoxicity of the test compounds was evaluated by adding 50 μl of CellTiter Glo (Promega) to the microplates containing the transfected 293FT cells. Luminescence was measured using a TopCount plate reader.
ITC.
Isothermal titration calorimetry (ITC) was performed at 25°C in 50 mM Tris (pH 8.0), 350 mM NaCl, and 1% DMSO by using a VP-ITC microcalorimeter (MicroCal Inc.; GE Health Sciences). Titrations were performed using 200 μM CANTD in the syringe and approximately 20 μM compound in the sample cell. After degassing, the compound solution was centrifuged at 15,000 × g for 15 min and was loaded in the sample cell. The precise compound concentration in the sample cell was assessed by high-performance liquid chromatography (HPLC) using a reference DMSO solution of the compound. Each titration consisted of 19 injections of 15 μl at 280-s intervals. Thermodynamic parameters were derived by fitting the binding isotherms to the single-site binding model algorithm, with stoichiometries (n), enthalpies (ΔH), and equilibrium dissociation constants (KD) allowed to float during nonlinear least-squares fits of the data. Typically, stoichiometries (n) were between 0.9 and 1.1. The starting concentrations of more-potent compounds were reduced to 5 μM in the sample cell for better assessment of the KD.
Selection of HIV-1 variants resistant to capsid inhibitors.
C8166 cells were infected at an MOI of 0.1 with WT HIV-1 2.12 (7) in complete RPMI medium (RPMI 1640, 10% FBS, 10 μg/ml gentamicin, and 10 μM β-mercaptoethanol) with twice to 5 times the EC50 of the capsid inhibitor. At each passage (3 to 4 days), microscopic evaluation of the cytopathic effect (CPE) was performed, and the culture supernatant was used to infect fresh C8166 cells, which were then maintained at the same or a higher concentration of inhibitor depending on the CPE. At passages where viral breakthrough was evident, the genomic DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen). The CA gene was amplified by PCR, and the fragments were cloned into the Zero Blunt TOPO plasmid (Invitrogen) and sequenced by automated sequencing.
Construction of recombinant HIV-1.
To create viruses carrying CA mutations, site-directed mutagenesis was carried out using the QuikChange site-directed mutagenesis kit (Stratagene). All mutations were confirmed by sequencing on an ABI Prism 3100 genetic analyzer (Applied Biosystems Inc.). Sequenced DNA fragments containing the confirmed mutation(s) were subcloned into the NL4-3 provirus by standard molecular biology techniques. Viral stocks were produced by transfecting 293 cells by the calcium phosphate method (20). The culture supernatant was collected after 3 days (37°C, 5% CO2); cell debris was removed; and aliquots were frozen at −80°C. The 50% cell culture infective dose (CCID50) was determined by monitoring the formation of syncytia in C8166 cells.
Jurkat cell replication capacity assay.
The replication capacity assay was carried out with Jurkat-LTR luciferase cells as described previously (7). Briefly, Jurkat cells expressing an HIV LTR luciferase construct were infected with virus at an MOI of 0.05 for 2 h at 37°C, washed, and seeded at 1 × 105 cells/well in 200 μl RPMI 1640 supplemented with 10% FBS and 10 μg/ml gentamicin in clear-bottom black 96-well microtiter plates. Every 3 to 4 days, the cells were mixed, and 100 μl medium was removed and replaced with fresh medium. Luciferase levels were determined by adding 50 μl/well BrightGlo (Promega) at days 7, 10, 12, and 14 postinfection and reading on the LUMIstar galaxy plate reader (BMG). Replication was not assessed at later times owing to the potential for variability over longer periods.
Assays for inhibition of virion release, infectivity, and assembly.
293T cells were transfected with the proviral HIV-1NL4-3 R9 expression construct in the presence of benzodiazepine (BD) and benzimidazole (BM) inhibitors (50-fold over the EC50s). Viral Gag expression, processing, virion release, and viral titers were assayed as described in detail elsewhere (40). Released virions were fixed with gluteraldehyde and osmium tetroxide, stained with uranyl acetate, embedded in epoxy resin, thin sectioned (thickness, 60 to 90 nm), poststained with Reynold's lead citrate, and imaged on a Hitachi H-7100 transmission electron microscope at a magnification of ×50,000, as described in detail elsewhere (39).
X-ray crystallography models.
A full description of the crystallization and diffraction data will be reported elsewhere (manuscript in preparation). For the superposition shown in Fig. 3, monomer A of the CANTD-BD 3 structure and monomer B of the CANTD-BM 4 structure were superposed on the unliganded CANTD (residues 1 to 146) of the stabilized hexameric CA structure (PDB identification code [ID] 3H47) (29). Monomer A of the CANTD-BD 3 structure was selected because it is the only one of the two monomers of the asymmetric unit that bound the BD 3 inhibitor (the other monomer is apo). Monomer B of the CANTD-BM 4 structure was selected because the bound inhibitor appears to be less affected by neighboring molecules of the crystal lattice. The alignments were based on 300 atoms constituting the backbone of 75 CANTD residues: Ala47 to Asn57, Ala64 to Leu83, Arg97 to Met118, and Ile124 to Tyr145. This selection focuses on the most immutable portions of CANTD, avoiding helices 1 and 2 and other highly flexible regions. The calculated root mean square distances (RMSDs) for the fitted CANTD-BD 3 and CANTD-BM 4 structures to the unliganded CANTD were both 0.44 Å.
Fig 3.

Structural comparisons illustrating the differing effects of BD and BM inhibitor binding on the CANTD conformation. (A) Overlay of the BD 3 (orange) and BM 4 (blue) complex structures. van der Waals surfaces illustrate how BM 4 protrudes further into the solvent, whereas BD 3 binds more deeply and induces the formation of a larger pocket, primarily by shifting CA-NC helices 1 and 2 and the Trp23 side chain. The “protruding region,” or the substituents on the inhibitors that extend outside the pocket, is indicated (see the text). (B) Overlay of the apo (white)-, BD 3 (orange)-, and BM 4 (blue)-bound CANTD structures detailing shifts of backbones and key residues. The bound inhibitors have been removed for clarity. Residues 1 to 146 of the stabilized hexameric CA structure (PDB ID 3H47) (29) were used as the reference apo structure. Residues of helices 1 and 2 and highly flexible portions of CANTD were omitted from the superposition calculations, as described in Materials and Methods.
For the liganded hexameric model shown in Fig. 9, monomer B of the CANTD-BM 4 structure was superposed on the unliganded CA monomer of the hexameric CA structure (PDB ID 3H47) (29). The superposition was based on 440 atoms constituting the backbone of 110 CA residues: residues 1 to 3, 10 to 24, 33 to 57, 64 to 82, and 97 to 144. This more comprehensive selection of residues includes the major secondary structural elements while avoiding highly flexible regions. A hybrid CA molecule was then made by fusing residues 1 to 140 of the CANTD-BM 4 structure with residues 141 to 219 of 3H47. The hexamer of this hybrid molecule was then generated by applying the appropriate crystallographic symmetry operations based on the hexagonal (P6) space group of 3H47.
Fig 9.

Models showing that BM inhibitors are expected to interfere with the formation of CANTD/CACTD interfaces in the CA hexamer. (A) Global view of BM 4 (sticks) bound to the CA hexamer (surface model, with different CA subunits in different colors, and N-terminal domains shown in darker shades). (B) Close-up view of a single CANTD/CACTD interface (boxed in panel A) showing how a BM 4 inhibitor bound to the CANTD (yellow) would be positioned to perturb the interface with an adjacent CACTD (red). The model was generated by superimposing the structure of the CANTD BM 4 complex onto the crystal structure of the HIV-1 CA hexamer (PDB ID 3H47) as described in Materials and Methods.
Protein structure accession numbers.
The PDB entries for CANTD-BD 3 and CANTD-BM 4 are 4E91 and 4E92, respectively.
RESULTS
Efficient HTS assay for inhibitors of HIV-1 CA assembly.
In principle, inhibition of CA tube assembly can be used to screen for small molecules that block capsid assembly, but this method generally requires high protein concentrations and must be performed under high-ionic-strength conditions (25, 26). We therefore developed an in vitro CA assembly assay that was more amenable to high-throughput screening (HTS).
Our assay employed a Gag fragment that spanned the CA and NC regions and took advantage of the ability of the nucleocapsid (NC) to bind tightly to TG-rich deoxyoligonucleotides (9). The use of d(TG)25 as a “scaffold” enabled CA tubes to form at much lower protein and salt concentrations than in previously reported assays. Assembly reactions were performed in neutravidin-coated 384-well plates using both biotin- and fluorescein-labeled d(TG25). The biotin-labeled d(TG25) oligonucleotide bound on the surface acts to nucleate the assembly of complexes, and further “polymerization” of CA-NC is driven by soluble fluorescein-labeled d(TG25). Unbound and unassembled species are washed away from captured assembly products at the end of the assembly reaction.
As shown in Fig. 1A, titration of increasing quantities of wild-type (WT) CA-NC in this assay led to a dose-dependent increase in the fluorescence signal up to a level of saturation that corresponded to complete loading of the oligonucleotide with CA-NC protein. In contrast, two control CA-NC proteins with amino acid substitutions previously shown to block capsid assembly in vitro and in vivo (A42D and W184A/M185A) (17, 40) were inactive in this assay.
Fig 1.

Validation of the capsid assembly assay. (A) Assay showing that wild-type HIV-1 CA-NC assembles in a concentration-dependent fashion, whereas the assembly-defective CA-NC mutants (the A42D and W184A M185A mutants) do not. CA-NC assembly was detected by capture and immobilization of a fluorescent oligodeoxynucleotide. (B) CAI, a known peptide inhibitor of HIV-1 capsid assembly (33), inhibits CA-NC assembly in a concentration-dependent fashion, whereas a scrambled peptide control (ctrl) does not. The assay was performed with wild-type CA-NC under standard assay conditions, and activities are reported as the percentage of inhibition of the control fluorescence signal.
The sensitivity of the capsid assembly assay to known inhibitors was confirmed using the CAI peptide as a positive control (33). As shown in Fig. 1B, increasing concentrations of the CAI peptide abrogated the assay signal, with an IC50 of 0.6 μM (Fig. 1B). In contrast, a control peptide with the same amino acid composition as CAI, but with a scrambled sequence, failed to inhibit CA-NC assembly, even at concentrations as high as 25 μM (Fig. 1B). Taken together, these results validate the assay and indicate that it recapitulates the formation and inhibition of capsid-like assemblies in vitro.
Identification of two classes of HIV-1 CA assembly inhibitors.
The CA assembly assay was used to screen the Boehringer Ingelheim corporate compound collection, producing >50 active hit clusters. Hits were triaged based on several criteria, including relative potency in the capsid assembly assay, lack of activity in an analogous assay based on assembly of a Gag CA-NC protein from Rous sarcoma virus (RSV Gag ΔMBD ΔPR protein), cytotoxicity, and assessment of chemical tractability. Compounds that survived this triage showed direct binding to CANTD as analyzed by NMR chemical shift perturbations and cocrystallization (see below) (data not shown). Based on these hit-to-lead studies, two structurally distinct families of compounds were chosen for lead optimization (Table 1): the benzodiazepines (BDs) and the benzimidazoles (BMs). Significant medicinal chemistry efforts (8; also manuscripts in preparation) led to the synthesis of the most potent inhibitors from the BD and BM series, BD 1 (half-maximal antiviral effective concentration [EC50], 70 ± 30 nM [n = 21]; half-maximal cytotoxic concentration [CC50], >28 μM) and BM 1 (EC50, 62 ± 23 nM [n = 53]; CC50, ≥20 μM), both of which displayed potent antiviral activity and a large window relative to cytotoxicity (CC50/EC50, >300-fold). These inhibitors, along with related compounds within the same two families (Table 1), were characterized further to establish their mechanisms of action.
Table 1.
Compounds used in this study and their antiviral activities
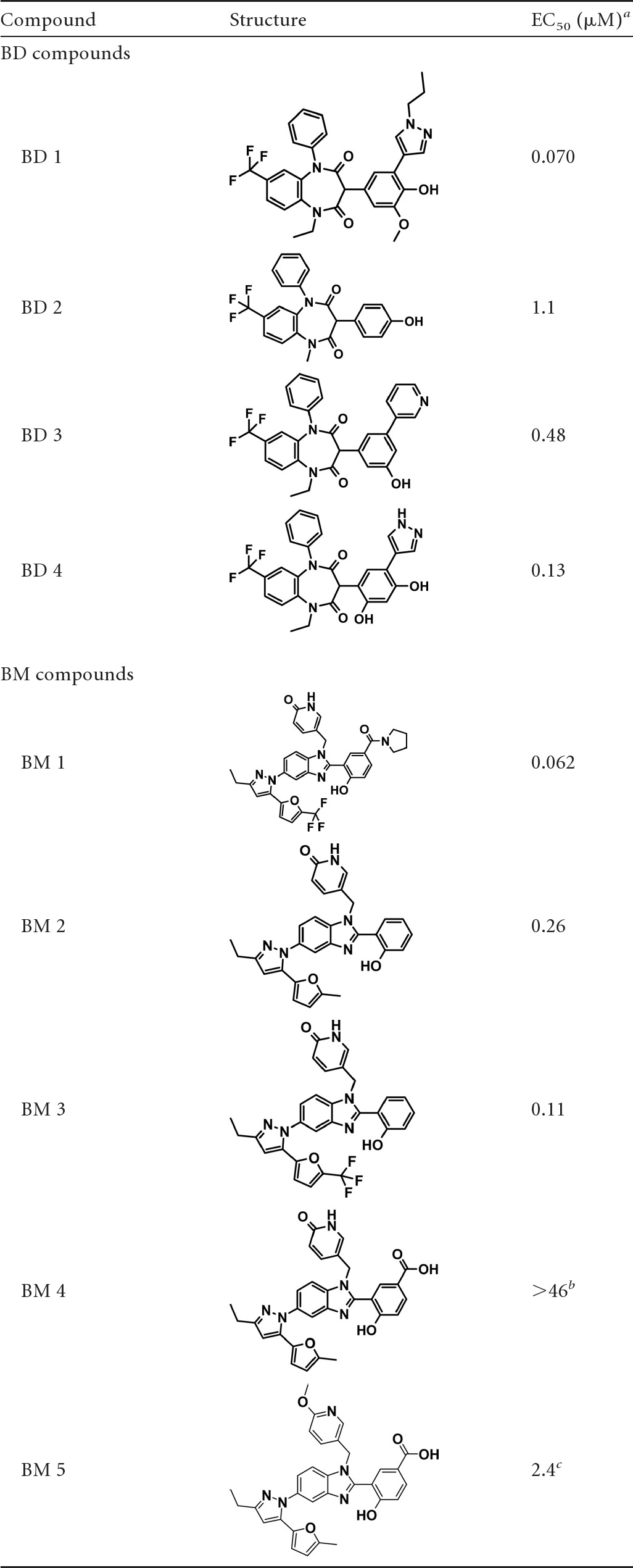
Activity determined in a standard antiviral replication assay (see Materials and Methods).
BM 4 was inactive in the viral replication assay but active in the capsid assembly assay.
BM 5 was active in the capsid assembly assay.
Mechanistic studies of the BD and BM inhibitors.
To ensure that the BD and BM compounds did not also inhibit the same targets as currently available antiretroviral agents, representative compounds from both the BD and BM series were tested for activity against HIV-1 NL4-3 strains that were resistant to the four major classes of antiretroviral inhibitors: nonnucleoside reverse transcriptase inhibitors (NNRTI) (RT mutations Y188L and V106A), nucleoside reverse transcriptase inhibitors (NRTI) (RT mutations K65R and M184V), protease (PR) inhibitors (PI) (PR mutations V32I I47V and L33F I54L), and integrase strand transfer inhibitors (INSTI) (IN mutation G140S Q148H). As shown in Table 2, BD 1, BM 2, and BM 3 inhibited the replication of the wild-type and all mutant viruses with equal potencies. In contrast, control experiments showed the expected pattern of reduced susceptibility to well-characterized antiretrovirals. These data indicate that the mechanism of action of the BD and BM compounds differs from that of the four major classes of antiretroviral agents.
Table 2.
Relative activities of capsid assembly inhibitors against drug-resistant viruses
| Inhibitor | EC50 (nM) for the WT | Fold change from the EC50 for the isogenic WT virusa with the following mutation (resistance class)b: |
||||||
|---|---|---|---|---|---|---|---|---|
| Y188L (NNRTI) | V106A (NNRTI) | K65R (NRTI) | M184V (NRTI) | V32I I47V (PI) | L33F I54L (PI) | G140S Q148H (INSTI) | ||
| BD 1 | 70 | 1.1 | 0.9 | 0.8 | 0.9 | 1.5 | 0.9 | 1.2 |
| BM 2 | 284 | 1.0 | 0.7 | 1.0 | 0.6 | 1.1 | 1.3 | 1.2 |
| BM 3 | 112 | 1.2 | 0.9 | 0.7 | 1.0 | 0.7 | 0.6 | 0.8 |
| Nevirapine | 18 | >83 | 130b | 0.3 | 0.5 | 1.4 | 1.4 | 1.3 |
| Lamivudine | 89 | 1.3 | 0.9 | 26.4 | >96 | 0.9 | 1.2 | 1.2 |
| Amprenavir | 35 | 0.6 | 0.8 | 1.2 | 0.5 | 6.9 | 8.3 | 1.3 |
| Raltegravir | 1.5 | 2.8b | 0.4 | 1.0 | 0.6 | 0.7 | 1.8 | 227 |
All values are averages for two independent experiments except for the values for V106A with nevirapine and Y188L with raltegravir, for which experiments were performed once. Significant fold change values are in boldface.
Resistance targets were RT for Y188L, V106A, K65R, and M184V; PR for V32I I47V and L33F I54L; and IN for G140S Q148H.
To determine the stage of the viral replication cycle targeted by the BM and BD compounds, we tested their effects on the efficiency of HIV-1 vector transduction when the compounds were applied either during virus production (late phase) or during infection (early phase). A vesicular stomatitis virus G glycoprotein (VSV-G)-pseudotyped HIV-1 vector containing a luciferase reporter under the control of the HIV-1 LTR was used in these studies, allowing discrimination between the inhibition of early replication events (i.e., virus entry through transcription) and late events (i.e., viral transcription through virus maturation). Control compounds behaved as expected in our assays: nevirapine, which inhibits the early-stage process of reverse transcription, reduced the reporter activity only when added to target cells. In contrast, lopinavir, which inhibits the late-stage processes of Gag proteolysis and viral maturation, inhibited vector transduction only when added to producer cells (Table 3). By these criteria, the BD and BM compounds both acted primarily at the late stage of the viral replication cycle, since both BD 1 and BM 2 inhibited replication when added during virus production. Furthermore, the EC50s obtained in the late-stage assay for BD 1 and BM 2 were similar to those obtained in standard, multiple-round HIV-1 replication assays (Table 3). In contrast, EC50s for these compounds were at least 10-fold higher when they were applied during the infection of target cells (early phase). Both compounds did exhibit weak inhibitory activity in the early phase (EC50, ∼3 μM), which could be due to effects on viral core stability and disassembly or, alternatively, to cytotoxicity. Regardless, the data establish that both the BD and BM compounds act primarily by inhibiting the late stage(s) of viral replication.
Table 3.
Inhibition of postentry (early) versus postintegration (late) stages of virus replication
| Compound | Value for single-cycle assaysa |
EC50 (μM) for multiple-cycle assays | ||
|---|---|---|---|---|
| Early EC50 (μM) | Late EC50 (μM) | Cytotoxicityb (CC50 [μM]) | ||
| BD 1 | 2.8 | 0.2 | >7.0 | 0.06 |
| BM 2 | 3.2 | 0.2 | 6.2 | 0.26 |
| Lopinavir | >0.5 | 0.01 | >0.5 | 0.009 |
| Nevirapine | 0.08 | >2.2 | >2.2 | 0.02 |
Boldface values denote the expected result for late- and early-stage activities of Lopinavir and Nevirapine, respectively.
Cytotoxicity was determined on virus producer cells (late phase).
BD and BM inhibitors bind the N-terminal domain of the HIV-1 CA protein.
Isothermal titration calorimetry (ITC) and 2-dimensional (2D)-NMR chemical shift perturbation analyses were performed to test whether the BD and BM compounds bound purified recombinant HIV-1 CA proteins and to map their binding sites. The NMR chemical shift perturbation analyses demonstrated that BD and BM inhibitors bound CANTD, with shifted residues clustered at the bases of helices 1, 2, 4, and 7 and at the flexible loops between helices 1 and 2 and helices 3 and 4 (data not shown). ITC binding isotherms showed that inhibitors from both families of compounds bound CANTD with 1:1 stoichiometries and submicromolar dissociation constants. For example, two BM compounds, BM 2 and BM 3, and the weak-binding BD compound BD 2 (see Table 1) bound CANTD with dissociation constants of 210 nM, 87 nM, and 690 nM, respectively (Table 4). These dissociation constants correlated well with the antiviral EC50s for these compounds (260, 110, and 1,100 nM, respectively [Table 1]). Binding of the BM compounds was enthalpically driven (ΔH, −18.3 and −18.7 kcal/mol, respectively), which is a favorable property for viral inhibitors (12). Unfortunately, ITC analyses could not be performed with more-potent compounds in the BD series owing to their limited solubility. Nevertheless, our data establish that both compound classes bind directly to the CANTD, independently of the CA C-terminal domain, CA dimerization, or higher-order CA assembly.
Table 4.
Energetics of inhibitor binding to CANTDa
| Compound | KD (μM) | ΔG (kcal/mol) | ΔH (kcal/mol) | −TΔS (kcal/mol) |
|---|---|---|---|---|
| BM 2 | 0.210 | −9.12 | −18.3 | 9.21 |
| BM 3 | 0.087 | −9.64 | −18.7 | 9.06 |
| BD 2 | 0.690 | −8.40 | −7.50 | −0.90 |
Inhibitor binding was quantified by isothermal titration calorimetry (see Materials and Methods).
Crystal structures of BD and BM inhibitors bound to the HIV-1 CANTD.
To explore the inhibitor binding modes in greater detail, representative compounds from each inhibitor family, BD 3 and BM 4 (see Table 1), were cocrystallized in complex with CANTD. The structures of both complexes were determined at high resolutions and were refined fully (for CANTD-BD 3, 1.7 Å resolution, R = 22.0%, and Rfree = 24.3%; for CANTD-BM 4, 1.8 Å resolution, R = 22.0%, and Rfree = 26.4%). Complete details of the crystallography will be reported elsewhere.
As shown in Fig. 2, both inhibitor types bind to a pocket located at the base of the central four-helix bundle of the CANTD, which is the same site described previously for CAP-1 (24). As was the case for the CAP inhibitors, binding of the BD or BM compounds displaces the side chain of Phe32 from the helical bundle to create a new pocket within the hydrophobic core. Although their binding sites overlap, there are interesting differences in the ways in which the two compounds interact with the protein and affect its structure (Fig. 2 and 3). Specifically, the BD compounds bind more deeply and enlarge the pocket to a greater extent, resulting in significant disruptions of the CANTD tertiary structure (Fig. 3A). In particular, the trifluoromethyl group of BD 3 binds very deeply, displacing the CA Trp23 side chain at the top of the pocket by more than 3 Å, which in turn contributes to a >1.5-Å displacement of the CA α1 helix. The BM compound BM 4 also perturbs the CA tertiary structure, but in this case the native CA interactions at the top of the pocket are much less disrupted (Fig. 3B).
Fig 2.

X-ray crystal structures of CANTD with the BD 3 (A) and BM 4 (B) inhibitors bound within an induced pocket at the base of the HIV-1 CANTD four-helix bundle. Note that BD 3 binds deeply within the pocket and forms two direct H-bonds (green) with the backbone nitrogen atoms of Phe32 and His62, as well as water-mediated hydrogen bonds with the backbone carbonyl oxygen atoms of Val24 and Val59. BM 4 binds less deeply and forms direct hydrogen bonds with the backbone nitrogen atom of Phe32 and with Nδ of His62. The inhibitors are shown in stick models with carbons in white, nitrogens in blue, oxygens in red, and fluorines in green. CA backbones and key side chains are shown in orange (BD 3 complex) and blue (BM 4 complex).
Additionally, both compounds reposition the flexible loop linking CA helices 3 and 4 (Fig. 3B), but in considerably different ways. BD 3 hydrogen bonds with the backbone NH of His62, thus positioning the imidazole side chain away from the pocket (Fig. 2A), while BM 4 interacts with the His62 imidazole, positioning it nearer to the pocket (Fig. 2B). Both inhibitor-bound loop conformations differ substantially from that of the apo-CA hexamer structure (29) and would be expected to perturb or inhibit the formation of the CANTD/CACTD interface.
Finally, the two classes of compounds protrude from the CANTD with significantly different trajectories, and the BM compounds extend much further outside the pocket than do the BD compounds (Fig. 3A). Structure-activity relationships within the protruding groups of the BM compounds revealed their importance for potency, suggesting that this region of the inhibitor may make additional interactions within the assembled capsid (or Gag) lattices. In contrast, the activities of the BD inhibitors were not significantly altered by chemical modifications beyond the directly attached phenyl group, implying that this part of the inhibitor does not make other significant contacts. Thus, although both classes of inhibitors bind the same pocket on CA, differences in the details of their binding modes suggest the possibility that they may have distinct effects on capsid assembly.
Differential effects of the BD and BM inhibitors on virus assembly and maturation.
The effects of the BD and BM inhibitors on Gag production, processing, and virus release were analyzed by Western blotting of cell- and virion-associated Gag proteins (Fig. 4). Virus was produced in the presence of high concentrations (50-fold over the EC50) of two different inhibitors from each family. As expected, in all cases, viral titers were dramatically reduced from that with the control DMSO treatment (Fig. 4A). Neither class of inhibitor had a significant effect on Gag expression or processing, as analyzed by Western blotting of intracellular and cell-associated viral proteins with antibodies that detected the MA and CA proteins (and their Gag precursors) (Fig. 4B). Virus release was analyzed by Western blotting to detect virion-associated CA proteins released into the culture supernatant. In this case, the two classes of inhibitors exhibited quite different effects. The BM compounds reduced virion release only modestly from that with the DMSO control, whereas the BD inhibitors reduced virion release to nearly undetectable levels. We therefore conclude that the BD inhibitors function primarily by blocking Gag assembly and virion release, whereas the BM inhibitors have a different mode of action.
Fig 4.

BM and BD compounds inhibit different stages of HIV-1 assembly and maturation. (A) HIV-1NL4-3 titers produced from cells treated with the designated inhibitors (concentration, 50-fold over the EC50) or with a DMSO control. (B) Western blots showing levels of virion-associated CA proteins released into the supernatant (top) or levels of Gag, CA, and MA proteins being produced in cells (center). Cellular tubulin levels are shown as a control for toxicity and loading levels (bottom). Note that virus release was nearly normal in cells treated with the two BM inhibitors but was severely impaired in cells treated with the two BD inhibitors. The experiments were performed twice, and similar results were obtained in both cases, although the absolute viral titers differed in the two experiments.
Electron microscopic (EM) analyses were performed to examine the morphologies of viral particles released in the presence of the two different classes of inhibitors. As expected, control virus produced in the presence of DMSO (Fig. 5A) yielded virions with a mixture of different core morphologies, but a high proportion had discernible conical cores (44% ± 9%, based on 1,005 virions scored in multiple fields). Consistent with the Western blot analysis, very few virions were produced in the presence of BD inhibitors, and most of the visible structures corresponded to amorphous cellular vesicles (Fig. 5B). The few virions that were present had a variety of gross morphological defects, including partially formed immature Gag shells and acentric electron-dense “cores” (see enlargements of individual virions below Fig. 5B for three examples). In contrast, virions produced in the presence of the BM series were much more prevalent and homogeneous (Fig. 5C) but only rarely had conical cores (2% ± 0.2% of 543 virions scored in multiple fields). Instead, they typically contained acentric, electron-dense complexes that lacked discernible capsids. These data indicate that the BM inhibitors act primarily by inhibiting the assembly of mature conical capsids.
Fig 5.

EM analyses showing the different viral assembly and maturation defects induced by BM and BD inhibitors. Images show transmission electron micrographs of thin-sectioned concentrated virions released from cells treated with DMSO (control) (A), BD 1 (B), or BM 3 (C). Enlargements of 3 individual virions are shown below each representative field. Inhibitor treatments were performed as for the experiments shown in Fig. 4. Similar results were also obtained with BD 4 (rare, highly aberrant virions) and BM 1 (acentric, nonconical cores). The experiments were performed twice, and similar results were obtained in both cases. Bars, 100 nm.
Selection of inhibitor resistance mutations.
To confirm that CA was the target of the inhibitors and to learn more about how the virus could adapt to inhibition, we selected for viruses that were resistant to BD or BM compounds. To select for resistance, HIV-1NL4-3 was cultured in C8166 cells in the presence of increasing concentrations of two representative compounds from each series for as many as 11 passages (see Table S1 in the supplemental material). The development of viral resistance resulted in increased virus replication, which led to cytopathicity. At later passages (between passages 7 and 11), whenever cytopathicity became evident, proviral DNA was cloned; the capsid gene was sequenced to identify candidate resistance mutations; and the inhibitor concentration was raised in subsequent passages to elicit additional resistance mutations.
In most cases, multiple different CA mutations were present when high-level resistance developed. Perhaps not surprisingly, many of the selected amino acid substitutions mapped to the inhibitor binding site (Fig. 6). Interestingly, however, distinct patterns of amino acid substitutions were selected with potent versus weaker inhibitors and with BD versus BM inhibitors. Specifically, the V36T and G61E substitutions were obtained only upon selection with BD inhibitors (Fig. 6A), whereas the K30R and S33G substitutions were obtained only upon selection with BM inhibitors (Fig. 6B). The G61E substitution was selected only by a weaker inhibitor from the BD series, BD 2, and was never obtained with the more potent compound BD 1. T58I was selected by both inhibitors from the BM series (BM 2 and BM 3) and was also observed as a minor substitution selected by BD 1. Finally, Val27 alterations to Ala or Ile were observed as minor substitutions for the BD series only. Table S1 in the supplemental material summarizes all major and minor CA amino acid substitutions obtained during selection. All major substitutions, as well as several of the minor substitutions, were chosen for further characterization, as described below.
Fig 6.

Identities and structural locations of viral resistance mutations selected by treatment with BD (A) and BM (B) inhibitors. Two compounds from each chemical series were used for the selection of resistant virus: BD 1 and BD 2 from the BD series and BM 2 and BM 3 from the BM series (see Table 1 for compound structures and activities). Resistance substitutions (highlighted in red; side chains shown in stick form) selected by these compounds are mapped onto the CANTD Cα trace (gray), with bound BD and BM inhibitors shown in orange (BD 3) and blue (BM 4).
Interestingly, inhibitors from both families also selected for amino acid substitutions in the CACTD, including G208R (major for BD 1 and BM 3; minor for BM 2) and E213G (a minor substitution for BM 2 only). At later passages and higher inhibitor concentrations, these CACTD substitutions were often found together with binding pocket substitutions, and the following double mutants were observed in different clones: the K30R/G208R, S33G/G208R, T58I/G208R, or T58I/E213G (BM) and V36T/G208R (BD) mutants.
Characterization of key inhibitor resistance mutations.
To quantify the degree of inhibitor resistance induced by different amino acid substitutions, all major single and double substitutions were introduced separately into the NL4-3 proviral clone, and the mutant viruses were tested for their sensitivities to capsid assembly inhibitors BD 1, BM 2, and BM 3. As shown in Table 5, all of the single substitutions that were selected by BD inhibitors resulted in significant BD drug resistance, ranging from 3-fold (G208R) to 25-fold (V36T). Similarly, single substitution mutations that were selected by BM inhibitors resulted in significant BM inhibitor resistance, ranging from 4-fold (K30R versus BM 2) to 22-fold (T58I versus BM 3). In most cases, resistance mutations selected against one inhibitor family also conferred resistance to the other, presumably reflecting their common binding sites. The demonstration that resistance is engendered by clonal mutations at a series of different CA residues provides formal proof that CA is the functional target of both the BD and the BM inhibitors.
Table 5.
Susceptibilities of viruses with resistance mutations to inhibitors of capsid assembly
| Compound | Fold change from the EC50 of the wild-type virus with the following mutationa: |
|||||||
|---|---|---|---|---|---|---|---|---|
| V27I (BD) | V36T (BD) | K30R (BM) | T58Ia (BM and BD) | G208R (BM and BD) | G208A | K30R G208R (BM) | V36T G208R (BD) | |
| BD 1 | 5 | 25 | 1 | 6 | 3 | 1 | 4 | >36 |
| BM 2 | 1 | 8 | 4 | 16 | 7 | 1 | 16 | 55 |
| BM 3 | 1 | 7 | 5 | 22 | 5 | 1 | 5 | 35 |
The class(es) of compound with which each mutation was selected is given in parentheses. T58I was a major substitution selected with BM compounds but a minor substitution selected with BD compounds. G208A is a natural polymorphism.
As has been seen in other drug resistance profiles, multiple mutations often synergized to produce high-level drug resistance. For example, although the most prevalent CACTD resistance substitution, G208R, conferred only modest (3- to 7-fold) resistance to either inhibitor family, this substitution synergized with V36T to produce ≥35-fold resistance to both inhibitor series. Interestingly, CA residue 208 is also highly variable among different virus isolates. Although Arg is found at this position in only 3 of 2,240 reported HIV-1 sequences, Ala substitutions are quite common, particularly in subtype B (44% of all sequences and 90% of subtype B sequences in the Los Alamos National Laboratory HIV Database [http://www.hiv.lanl.gov]). We found, however, that the G208A substitution in HIV-1NL4-3, a subtype B-derived virus, did not confer resistance to any of the capsid assembly inhibitors, implying that viral strains with Ala at position 208 will not exhibit intrinsic resistance to BD or BM inhibitors (Table 5).
Most BD and BM resistance mutations reduce viral replicative capacity.
Drug resistance mutations often reduce viral fitness (31). We therefore tested whether this was also the case for the BD and BM resistance substitutions. Viral fitness was assessed in a Jurkat-LTR-luciferase reporter T cell line that provided a more stringent test of replicative capacity than the highly permissive cell lines used for the selection of resistant virus. As shown in Fig. 7, BD and BM resistance substitutions invariably reduced viral replication capacity from that of the wild-type virus, showing impairments that were modest (40 to 80% of wild-type replication for V27I), moderate (5 to 40% for V36T, K30R, T58I, K30R/T58I, and E213G), or profound (less than 5% for G208R, V36T/G208R, and K30R/G208R). The S33G resistance substitution could not be tested in this assay because infectious virus was not produced from the mutated proviral clone, implying that this substitution severely impairs viral replicative capacity. Thus, the development of resistance to either family of capsid inhibitors can be expected to reduce viral fitness.
Fig 7.

Inhibitor resistance substitutions reduce viral fitness. Shown are levels of luciferase produced by Jurkat LTR-luciferase reporter cells 14 days after infection with wild-type HIV-1 or with viruses carrying the designated mutations within the CA protein that confer resistance to BD inhibitors or BM inhibitors. The major G208A polymorphism was also tested. Luciferase activities are expressed as relative replicative capacities, with that for the wild-type virus set at 100%. Replicative capacities were measured at days 7, 12, and 14. The results for each mutant were similar on days 12 and 14. Replicative capacity was not assessed at additional times owing to the potential for variability over longer periods. Each value represents the average for 6 replicates from a typical experiment.
Effects of resistance mutations on inhibitor binding.
To test whether inhibitor resistance mutations reduced inhibitor binding, recombinant CANTD proteins carrying representative CA inhibitor resistance substitutions were purified and used in ITC experiments to quantify the binding of the BM 2, BM 5, BM 6, and BM 7 inhibitors. As shown in Table 6, and in Table S2 in the supplemental material, three of the substitutions; S33G, V27A, and V36T, reduced the inhibitor binding affinity between 6- and 21-fold. For BM 2, the decrease in binding energy was driven by reductions in binding enthalpy, indicating that these substitutions removed favorable interactions (see Table S3 in the supplemental material). Similar reductions in binding affinity were seen for each mutant with all related BM compounds that were tested (see Table S2). Given that all three of these residues are located within the BM inhibitor binding site, it is likely that the mutations confer resistance, at least in part, by directly reducing inhibitor binding. Two other resistance substitutions, K30R and T58I, were of interest because they did not reduce the CANTD binding affinities of the BM compounds (Table 6; see also Table S2). Thus, the 4- to 22-fold levels of resistance conferred by these substitutions (Table 5) were not attributable to reduced inhibitor binding affinity, implying that they act via an indirect mechanism.
Table 6.
Reduction in affinity for BM compounds by resistance substitutions
| Compound | Fold change from the KD of the compound for wild-type CANTDa with the following mutation: |
||||
|---|---|---|---|---|---|
| S33G | V27A | V36T | T58I | K30R | |
| BM 2 | 13 | 6 | 16 | 0.9 | NDb |
| BM 5 | 14 | 10 | 18 | 1.1 | 0.9 |
The KDs of BM 2 and BM 5 for wild-type CANTD were 210 and 170 nM, respectively.
ND, not determined.
Indirect resistance mutations promote CA-NC assembly in vitro.
We hypothesized that resistance mutations that did not directly affect inhibitor binding might act indirectly by increasing the efficiency of capsid assembly and/or capsid stability, thereby counteracting the detrimental destabilizing effects of inhibitor binding. To investigate this possibility, we took advantage of previous reports that amino acid substitutions that increase HIV-1 core stability often reduce the ionic strength requirements for CA-NC assembly in vitro (6, 10). We therefore examined the assembly properties of purified recombinant CA-NC proteins that carried either the aforementioned T58I resistance substitution or the G208R substitution, which is located in the CACTD and is therefore distant from the inhibitor binding site. The wild-type and mutant CA-NC proteins were tested for their abilities to assemble in vitro under suboptimal (150 mM) salt concentrations, and the “direct” S33G resistance substitution was used as a control.
As predicted, the wild type CA-NC protein assembled efficiently in buffers that contained 350 mM NaCl (Fig. 1), but not at 150 mM NaCl (Fig. 8). Similarly, the control CA-NC mutant (the S33G mutant) also failed to assemble detectably under these low-salt conditions. In contrast, low-salt CA-NC assembly was dramatically stimulated by both of the resistance mutations located outside the inhibitor binding pocket (T58I and G208R). Thus, both of these substitutions enhanced CA-NC assembly efficiency and/or stability in vitro, consistent with the idea that they impart inhibitor resistance by “counteracting” the capsid destabilization that accompanies BM inhibitor binding.
Fig 8.

Inhibitor resistance mutations can enhance CA-NC assembly in vitro. The assembly of wild-type and mutant CA-NC proteins was assayed as shown in Fig. 1, except that the assembly conditions were more stringent (150 mM NaCl). CA-NC/oligonucleotide ratios were maintained across the 2-fold dilution series, and “1×” represents the concentrations used in the standard assembly assay.
DISCUSSION
There is a growing need for drugs that act against new HIV-1 targets. Toward this end, we developed a high-throughput in vitro capsid assembly assay and used it to screen a compound library to identify potential inhibitors of capsid assembly. Subsequent optimization of two distinct chemical series produced very potent inhibitors of viral replication. These compounds (i) bind CANTD, (ii) are fully active against viruses that are resistant to other classes of antiretroviral drugs, (iii) select for resistance mutations that map to CA, (iv) act during the late phase of the viral replication cycle, and (v) inhibit virus assembly. We therefore conclude that both compound classes are bona fide capsid assembly inhibitors.
X-ray crystal structures of CANTD in complex with representative compounds from the BD and BM families revealed that these inhibitors bind to the same site, but with distinct binding modes that lead to different effects on virus assembly. BD compounds bind more deeply within the four-helix bundle, and the trifluoromethyl group induces a large displacement of the α1 helix, which normally makes intersubunit contacts within the mature CANTD hexameric and pentameric rings (29, 30). This displacement may, therefore, be incompatible with mature CA ring formation. The α1 displacement may also be incompatible with the formation of Gag hexamers that make up the immature virion lattice. Although a high-resolution structure of the Gag hexamer is lacking, modeling studies have suggested that the Gag and CA hexamers may be similar (3, 42), and recent H/D exchange studies support the idea that intersubunit packing within the mature and immature hexamers is similar, albeit with some reorientation of α1 within the hexamer interface (27). The dramatic effects of the BD compounds on both virion production and capsid morphology (Fig. 5B) are consistent with the idea that these inhibitors affect the formation of both immature Gag and mature CA hexamers.
In contrast, BM inhibitors exert their effects primarily by inhibiting the assembly of the mature viral capsid. These inhibitors insert less deeply into the helix bundle and disrupt the surrounding tertiary structure less extensively, but they protrude significantly further outside the CANTD. The high-resolution structures of CA hexamers and pentamers (29) reveal that this region of the CANTD makes contact with the CACTD of the neighboring unit within the hexamer. Indeed, when a BM compound is modeled into the hexamer structure (Fig. 9), the protruding portion of the inhibitor clashes with the neighboring CACTD domain, particularly with the conserved residues Arg162, Asp163, and Asp166. Moreover, BM inhibitor binding also displaces the CANTD Phe32 and His62 side chains and reorients the loop between helices 3 and 4. All of these changes are predicted to destabilize the CANTD/CACTD interface, and therefore it is likely that this is how the BM compounds prevent or disrupt mature capsid assembly (Fig. 5C). In summary, our data show that the BM and BD capsid assembly inhibitors have distinct binding modes within the four-helix bundle of CANTD that apparently translate into distinct effects on virion morphology.
Consistent with their differing binding modes, the profile of resistance substitutions selected by the BM and BD inhibitors are also different. Mutations conferring resistance to both classes of compound had two different types of effects. Not surprisingly, substitutions within the inhibitor binding site decreased the affinity of inhibitor binding, consistent with the notion that these mutations exert their effects directly by reducing inhibitor binding. Most of these resistance mutations reduced viral fitness, implying that the integrity of the inhibitor binding pocket is important for viral replication.
More interestingly, other resistance substitutions were located slightly outside (T58I) or much more distant (G208R) from the inhibitor binding site and apparently did not affect inhibitor binding affinity. Remarkably, both these substitutions enhanced CA-NC assembly in vitro, implying that these changes conferred resistance by counteracting the destabilizing effects of the binding of the inhibitor to the viral capsid. We speculate that the G208R mutation may stabilize the capsid by making favorable contacts across the local 3-fold axis between adjacent hexamers (4). Although a precise structure of the trimer interface is not yet available, it is believed that the Gly208 residue sits near the Glu231 residue of a neighboring subunit within the trimer. An E231Q mutation stabilizes the viral capsid (4), and it is therefore conceivable that the G208R mutation also exerts a similar stabilizing effect by helping to neutralize the Glu231 side chain. Increases in capsid stability have been reported to inhibit viral replication (10), and both of the indirect resistance substitutions reduced viral replication, with a >95% decrease observed for G208R (Fig. 7).
Viral fitness was reduced by each of the BM and BD resistance mutations tested (Fig. 7), suggesting that there may be a high barrier to the development of resistance against capsid assembly inhibitors binding within this site in vivo. It is also noteworthy that most of the resistance substitutions appear to be rare in natural HIV-1 isolates. For example, S33G, E213G (both selected by BM compounds only), V27A, and G208R (selected by both chemotypes) are each present in fewer than 5 of the 2,240 HIV-1 sequences in the Los Alamos database, and the K30R substitution is present in only approximately 2% of sequences. Two of the resistance substitutions did correspond to known polymorphisms: V27I (selected by BD compounds only) is present in 49%, and T58I (selected primarily by BM compounds) is present in 18%, of the database sequences. However, V27I conferred only low-level BD resistance and did not confer cross-resistance to BM compounds (Table 5). Interestingly, T58I was not observed among subtype B sequences and appears to cosegregate with the I54M polymorphism in subtype C viruses. The significance of this observation is not known, but it may represent a compensatory change necessary for efficient replication because, at least in the NL4-3 background, the T58I mutant replicated poorly (Fig. 7). Finally, although the BD and BM compounds selected different patterns of resistance mutations, those selected against one inhibitor class typically also conferred moderate resistance to the other compound class. This is presumably because the inhibitor binding sites overlap and because inhibitor binding likely destabilizes the viral capsid in both cases.
Compounds that bind to a different site on the outer face between helices 4 and 7 of the CANTD were recently shown to reduce capsid stability and inhibit HIV-1 replication (2, 32). These inhibitors promote the disassembly of purified capsids in vitro and during early stages of virus entry (32). They therefore exhibit postentry defects in single-cycle replication assays, in addition to inhibiting late stages of viral replication (2). Our BD and BM compounds exhibited only modest early-stage effects, although it is conceivable that these effects might be even greater when entry is mediated by wild-type HIV-1 Env (rather than by VSV-G, as in our single-cycle experiments). Nevertheless, the major inhibitory effects occurred during virus assembly, which is consistent with the observation that CAP inhibitors also affect only the late events during replication (37).
In conclusion, we have shown that the HIV-1 CA protein, and in particular the CAP inhibitor binding site within the N-terminal domain, is a tractable inhibitor binding target and that several different classes of inhibitors can bind tightly within this pocket. However, the highly hydrophobic nature of the pocket, as well as the apparent flexibility that allows it to adjust to the binding of different inhibitors, makes it a challenging target for inhibitor optimization. We have disclosed two families of very potent capsid assembly inhibitors. Although chemically tractable, these series were not advanced as drug candidates because of intrinsic limitations in biopharmaceutical properties, such as solubility and metabolic stability. In spite of these challenges, CA is now a validated target for therapeutic intervention and merits additional drug discovery efforts.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pierre Bonneau, Michael Bös, Paul Anderson, Richard Bethell, and Michael Cordingley for leadership and guidance in structural research, chemistry, and biology; Louie Lamorte for input in the writing of the manuscript and the development of the VSV-G-pseudotyped viral system described in this work; Mireille Cartier for sequence alignments and analysis of Los Alamos database sequences of CA; and Volker Vogt for the RSV Gag DMBD ΔPR protein expression construct.
W.I.S. acknowledges financial support for this project from Boehringer Ingelheim and from NIH grant P50 GM082545.
Footnotes
Published ahead of print 11 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Bartonova V, et al. 2008. Residues in the HIV-1 capsid assembly inhibitor binding site are essential for maintaining the assembly-competent quaternary structure of the capsid protein. J. Biol. Chem. 283:32024–32033 [DOI] [PubMed] [Google Scholar]
- 2. Blair WS, et al. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 6:e1001220 doi:10.1371/journal.ppat.1001220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Briggs JA, et al. 2009. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U. S. A. 106:11090–11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byeon IJ, et al. 2009. Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Clercq E. 2010. Antiretroviral drugs. Curr. Opin. Pharmacol. 10:507–515 [DOI] [PubMed] [Google Scholar]
- 6. Douglas CC, Thomas D, Lanman J, Prevelige PE., Jr 2004. Investigation of N-terminal domain charged residues on the assembly and stability of HIV-1 CA. Biochemistry 43:10435–10441 [DOI] [PubMed] [Google Scholar]
- 7. Doyon L, Tremblay S, Bourgon L, Wardrop E, Cordingley MG. 2005. Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir. Antiviral Res. 68:27–35 [DOI] [PubMed] [Google Scholar]
- 8. Fader LD, et al. 2011. Discovery of a 1,5-dihydrobenzo[1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg. Med. Chem. Lett. 21:398–404 [DOI] [PubMed] [Google Scholar]
- 9. Feng YX, Li T, Campbell S, Rein A. 2002. Reversible binding of recombinant human immunodeficiency virus type 1 Gag protein to nucleic acids in virus-like particle assembly in vitro. J. Virol. 76:11757–11762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freed EO. 1998. HIV-1 Gag proteins: diverse functions in the virus life cycle. Virology 251:1–15 [DOI] [PubMed] [Google Scholar]
- 12. Freire E. 2008. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 13:869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gamble TR, et al. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294 [DOI] [PubMed] [Google Scholar]
- 14. Gamble TR, et al. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853 [DOI] [PubMed] [Google Scholar]
- 15. Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83 [DOI] [PubMed] [Google Scholar]
- 16. Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79 [DOI] [PubMed] [Google Scholar]
- 17. Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ganser-Pornillos BK, Yeager M, Sundquist WI. 2008. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18:203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gitti RK, et al. 1996. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273:231–235 [DOI] [PubMed] [Google Scholar]
- 20. Graham FL, van der Eb AJ. 1973. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 52:456–467 [DOI] [PubMed] [Google Scholar]
- 21. Hansen MS, et al. 1999. Integration complexes derived from HIV vectors for rapid assays in vitro. Nat. Biotechnol. 17:578–582 [DOI] [PubMed] [Google Scholar]
- 22. Ivanov D, et al. 2007. Domain-swapped dimerization of the HIV-1 capsid C-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 104:4353–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwakuma T, Cui Y, Chang LJ. 1999. Self-inactivating lentiviral vectors with U3 and U5 modifications. Virology 261:120–132 [DOI] [PubMed] [Google Scholar]
- 24. Kelly BN, et al. 2007. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 373:355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lanman J, Sexton J, Sakalian M, Prevelige PE., Jr 2002. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 76:6900–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li S, Hill CP, Sundquist WI, Finch JT. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409–413 [DOI] [PubMed] [Google Scholar]
- 27. Monroe EB, Kang S, Kyere SK, Li R, Prevelige PE., Jr 2010. Hydrogen/deuterium exchange analysis of HIV-1 capsid assembly and maturation. Structure 18:1483–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moreno S, et al. 2010. The future of antiretroviral therapy: challenges and needs. J. Antimicrob. Chemother. 65:827–835 [DOI] [PubMed] [Google Scholar]
- 29. Pornillos O, et al. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quinones-Mateu ME, Moore-Dudley DM, Jegede O, Weber J, Arts EJ. 2008. Viral drug resistance and fitness. Adv. Pharmacol. 56:257–296 [DOI] [PubMed] [Google Scholar]
- 32. Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sticht J, et al. 2005. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 12:671–677 [DOI] [PubMed] [Google Scholar]
- 34. Sundquist WI, Hill CP. 2007. How to assemble a capsid. Cell 131:17–19 [DOI] [PubMed] [Google Scholar]
- 35. Sundquist WI, Krausslich H-G. (ed). 13 March 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. [Epub ahead of print.] doi:10.1101/cshperspect.a006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taiwo B, Hicks C, Eron J. 2010. Unmet therapeutic needs in the new era of combination antiretroviral therapy for HIV-1. J. Antimicrob. Chemother. 65:1100–1107 [DOI] [PubMed] [Google Scholar]
- 37. Tang C, et al. 2003. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327:1013–1020 [DOI] [PubMed] [Google Scholar]
- 38. Ternois F, Sticht J, Duquerroy S, Krausslich HG, Rey FA. 2005. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat. Struct. Mol. Biol. 12:678–682 [DOI] [PubMed] [Google Scholar]
- 39. von Schwedler UK, et al. 1998. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 17:1555–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Worthylake DK, Wang H, Yoo S, Sundquist WI, Hill CP. 1999. Structures of the HIV-1 capsid protein dimerization domain at 2.6 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 55:85–92 [DOI] [PubMed] [Google Scholar]
- 42. Wright ER, et al. 2007. Electron cryotomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 26:2218–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.