

Abstract
The structure of the bacteriophage SPP1 capsid was determined at subnanometer resolution by cryo-electron microscopy and single-particle analysis. The icosahedral capsid is composed of the major capsid protein gp13 and the auxiliary protein gp12, which are organized in a T=7 lattice. DNA is arranged in layers with a distance of ∼24.5 Å. gp12 forms spikes that are anchored at the center of gp13 hexamers. In a gp12-deficient mutant, the centers of hexamers are closed by loops of gp13 coming together to protect the SPP1 genome from the outside environment. The HK97-like fold was used to build a pseudoatomic model of gp13. Its structural organization remains unchanged upon tail binding and following DNA release. gp13 exhibits enhanced thermostability in the DNA-filled capsid. A remarkable convergence between the thermostability of the capsid and those of the other virion components was found, revealing that the overall architecture of the SPP1 infectious particle coevolved toward high robustness.
INTRODUCTION
The large majority of bacterial viruses (phages or bacteriophages) have a double-stranded DNA (dsDNA) genome and are comprised of an icosahedral capsid, or head, which protects the viral genome, and of a tail structure, a device that delivers the genome to the host cell. The tail is attached to the unique portal vertex of the capsid. DNA entry and exit from the capsid occur through the portal system. The overall tailed-phage architecture is well suited both to protect the viral dsDNA from environmental insult and to deliver it to the host cytoplasm. Tailed phages are the most abundant biological entities found in the biosphere: their total number was estimated to be on the order of 1031 on Earth (58).
The capsids of tailed phages and herpesviruses follow a common assembly pathway. An icosahedral procapsid is built by the polymerization of the major capsid protein initiated at the dodecameric portal protein (21). The viral genome is then packed at a high density inside the DNA-free precursor particle. The capsid normally undergoes expansion during packaging, leading to an increase in the internal volume with concomitant changes to its shape and physicochemical properties (9). In a number of phages, this large reorganization of the major capsid protein creates binding sites for decoration proteins (also designated auxiliary proteins) that might cement their structure (50). After the termination of DNA packaging, the assembly of the viral particle ends either by the addition of a preformed long tail or by the assembly of a short tail at the portal vertex (9). The dsDNA inside the sealed capsid can reach concentrations of around 500 mg/ml, allowing the large the amount of genetic material to be transported by the viral particle. The pressure exerted by the tightly packed dsDNA on the capsid protein shell can exceed 6 MPa (49).
Studies of the capsid assembly pathway and structures of capsid proteins clearly established that tailed bacteriophages and herpesviruses are evolutionarily related, thus defining a viral lineage (3, 4). To date, all structurally characterized capsids of viruses from this lineage share a structural motif typified by the fold of the major capsid protein gp5 from the phage HK97 (13, 22, 25, 57). gp5 establishes quasiequivalent intersubunit interactions, forming pentamers that define vertices and hexamers found at the faces of the icosahedron. The protein subunit has a mixed α/β structure comprising the globular domain A, protruding toward centers of hexamers and pentamers; the elongated periphery domain P; and the long E loop, involved in interconnections of pentamers and hexamers (57). This fold, although combined with some extensions or additional domains, is conserved in different tailed phage and herpesvirus capsids and serves as a major building block of the icosahedral capsids (1, 3, 18, 20, 29, 30, 32, 36, 38, 57). The walls of the mature capsids are remarkably thin (18 Å in HK97 [57] and 20 Å in lambda [32]), in contrast to numerous other plant and animal icosahedral viruses, which are built by protein subunits with a jellyroll motif, leading to capsid shells of around 40 Å in thickness (6, 24).
Proteins with the HK97 fold show a conserved core-bonding pattern (see references 1, 3, 18, 20, 29, 31, 32, 36, 38, and 57 and references therein). Additional connections between subunits can take place through intersubunit covalent cross-linking (phage HK97 [57]), through the addition of extra domains that provide new interactions between subunits (phi29 and P22), and by the binding of auxiliary proteins to the capsid lattice (phi29, lambda, herpes simplex virus 1 [HSV-1], T4, L, T5, epsilon 15, and N4) (43). The binding sites for auxiliary proteins are normally created or exposed during the rearrangement of capsid subunits when the procapsid expands to yield the mature capsid structure during genome packaging (32, 50).
Bacteriophage SPP1 is a siphovirus that infects the Gram-positive bacterium Bacillus subtilis (2). Capsid assembly is initiated by the formation of the procapsid, a DNA-free, spherically shaped protein shell ∼55 nm in diameter. The procapsid comprises the major capsid protein gp13, the internal scaffolding protein gp11, the portal protein gp6, and a few copies of the minor protein gp7 (16). This is followed by the assembly of the DNA-packaging motor at the portal vertex of the shell. The motor pumps the SPP1 dsDNA into the capsid (8, 39). It consists of gp6, gp1 (SPP1 DNA binding protein), and gp2 (viral ATPase/endonuclease). The wild-type packaged chromosome is a linear ∼45.9-kbp ± 2.5-kbp dsDNA molecule with terminal sequence redundancy. Its size variability results from the imprecision of the SPP1 headful packaging mechanism (51). The packaging of DNA into the procapsid is accompanied by the exit of the internal gp11 scaffold. The expansion of the gp13 procapsid shell into the ∼610-Å-diameter capsid creates or exposes the binding sites for the auxiliary protein gp12 on its outer surface (27). Upon the termination of DNA packaging, gp1 and gp2 detach from the capsid; this is followed by the sequential attachment of gp15, gp16, and the tail to the portal vertex to complete the assembly of the infectious virion (35, 41). Most studies so far have concentrated on the earlier stages of the phage assembly pathway, particularly from the procapsid to the full capsid. Here we are aiming to reveal and understand conformational changes in the later stages of bacteriophage assembly and to assess capsid thermostability.
We have determined three-dimensional (3D) reconstructions of the bacteriophage SPP1 capsid at different states after viral genome packaging (Fig. 1) and analyzed their stabilities. Structures of capsids with and without gp12 allowed the delineation of the boundaries of the auxiliary protein and its location in the capsid. Structure-based modeling and bioinformatics comparisons with major capsid proteins of other siphoviruses identified the HK97 fold in gp13, allowing us to produce a pseudoatomic model of the SPP1 major capsid protein and its arrangement in the T=7 capsid.
Fig 1.

Schematic representation of the final steps of bacteriophage SPP1 assembly and DNA release from virions. The assembly of structures lacking gp12 is outlined at the top of the scheme. Sizes are not scaled, to emphasize the locations of the minor capsid components. gp6, portal protein; gp13, major capsid protein; gp12, auxiliary protein; gp15 and gp16, head completion proteins (the adaptor and the stopper).
MATERIALS AND METHODS
Sample preparation.
Bacteriophage SPP1 wild-type particles (full phage [FP] and empty phage [EP]) and SPP1 suppressor-sensitive mutants (SPP1sus9 and SPP1sus9del12) were multiplied in B. subtilis strains YB886 (sup+) and BG295 (sup-3), respectively, as described previously (16). SPP1sus9del12 was constructed by a genetic cross between SPP1sus9, defective in a tail gene (5), and SPP1del12, which carries deletion-disrupting gene 12 (P. Tavares, unpublished data).
SPP1 particles, DNA-filled capsids (H), and DNA-filled capsids lacking gp12 (HΔ12) were produced by the infection of the nonpermissive host B. subtilis YB886 with SPP1wt, SPP1sus9, and SPP1sus9del12, respectively. Phage lysates were cleared by centrifugation (11,000 × g for 15 min at 4°C), and viral particles were then sedimented by overnight centrifugation at 11,000 × g (4°C). The pellet was resuspended in TBT buffer (100 mM Tris-Cl, 100 mM NaCl, 10 mM MgCl2 [pH 7.5]) and treated with 1 μg/ml DNase (Sigma) and 100 μg/ml RNase (Sigma) for 1 h at room temperature, followed by centrifugation in a discontinuous CsCl gradient (28). Viral particles were dialyzed against 100 mM Tris-Cl (pH 7.5)–10 mM MgCl2, applied onto a Resource Q anion-exchange column (GE Healthcare), and eluted with an NaCl gradient. Virions and DNA-filled capsids eluted at a concentration of ∼250 mM NaCl. They were dialyzed against 0.5× TBT buffer and stored at 4°C until preparation for cryo-electron microscopy (cryo-EM). Particle protein concentrations and compositions were determined as described previously (56). The presence or absence of gp12 was assessed by Western blotting with anti-gp12 serum (not shown).
Viral particle thermostability assays.
Capsid samples (∼0.4 mg/ml protein) were diluted 1,600-fold in TBT or in TBEDTA buffer (100 mM Tris-Cl, 100 mM NaCl, 10 mM EDTA [pH 7.5]) containing SYBR gold (diluted 3,000-fold from a 10,000-fold stock solution; Invitrogen) to quantify free DNA in solution. SYBR gold exhibits a >1,000-fold fluorescence enhancement upon binding to nucleic acids. Reaction mixtures were made in triplicate in a 96-well fast PCR plate at a final volume of 10 μl. The temperature gradient was carried out in the range of 25°C to 95°C at 3°C/min with a StepOnePlus real-time PCR system (Applied Biosystems). Fluorescence was recorded as a function of temperature in real time (excitation with a blue light-emitting diode [LED] source and emission filtered through a Joe emission filter). The capsid disruption temperature (Tdis), assessed by the release of DNA, was calculated with StepOne software v2.2 as the maximum of the derivative of the resulting SYBR gold fluorescence curves. The experimental conditions to monitor protein denaturation were identical, except that capsids were diluted only 10-fold and SYPRO orange (diluted 1,000-fold from a 5,000-fold stock solution; Invitrogen) was used as a fluorescent probe (excitation with a blue LED source and emission filtered through a 5-carboxy-X-rhodamine [ROX] emission filter). This dye becomes highly fluorescent when bound to protein hydrophobic sites, which become accessible upon thermal unfolding (19).
Wild-type SPP1 and SPP1del12 virions (0.5 × 1011 to 2.5 × 1011 PFU/ml) were challenged at different temperatures in a PCR machine with a hot lid, in blocks preheated to the target temperature. Incubations were carried out for 15 or 120 min. After cooling for 10 min on ice, the samples were split for titration with B. subtilis YB886 and for a DNase protection assay. The latter sample was treated with Benzonase for 30 min at 37°C, followed by deproteinization with stop buffer (10 mM EDTA, 0.5% SDS, 50 mg/ml proteinase K [Roche]) and incubation at 65°C (14). DNA was resolved in 1% agarose gels and stained with ethidium bromide.
Electron microscopy.
Wild-type phage SPP1 particles and DNA-filled capsids were plunge frozen, as reported previously (41, 48), on holey carbon film grids (R2x2 Quantifoil [Micro Tools GmbH, Jena, Germany] or C-flat [Protochips, Raleigh, NC]). We used a 626 cryo-specimen holder (Gatan, Inc., Pleasanton, CA) for imaging. Data were collected by using a Jeol 2200FS electron microscope equipped with an in-column energy filter (omega type) and a field emission gun (FEG) operating at 200 keV. Viral particles were imaged at a ×50,000 nominal magnification using either a 4,096-by-4,096 pixel charge-coupled-device (CCD) camera (UltraScan 4000; Gatan, Inc., Pleasanton, CA) or photographic film (SO-163; Kodak, Rochester, NY). To reduce radiation damage, a low-dose imaging procedure was used during data collection. Images were acquired with ca. 20 electrons/Å2 dose in the defocus range of 0.4 to 1.85 μm; the CCD pixel size corresponded to 2.1 Å on the specimen scale. Images acquired on film were digitized by using a Zeiss scanner with a step size of 1.49 Å/pixel. The number of particles collected for each sample is given in the Table S1 in the supplemental material. Data for the FP and EP capsids were collected from 48 films. Images of H and HΔ12 capsids were collected with a CCD camera. A total of 170 CCD frames were analyzed for H capsids, and 80 CCD frames were used for HΔ12.
Image processing.
Four data sets of individual viral particle images (HΔ12, H, FP, and EP) were normalized to the same standard deviations, and frequencies lower than 1/300 Å were suppressed. All images were corrected for the effect of the microscope contract transfer function (CTF). We used CTFFIND3 to determine the CTF and applied corrections in IMAGIC for images taken on films while using CTFIT (37) to determine parameters of the CTF and for the correction of images taken by the CCD camera. BOXER from EMAN was used for automated particle picking (37). Subsequent processing was done by using Imagic-5 (54). Data sets were centered to the rotationally averaged total sum of images in three rounds, using the refined total sum of images as a reference. For classification, we used a multivariation statistical analysis implemented in IMAGIC-5 (54). An initial model of the viral particle under analysis was obtained by using a few classes that revealed the lowest error at the angular search with an angular orientation that did not coincide with rotational axes in the asymmetric unit. The refinement of the alignment for each set was done by using reprojections of the corresponding initial models. The angular reconstitution technique was used to determine the relative orientations of the single images, and these orientations were then used to calculate the 3D reconstruction. The TRUE-THREE command (23) in IMAGIC and BKPR (40) was used to reconstruct the 3D maps, assuming icosahedral symmetry for the particles. The best images (showing the highest correlations with reference map projections) were then selected to calculate the next 3D map, used as the new reference for the next round of refinement. Iterative origin/orientation refinement cycles were performed until the images converged. The final 3D map was reconstructed from the best virus images, which was ∼60% of all images in each set. The effective resolution of the maps was determined according to a 0.5 Fourier shell correlation function (FSC) between maps calculated from independent half-data sets (see Fig. S1 and Table S1 in the supplemental material) (46).
The similarity in sequences of secondary elements between the SPP1 major capsid protein gp13, HK97, and T4 allowed the use of structures of capsid proteins from HK97 and T4 to obtain a comparative model with MODELLER (53). The final model was obtained from an alignment of HK97 (Protein Data Bank [PDB] accession number 1OHG) and the A domain, P domain, and E loop of gp24 from T4 (PDB accession number 1YUE). A later model obtained from HK97 (PDB accession number 1OHG) and T7 (PDB accession number 2XVR) was similar to the original models. Segmentation of maps was obtained by using the Segger option in Chimera (45). Seven copies of the subunit model were generated, and these copies were fitted as rigid bodies into the cryo-EM electron density by using Chimera to produce the asymmetric unit with seven gp13 subunits. The α-helices and β-sheets were then moved manually into the corresponding areas of the cryo-EM map. The fit was refined by the Flex-EM program (53), using molecular dynamics optimization and keeping the helices and sheets as rigid bodies.
The 3D maps were surface rendered and displayed with a 1-standard-deviation (1σ) threshold in Chimera (44), which accounted for a ∼100% particle volume.
Protein structure accession numbers.
The three-dimensional density maps have been deposited into the EBI-MSD EMD database with accession numbers EMD-2049 for the structure of FP, EMD-2052 for the structure of EP, EMD-2050 for HΔ12, and EMD-2051 for H particles.
RESULTS
Visualization of the mature SPP1 capsid at different stages of the viral cycle.
The structure of the SPP1 capsid was analyzed at four different states after expansion (Fig. 1) by cryo-electron microscopy of frozen-hydrated samples (see Materials and Methods). Tailless mature capsids filled with DNA were purified from bacteria infected with an SPP1 conditional lethal mutant phage defective in tail assembly. Two types of capsids were investigated: the first lacks auxiliary protein gp12 (HΔ12) (Fig. 2A), while the other carries this protein (H) (Fig. 2B). Capsids of infectious phage particles (FP, for full phage) (Fig. 2C, full circle) and of a subpopulation of particles that ejected their genomes (EP, for empty phage) (Fig. 2C, dashed circle) were also imaged. These empty particles most likely lost their DNA via adsorption to bacterial debris. Visual inspection of cryo-micrographs allowed us to detect DNA inside filled capsids (Fig. 2) and to confirm the lack of tails in samples of tailless mutants but provided no clear distinction between wild-type heads and particles lacking gp12. The images of the H, HΔ12, FP, and EP particles demonstrate that the capsids have the same general appearance and are ∼600 Å in size. Together, the specimens analyzed provide a range of structures spanning the last steps of SPP1 assembly and chromosome ejection from the virus particle entering the infection process.
Fig 2.

Cryo-electron microscopy of SPP1 capsid and phage particles. (A) Images of HΔ12. (B) Images of H. (C) Images of FP and EP. Examples of phage capsids filled with DNA are circled (white line), while the capsid without DNA is shown in a dashed white circle. (D) Class averages of all four samples shown in the same scale as quarters of capsid images. (E) Enlarged Fourier transforms from class averages of HΔ12 (top half) and FP (bottom half), indicating that there are no changes in distances for DNA packing in decorated and nondecorated capsids.
Organization of DNA inside the SPP1 capsid.
The images of single capsids or averaged images of the capsids of the HΔ12, H, and FP capsids show the consistent presence of density layers or a granular pattern within the capsid shells, which are attributed to dsDNA (Fig. 2D, and see Fig. S2 in the supplemental material). To assess the distances between the layers and the features of the granularity seen inside the capsids, we calculated Fourier transforms from classes with the distinct appearance of a packed genome (Fig. 2E, and see Fig. S2 in the supplemental material). They represent different types of projections of the dsDNA packed within the capsids. A comparison of the diffraction patterns demonstrates the presence of identical rings for all different projections which correspond to a distance of 24.5 Å between dsDNA layers (see Fig. S3 in the supplemental material). This distance found in the averages of similar capsid images (Fig. 2D) and the size of the packaged DNA molecule assessed by pulsed-field gel electrophoresis (data not shown) are identical in all chromosome-filled capsids, showing that neither gp12 nor tail attachment changes DNA packing detectably.
Stability of SPP1 capsids.
A major role of capsids is to protect the viral genome from an aggressive environment and to withstand the high internal pressure from the tight DNA packing (49). To investigate the role of the auxiliary protein gp12 in cementing the SPP1 capsid structure and to evaluate the robustness of HΔ12 and H capsids in general, we analyzed their stabilities at elevated temperatures. The capsids were challenged with a linear temperature gradient (3°C/min), and the accessibility of phage dsDNA in solution was quantified by using SYBR gold (Fig. 3A), a probe that fluoresces upon binding to nucleic acids. It is important that the fluorescence of SYBR gold bound to free DNA (Fig. 3A, black squares) showed a steep decrease with increasing temperature; the same reduction of fluorescence was observed for the DNA in the presence of magnesium (10 mM in the TBT phage buffer) (Fig. 3A, black open squares). A steep downward slope in SYBR gold fluorescence was observed, because the probe fluorescence is highly dependent on temperature. HΔ12 (Fig. 3A, open red triangles) and H capsids (open blue circles) in TBT buffer showed overlapping curves characterized by a low fluorescence signal compared to that of free DNA up to a temperature of ∼65°C. This was followed by a peak of fluorescence at ∼71°C (Fig. 3A, red arrowheads), which can be best assessed from the inset plot of the opposite first derivative of the curves for HΔ12 and H (Fig. 3A). The signal at higher temperatures approached the level observed for an identical amount of free SPP1 DNA in the same buffer (Fig. 3A, open square curve), indicating that DNA was released from HΔ12 and H capsids. Both types of capsids demonstrated similar behaviors, disrupting at nearly the same temperatures (Tdis of ∼71°C). If magnesium was replaced by 10 mM EDTA, which chelates divalent cations, capsids were disrupted (Fig. 3A, inset electron micrograph), releasing their DNA at a temperature below 40°C, as demonstrated by the similar fluorescence levels observed for both capsid preparations (Fig. 3A, filled red triangles and filled blue circles) and free DNA fluorescence (black filled squares) throughout the complete temperature gradient.
Fig 3.
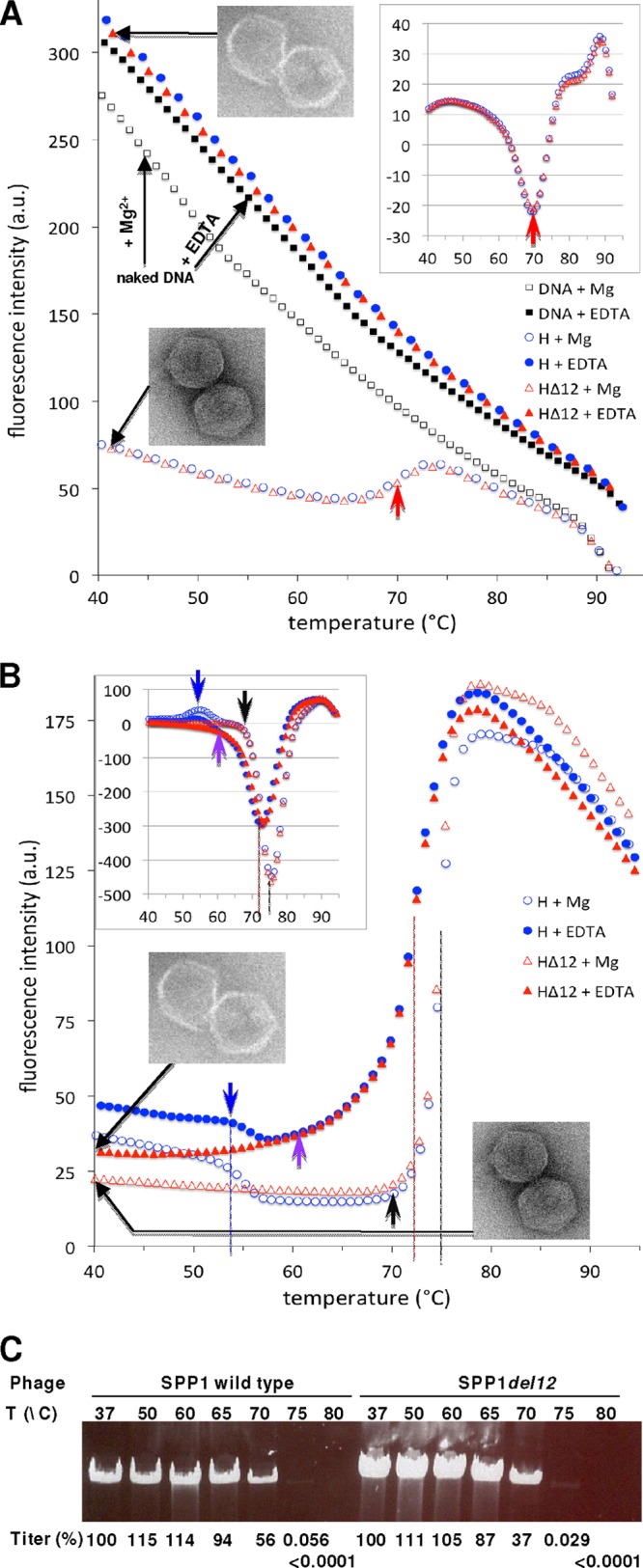
Thermostability of SPP1 particles. (A) Release of DNA from HΔ12 (red open triangles) and H (open blue circles) capsids challenged with a linear temperature gradient (3°C/min) in TBT buffer. Capsids incubated in the presence of 10 mM EDTA (TBEDTA buffer), which leads to their physical disruption (51), were also tested (filled red triangles and filled blue circles). Only every second point of the curves for HΔ12 and H capsids is shown, for better visualization of the curve overlap. DNA in solution was quantified by strong fluorescence emission upon SYBR gold binding to nucleic acid. Free SPP1 DNA (∼10 ng) (black squares) was used as a control for the normalization of the SYBR gold signal, which is affected by both temperature and magnesium. The graph inset shows the opposite of the first derivatives of the fluorescence signal, calculated with StepOne software v2.2. The top inset shows an electron micrograph of the viral particles treated with EDTA. The bottom inset shows an electron micrograph of the viral particles in TBT phage buffer. Red arrows indicate DNA release from capsids. (B) Fluorescence-based thermal shift assay of HΔ12 and H capsids. Samples were treated as described above for panel A, except that 160-fold more material was used. Fluorescence measures the binding of the SYPRO orange probe to exposed protein hydrophobic regions. The inset shows the opposite of the first derivatives of the fluorescence signal. Data symbols are as described above for panel A. Black arrows indicate a starting point of the gp13 denaturation in TBT phage buffer (intact DNA-filled capsids), purple arrows indicate a starting point of the gp13 denaturation in TBEDTA buffer (disrupted capsids), and blue arrows indicate a reduction of the fluorescence signal attributed to an uncharacterized structural change of gp12 in H capsids. (C) DNase protection assay and viability of wild-type SPP1 and SPP1del12 infectious virions challenged to the temperatures indicated for 15 min. Phage viability is shown as a percentage of the initial virion input (∼1011 PFU/ml), determined from an average of three independent experiments. The initial input of infective phages determined from titrations of control samples (incubation at 37°C) was 2.2-fold higher for SPP1del12 (100% at 37°C, equal to 1.4 × 1011 ± 0.25 × 1011 PFU/ml; n = 5) than for wild-type virions (100% at 37°C, equal to 0.6 × 1011 ± 0.12 × 1011 PFU/ml; n = 5). The difference in the inputs of viral particles correlates with a larger amount of DNA observed in the DNase protection assay. a.u., arbitrary units.
We then investigated whether the capsid disruption results from an unfolding of the capsid protein(s). The thermal denaturation of HΔ12 and H capsids was monitored by a fluorescence-based thermal shift assay using the fluorescence signal of SYPRO orange upon binding to exposed hydrophobic regions of proteins. The heating conditions of the capsids were identical to the those of first set of experiments with DNA. A sharp increase in fluorescence was observed at 75°C, compatible with a highly cooperative protein denaturation event for intact structures in TBT buffer (Fig. 3B, black dashed line with open triangles and open circles). When capsids were treated with EDTA, leading to their physical breakage at low temperatures and, consequently, to DNA release (51) (Fig. 3A, filled triangles and filled circles), the increase of SYPRO orange fluorescence started at ∼60°C (Fig. 3B, purple arrow), and the peak of its increase was significantly less steep than the peak at 72°C (Fig. 3B, red dashed line). A minor fluorescence-negative peak, found only for the H capsids (Fig. 3B, arrows and dashed line in blue), was observed at ∼54°C and is attributed to the denaturation of gp12. Apart from this difference, the H and HΔ12 capsids exhibited identical resistances to thermal disruption (Fig. 3A) and to thermal protein unfolding (Fig. 3B) under both buffer conditions, showing that gp12 does not provide additional stability to SPP1 capsids. The observed protein denaturation profile shown in Fig. 3B is attributed essentially to the unfolding of the gp13 polypeptide chains, which account for 87% of the total protein mass of the capsid.
SPP1 virions were also challenged with high temperatures, followed by a DNase protection assay and scoring for a loss of infectivity (Fig. 3C). Phage particles remained fully viable at 65°C, while their titer was reduced to 35% after incubation at 70°C for 15 min and dropped by 3 orders of magnitude at 75°C. A longer incubation for 2 h showed that ∼50% of the phages remained infectious after incubation at 65°C, but less than 1% survived incubation at 70°C (data not shown). The reduction of virion viability showed an excellent correlation with the release of DNA from phage particles, as measured by its accessibility to DNase digestion (Fig. 3C). Note that the initially larger amounts of DNA protected in SPP1del12 correlate with a 2.2-fold-higher initial input of infectious phage particles of this mutant than that of wild-type particles in the experiment, as assessed by the scoring of infective virions in the control and heat-treated samples (Fig. 3C). The thermal stabilities of the phage capsid (Fig. 3A) and of the overall virion (required for infectivity) (Fig. 3C) are thus very similar. The same results were obtained for the complete SPP1del12 and wild-type SPP1 virions, showing that gp12 does not have a role in SPP1 infectivity under laboratory conditions or in the stability of virus particles.
Structures of the mature icosahedral capsids at the late stages of the viral cycle.
The SPP1 capsid is composed of gp13 and gp12 organized with icosahedral symmetry, while gp6, gp15, gp16, and probably gp7 are located at the portal vertex. During the 3D reconstruction of the capsid, when icosahedral symmetry was imposed, all phage components that had no such symmetry were averaged out, including the portal complex that was seen in images of phages that lost their DNA (Fig. 2C, arrowhead). We determined the structures of the HΔ12, H, FP, and EP capsids to resolutions of 15.1 Å, 11.7 Å, 8.8 Å, and 10.5 Å, respectively (Fig. 4, and see Fig. S1 and Table S1 in the supplemental material). A comparison of the 3D EM maps indicated that capsids do not undergo significant changes during the final steps of phage assembly and upon DNA release from the particles. Their overall average diameter remained the same, 610 Å, and the protein shell of all four capsids is ∼27 Å thick. The capsids are composed of hexameric and pentameric capsomers formed by the major capsid protein gp13. In the SPP1 phage, the hexamers are about 140 Å, and the pentamers are 110 Å in diameter. SPP1 pentamers are located in the vertices of the capsid, providing the angular appearance of the particles (Fig. 4). There was no observable difference in the shape of the empty capsid (EP) compared to the shape of the full capsid (FP), unlike the case for HK97 (17). Hexameric capsomers that form the flat faces of the icosahedron deviate from an exact 6-fold symmetry and are better described by pseudo-2-fold symmetry. A similar distortion of the hexamers was observed previously for P22 procapsids (52). The overall capsid structure is characterized by T=7 laevo icosahedral symmetry (Fig. 4). All 3D maps of SPP1 capsids with a packaged genome clearly revealed inner shells of dsDNA that were absent in the EP capsid (see Fig. S3 in the supplemental material). A comparison of the EP structure with the organization of the other capsids showed that the release of DNA does not affect the arrangement of protein subunits in the capsid. However, empty phage particles were frequently found to be partially disrupted, suggesting that their capsids are structurally more fragile.
Fig 4.
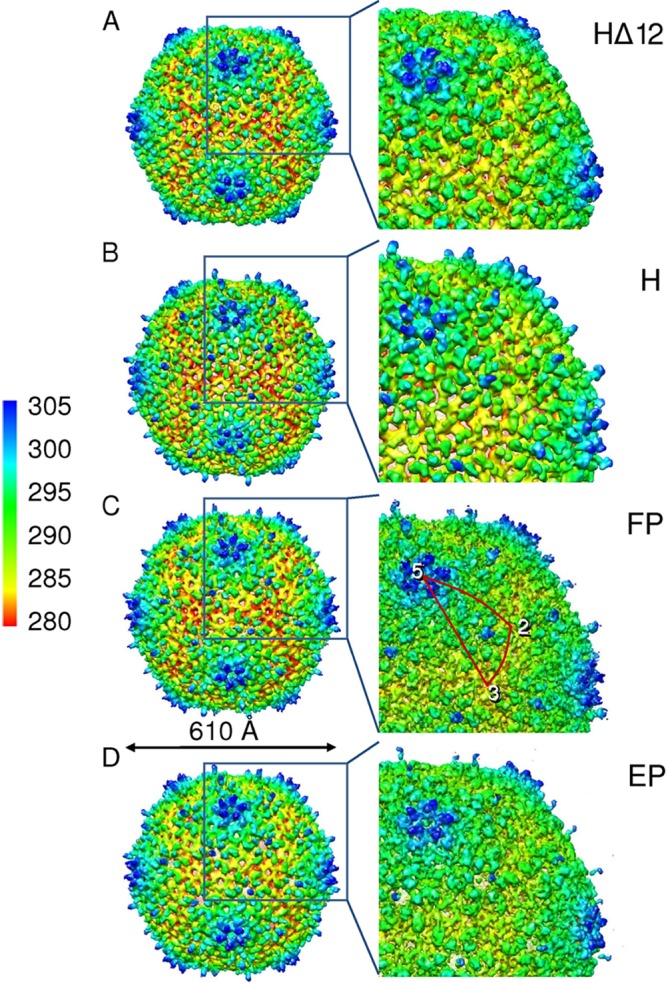
Three-dimensional reconstructions of the SPP1 capsids. (A) Surface representation of HΔ12 with radial coloring. Color coding is shown on the right side of the figure. (B to D) Structures of H (B), FP (C), and EP (D). The right column shows enlarged areas of reconstructions in the left column. The best resolution was achieved for the FP (C). The outline in red (C) shows the asymmetric unit of the capsid.
Localization of auxiliary protein gp12.
A comparison of gp12-deficient capsid HΔ12 with the other capsids allowed the identification of the position of gp12 in 3D icosahedral reconstructions. The H, FP, and EP capsids differ from HΔ12 by the presence of spikes protruding from the center of their hexamers, which were assigned to gp12 (Fig. 5A). The hexamers in H were filtered to a 15-Å resolution for comparison with the HΔ12 hexamers. Both structures were aligned and then superimposed based on the highest local correlation coefficient (Fig. 5). The perfect overlap of the two structures (except in the center of hexamers) implies that, at least at a ∼15-Å resolution, gp13 does not change significantly upon the binding of gp12. The difference between the two hexamers identifies the gp12 density that is shown in Fig. 5D and E (light purple). The gp12 spikes have a cylindrical shape with a ∼35-Å height and a ∼15-Å diameter. The center of hexamers where gp12 is anchored in the H map is flat and closed in HΔ12 capsids (Fig. 5C). It was estimated previously that there are 180 gp12 molecules bound to the SPP1 capsid, indicating that each of the 60 hexamers has three copies of the 6.6-kDa gp12 in its center (15; M. Zairi, unpublished results). The molecular mass of the spike assessed from the difference map is only ∼10 kDa, which is not sufficient to accommodate three gp12 subunits. While the bottom part of the spike has well-defined densities, its outer peripheral end is blurry in 3D reconstructions (Fig. 5A, left), suggesting that the distal spike end is flexible, accounting for the lower mass of gp12 estimated from the reconstructions. The prediction of secondary elements of gp12 implies that the protein has potential α-helices at its ends, but a large central part of the protein does not show any defined structural organization (data not shown). We have docked three coiled-coil helices into the spikes of our reconstructions; however, the quality of the fit was low, since the diameter of the coiled-coil trimer (∼23 Å) was not compatible with the diameter of gp12 (∼15 Å).
Fig 5.

Identification of the gp12 auxiliary protein position. (A) Localization of gp12 in the capsids. The left column shows cross sections of each reconstruction in the area where extra density is observed in three structures containing gp12 compared to the gp12-deficient capsid. The right column shows the same images, with the position of gp12 highlighted by light magenta ovals. The blue arrows point to the capsid shell, while the gold arrows point to layers of DNA inside the capsids. (B) Surface rendering of the H hexameric capsomer. (C) Surface rendering of the HΔ12 hexameric capsomer. (D) The difference between two hexameric capsomers is shown in light magenta. The green surface shows the common organization of the capsid hexamers. (E) Cutaway view of the difference map overlaid with the HΔ12 hexameric capsomer. All maps for comparison with gp12 were filtered to a resolution of 15 Å, equivalent to the HΔ12 map.
Modeling of the pseudoatomic structure of the major capsid protein gp13.
The subnanometer resolution of the SPP1 FP capsid structure allowed us to delineate single gp13 subunits in EM maps. The extraction of single gp13 molecules from the maps by segmentation using Chimera automated procedures provided approximate boundaries of single subunits. An asymmetric unit of the SPP1 capsid containing six gp13 subunits from the hexamer and one subunit from the pentamer (Fig. 6A) was then extracted, and subunits were aligned and averaged to provide an averaged subunit density map. A Helixhunter (30) search clearly identified positions of three helices in the map (Fig. 5B, green rods), with orientations and sizes exhibiting the fingerprint of helices found in the major capsid protein fold of phage HK97. Since there is no high-resolution gp13 structure available, we opted to use bioinformatics to produce a pseudoatomic model for gp13. It is known that in spite of the low primary sequence homology of major capsid proteins within tailed bacteriophages, the HK97 fold signature is well conserved. The sequences of secondary-structure elements within the capsid proteins are thus expected to be similar, and these elements could be aligned and used to construct a structural model. We therefore carried out secondary structure predictions for a set of major capsid protein sequences, from tailed phages with available X-ray or EM 3D structures, using several different programs (Psipred [7], Jpred [12], SSpro [11], and PROF [42]) and aligned the amino acid sequences according to their consensus secondary-structure elements. A subset of the alignments for capsid proteins of the siphoviruses HK97, SPP1, T5, and lambda is shown in Fig. S4 in the supplemental material. The robustness of the prediction methods was assessed by comparing the predicted secondary structure of gp5 from phage HK97 with its atomic structure. The conserved order of predicted secondary structure elements, despite low amino acid sequence homology, supports a common fold for the capsid proteins of the four phages. This was particularly evident for three main helices, α3, α5, and α6, in gp5 of HK97 and the predicted long helices in gp13 of SPP1 (see Fig. S4 in the supplemental material).
Fig 6.
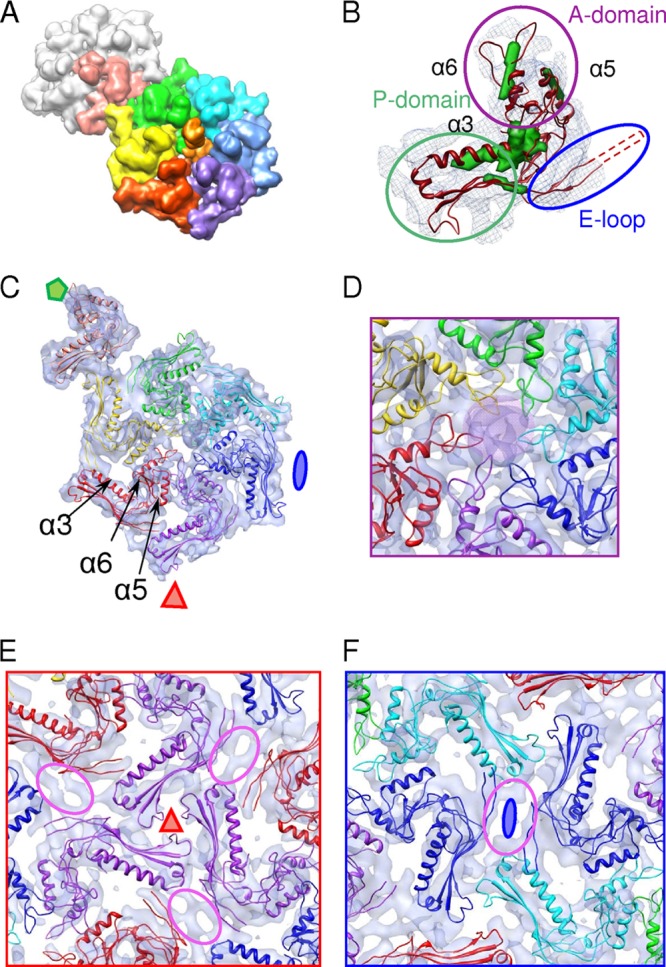
Segmentation of the asymmetric unit in the FP capsid and modeling of the pseudoatomic structure of the gp13 capsid protein. (A) The asymmetric unit of the 8.8-Å-resolution map was segmented to reveal seven capsid protein subunits. The unit contains an entire hexamer and one pentamer subunit. Six subunits that form the hexameric capsomer of the phage shell are shown in rainbow colors. The capsid protein that participates in the formation of the pentamers is shown in salmon, and the remaining subunits of the pentamer are shown in gray. gp12 at the center of the hexamer is depicted in orange. (B) Single gp13 subunit obtained by the averaging of six density maps corresponding to proteins forming the hexamers of the FP capsid. The green densities within the gray mesh reveal putative positions of α-helices, assigned with Helixhunter, within one subunit. The initial atomic model of the gp13 protein obtained by the modeling of its ternary structure based on the prediction of the secondary elements (see Fig. S4 in the supplemental material) is shown in red. The fit indicates a good agreement between densities assigned to helices by Helixhunter and the fitting of the modeled atomic gp13 structure (backbone chain). (C) Pseudoatomic model fitted into the asymmetric unit of the FP capsid by Flex-EM. The colors correspond to the colors described above for panel A. (D) Zoomed-in view along the central axis of the hexamer. gp12 is shown as a purple overlay in the center. (E) View along the 3-fold axis. (F) View along the 2-fold axis. Intracapsomer contacts near 2-fold axes could possibly be formed by the E loop of a gp13 subunit and the P domain of an adjacent subunit within the same hexamer, as found previously for the phage T7 capsid (1, 26). Unassigned densities are encircled in purple.
A rigid-body fit of crystallographic structures of the HK97 (Fig. 6B, dark red) and T4 capsid proteins into the map of SPP1 FP yielded cross-correlation coefficients (ccc) of 0.5 and 0.47, respectively. However, some features of the fitted structures were clearly out of register from the EM density map, indicating that a flexible fit of a pseudoatomic model should be done. An initial model of the SPP1 gp13 protein based on the conserved features of major capsid proteins from phages HK97 (gp5 [PDB accession number 1OHG]), T4 (vertex protein gp24 [PDB accession number 1YUE]), and T7 (gp10A [PDB accession number 2XVR]) was constructed by using Modeller (53) (see Materials and Methods). This structure was then docked into each of the seven subunits in the asymmetric unit from the SPP1 capsid by flexible fitting to refine the pseudoatomic model of gp13. The refined model of gp13 showed a significantly improved correlation with the map (ccc raised to 0.72) (see Fig. S5 in the supplemental material). The docking confirmed the good correspondence of the fold in the A and P domains of gp13 from SPP1 relative to that of gp5 from phage HK97, as illustrated in Fig. 6B and C and Fig. S5 in the supplemental material for the fit in the 8.8-Å resolution maps of the FP capsid, and correlated the 3D fold to the SPP1 polypeptide chain sequence (see Fig. S4 in the supplemental material). The end of the E loop could not be traced in the cryo-EM density map (Fig. 6B and C), suggesting some divergence in the gp13 structure relative to the HK97 gp5 structure.
gp13 structure in the SPP1 capsid.
In SPP1, like in HK97, the A domains form the center of pentamers and hexamers, engaging in extensive intracapsomer contacts and delimiting a central flat region in the gp13 hexamers where gp12 is located (Fig. 4 to 6). The fit of the pseudoatomic model of gp13 in the FP capsid shows that the loops of the A domains of gp13 forming the center of the hexamer could provide the contacts with gp12 spikes in the wild-type phage while filling the space in HΔ12 capsids and keeping the entire capsid shell tightly closed (Fig. 5E and 6D).
Subunits establish equivalent contacts within pentamers whose A domains project significantly outward from the capsid surface, compared to the more flat organization of hexamers. The skewed shape of the hexamers resulting in their pseudo-2-fold symmetry leads to subtle variations in the subunit contacts between the densities attributed to helix α5 and part of a putative loop from the neighboring subunit that connects the α5 helix to the α6 helix in the A domain (Fig. 6D). Additional intracapsomer interactions were observed between the peripheral P domain and regions of density extending from the beginning of the E loop, which could not be assigned in our model, probably because the SPP1 fold in this region differs from those of other major capsid proteins.
The extended peripheral P domain establishes significant intercapsomer interactions (Fig. 6E and F). The distal end of the P domain from one subunit contacts the joint between the P domain and E loop from another subunit of an adjacent capsomer. The skewed shape of the hexamer implies that the network of interactions defines slight differences between quasi-3-fold and icosahedral 3-fold symmetry axes in the SPP1 capsid (Fig. 6E).
DISCUSSION
We determined reconstructions of the T=7 isometric capsids of the siphovirus SPP1 at three different late stages of phage assembly and of capsids from virions that had released their DNA (Fig. 1). A comparison showed that the angular icosahedral shell formed by the major capsid protein gp13 remains unchanged upon the binding of the auxiliary protein gp12, positioned at the center of each of the 60 gp13 hexons; upon tail binding to the portal vertex; and upon the release of DNA, which leaves the capsid interior empty (Fig. 1 and 4). The length of the DNA packaged and its density inside filled capsids (Fig. 2, and see Fig. S2 and S3 in the supplemental material), corresponding to ∼0.58 bp/nm3, were also identical. This is consistent with the constant diameter of capsids at the final steps of SPP1 assembly and the same distances between concentric layers of DNA irrespective of the binding or not of gp12 to the capsid lattice (Fig. 2D, and see Fig. S2 and S3 in the supplemental material). All reconstructions of the mature capsids reveal at least four concentric layers of DNA (see Fig. S3 and Table S1 in the supplemental material), equally spaced by 24.5 Å, as seen previously for other phages (10, 20, 33). The observation that only peripheral DNA layers are found in averaged images and 3D reconstructions shows that DNA has a different or variable organization at more central positions in the capsid. This feature is common to phages that do not have a central core to organize DNA spooling during viral genome packaging (34).
The novel thermal stability assays used here to assess the release of DNA from SPP1 capsids and for capsid protein denaturation showed that DNA-filled capsids withstand temperatures above 70°C and that the gp13 fold is stabilized in the capsid assembled state. The disruption of the capsid with the concomitant release of DNA at a Tdis of ∼71°C (Fig. 3A) was rapidly followed by a highly cooperative unfolding of gp13 (melting temperature [Tm], ∼75°C) (Fig. 3B). The predisruption of DNA-filled capsids by the chelation of divalent cations with EDTA, which occurs at temperatures below 40°C (51) (Fig. 3A, filled circles), showed a less cooperative and lower-Tm gp13 denaturation behavior (Fig. 3B). The structure of the major capsid protein is thus stabilized in the DNA-filled capsid. Remarkably, the infectivity of SPP1 particles exhibited a temperature dependence very similar to that observed for capsid integrity (Fig. 3C). This implies a high stability of all the components that are essential for the infectious viral particle: the DNA-filled capsid; the tail, responsible for DNA delivery to the host; and the head-to-tail interface between the two structures. This situation differs from that of phage HK97 particles, which lose DNA at temperatures above 50°C, but their covalently cross-linked capsid resists temperatures above 90°C, as assessed by calorimetry studies (17).
Auxiliary proteins of tailed phages and herpesviruses are structurally diverse, in contrast to the highly conserved HK97 fold of their major capsid proteins. The wealth of data suggests that intercapsomer bonding is a structurally weak point in viral capsids of the tailed-phage–herpesvirus lineage that calls for reinforcement strategies as the binding of auxiliary cementing proteins at the 2- and 3-fold axes of the capsid (see blue shading in Table S2 in the supplemental material). In contrast, no function in capsid stability was found for auxiliary proteins that bind to the center of hexamers, like gp12 of SPP1 (see green shading in Table S2 in the supplemental material). This likely reflects that the intracapsomer bonding of HK97-like major capsid proteins is strong, being exploited by the phage to attach auxiliary proteins at the center of their hexamers, which will be exposed at the capsid surface in a regular icosahedral arrangement. Such a display strategy might provide adhesion properties to viral particles for nonspecific attachment to bacteria and other surfaces or may serve other functions yet to be established. A potential adhesion role of gp12 would be to adsorb nonspecifically the phage particles onto surfaces until an interaction with a suitable host is possible, rather than promoting infection. The specific and strong binding to the identified host B. subtilis, priming infection, is mediated by the tail adsorption apparatus protein gp21 (55), localized at the distal tail end, nearly 160 nm away from the capsid in viral particles.
The subnanometer resolution of the SPP1 FP capsid allowed the segmentation of individual subunits in the EM maps whose densities showed structural similarity to the HK97 fold (Fig. 6, and see Fig. S5 in the supplemental material). Segmentation, homology modeling, and subsequent flexible fitting were used to construct a pseudoatomic model accounting for most of the gp13 polypeptide chain fold in the seven subunits found in the capsid asymmetric unit (Fig. 6C). The application of icosahedral symmetry to this unit led to a model of the T=7 capsid containing 420 subunits of gp13. The gp13 fold exhibits the features conserved among major capsid proteins having an HK97 fold, with its “A domain” building the center of capsomers and the “P domain” carrying out intercapsomer contacts around the 3-fold symmetry axes (Fig. 6). This core gp13 fold is closely related to those of major capsid proteins of phages HK97 (22, 57) and T7 (26), besides some differences in quasiequivalent interactions and in intersubunit contacts among A domains of the same capsomer and P domains of different capsomers. The electron density of gp13 in the SPP1 capsid reconstruction, like that of gp10A of phage T7 (1, 26), does not account for the complete length of the HK97-fold “E loop” (Fig. 6B and C, and see Fig. S5 in the supplemental material). An extra unassigned density underneath and connected to the E loop protrudes toward the P-domain end of the neighbor subunit of the same capsomer (Fig. 6E and F, purple ovals). Such a density is also found in T7 and crosses over the adjacent P domain. At the present resolution, we cannot establish if the gp13 polypeptide chain reaches the P domain of the subunit from another capsomer, as was found previously for intercapsomer interactions between the E loop and P domain leading to formation of covalent cross-links in HK97 (57) and strong electrostatic interactions in phage epsilon 15 (29). The most extensive intercapsomer densities in SPP1 are established between the extremity of the gp13 P domain and the large base of the P domain from the subunit of a neighboring capsomer. The contact area extends to the subunit E-loop-anchoring point, where an additional density between capsomers is found. Interestingly, this region is marked in phage SSP7 by the presence of the “F loop,” engaged in intercapsomer interactions (36).
The gp13 polypeptide chain is 36 amino acids longer than the mature HK97 major capsid protein, but these additional amino acids are found in short insertions distributed between the landmark secondary structure elements of the HK97 fold and at the protein termini rather than by the formation of an additional domain (see Fig. S4 in the supplemental material). The SPP1 major capsid protein (324 amino acids) thus represents a variation of the basic fold that uses neither chemical intersubunit cross-linking nor elaborated additional domains to build a highly stable capsid. SPP1 combines a “classical” HK97 major capsid fold, whose organization is maintained throughout the late steps of viral assembly and after genome release, with a new type of auxiliary protein. The synergy of the capsid and other phage components ensures thermostability and a high robustness of the overall infective viral particle. Near-atomic-resolution information will be required to uncover the subtle structural features that cement the intercapsomer network of contacts to withstand high internal pressure (>47 atm [47]) and high temperature (65°C) (Fig. 3). Although SPP1 has not developed the sophisticated cross-linking chemistry and chainmail found for HK97, the SPP1 infectious particle is 15°C more thermostable than the HK97 virions because of a remarkably stable combination of the virion components. The weak links between the HK97 components allow DNA leakage at much lower temperatures in spite of the robustness of its capsid. Although the phage particles are rarely challenged with temperatures of around 65°C, the thermostability of the SPP1 virions might reflect a more general feature of the phages important for resistance to different harsh environmental conditions. Virus particles are designed to be highly stable, resisting aggression in the environment when standing (sometimes for long periods) in the soil, in water, inside a multicellular organism, or in other environments while waiting to infect the host. Thus, there is a clear advantage to being resistant to various environmental components such as pH, temperature, and salt. B. subtilis is a bacterium of the soil that can grow and become infected very efficiently at temperatures close to 46°C. The structural stability of different components of the SPP1 virion is a likely driving force of virus particle design throughout evolution so that a virion can withstand large physicochemical variations in the environment during the frequently long periods that it has to wait in order to infect a suitable host.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to R. Lurz (MPI-MG, Berlin, Germany) for the electron micrographs shown in Fig. 3. We thank D. Houldershaw and R. Westlake for computer support. We acknowledge Mohamed Zairi for communication of unpublished results and discussions on gp12 structure and function. We thank Asita C. Stiege for anti-gp12 serum.
P.S. was funded by the Wellcome Trust (grant to A. A. Antson, York University). We acknowledge the BBSRC (BB/F012705/1, EVO), CNRS, and ANR-06-BLAN-0168 (P.T.) for financial support.
Footnotes
Published ahead of print 18 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Agirrezabala X, et al. 2007. Quasi-atomic model of bacteriophage t7 procapsid shell: insights into the structure and evolution of a basic fold. Structure 15:461–472 [DOI] [PubMed] [Google Scholar]
- 2. Alonso JC, Tavares P, Lurz R, Trautner TA. 2006. Bacteriophage SPP1, p 331–349 In Calendar R. (ed), The bacteriophages. Oxford University Press, New York, NY [Google Scholar]
- 3. Baker ML, Jiang W, Rixon FJ, Chiu W. 2005. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 79:14967–14970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bamford DH, Grimes JM, Stuart DI. 2005. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 15:655–663 [DOI] [PubMed] [Google Scholar]
- 5. Becker B, et al. 1997. Head morphogenesis genes of the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 268:822–839 [DOI] [PubMed] [Google Scholar]
- 6. Benson SD, Bamford JK, Bamford DH, Burnett RM. 1999. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell 98:825–833 [DOI] [PubMed] [Google Scholar]
- 7. Bryson K, et al. 2005. Protein structure prediction servers at University College London. Nucleic Acids Res. 33:W36–W38 doi:10.1093/nar/gki410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Camacho AG, Gual A, Lurz R, Tavares P, Alonso JC. 2003. Bacillus subtilis bacteriophage SPP1 DNA packaging motor requires terminase and portal proteins. J. Biol. Chem. 278:23251–23259 [DOI] [PubMed] [Google Scholar]
- 9. Casjens S, Hendrix R. 1988. Control mechanisms in dsDNA bacteriophage assembly, p 15–91 In Calendar R. (ed), The bacteriophages, vol 1 Oxford University Press, New York, NY [Google Scholar]
- 10. Cerritelli ME, et al. 1997. Encapsidated conformation of bacteriophage T7 DNA. Cell 91:271–280 [DOI] [PubMed] [Google Scholar]
- 11. Cheng J, Randall AZ, Sweredoski MJ, Baldi P. 2005. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 33:W72–W76 doi:10.1093/nar/gki396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cole C, Barber JD, Barton GJ. 2008. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36:W197–W201 doi:10.1093/nar/gkn238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conway JF, et al. 2001. Virus maturation involving large subunit rotations and local refolding. Science 292:744–748 [DOI] [PubMed] [Google Scholar]
- 14. de Frutos M, Brasiles S, Tavares P, Raspaud E. 2005. Effect of spermine and DNase on DNA release from bacteriophage T5. Eur. Phys J. E Soft Matter 17:429–434 [DOI] [PubMed] [Google Scholar]
- 15. Dröge A. 1998. Capsidmorphogenese des BakteriophagenSPP1. Tecnical University, Berlin, Berlin, Germany [Google Scholar]
- 16. Droge A, Tavares P. 2000. In vitro packaging of DNA of the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 296:103–115 [DOI] [PubMed] [Google Scholar]
- 17. Duda RL, et al. 2009. Structure and energetics of encapsidated DNA in bacteriophage HK97 studied by scanning calorimetry and cryo-electron microscopy. J. Mol. Biol. 391:471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Effantin G, Boulanger P, Neumann E, Letellier L, Conway JF. 2006. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361:993–1002 [DOI] [PubMed] [Google Scholar]
- 19. Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. 2006. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357:289–298 [DOI] [PubMed] [Google Scholar]
- 20. Fokine A, et al. 2005. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. U. S. A. 102:7163–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu CY, Prevelige PE., Jr 2009. In vitro incorporation of the phage Phi29 connector complex. Virology 394:149–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gertsman I, et al. 2009. An unexpected twist in viral capsid maturation. Nature 458:646–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harauz G, van Heel M. 1986. Exact filters for general geometry three dimensional reconstruction. Optik 73:146–156 [Google Scholar]
- 24. Harrison SC. 2001. The familiar and the unexpected in structures of icosahedral viruses. Curr. Opin. Struct. Biol. 11:195–199 [DOI] [PubMed] [Google Scholar]
- 25. Helgstrand C, et al. 2003. The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 A resolution. J. Mol. Biol. 334:885–899 [DOI] [PubMed] [Google Scholar]
- 26. Ionel A, et al. 2011. Molecular rearrangements involved in the capsid shell maturation of bacteriophage T7. J. Biol. Chem. 286:234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Isidro A, Henriques AO, Tavares P. 2004. The portal protein plays essential roles at different steps of the SPP1 DNA packaging process. Virology 322:253–263 [DOI] [PubMed] [Google Scholar]
- 28. Jakutyte L, et al. 2011. Bacteriophage infection in rod-shaped Gram-positive bacteria: evidence for a preferential polar route for phage SPP1 entry in Bacillus subtilis. J. Bacteriol. 193:4893–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang W, et al. 2008. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature 451:1130–1134 [DOI] [PubMed] [Google Scholar]
- 30. Jiang W, Baker ML, Ludtke SJ, Chiu W. 2001. Bridging the information gap: computational tools for intermediate resolution structure interpretation. J. Mol. Biol. 308:1033–1044 [DOI] [PubMed] [Google Scholar]
- 31. Jiang W, et al. 2003. Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat. Struct. Biol. 10:131–135 [DOI] [PubMed] [Google Scholar]
- 32. Lander GC, et al. 2008. Bacteriophage lambda stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryo-EM. Structure 16:1399–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lander GC, et al. 2006. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 312:1791–1795 [DOI] [PubMed] [Google Scholar]
- 34. Leforestier A, Livolant F. 2010. The bacteriophage genome undergoes a succession of intracapsid phase transitions upon DNA ejection. J. Mol. Biol. 396:384–395 [DOI] [PubMed] [Google Scholar]
- 35. Lhuillier S, et al. 2009. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc. Natl. Acad. Sci. U. S. A. 106:8507–8512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X, et al. 2010. Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat. Struct. Mol. Biol. 17:830–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128:82–97 [DOI] [PubMed] [Google Scholar]
- 38. Morais MC, et al. 2005. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of phi29. Mol. Cell 18:149–159 [DOI] [PubMed] [Google Scholar]
- 39. Oliveira L, Alonso JC, Tavares P. 2005. A defined in vitro system for DNA packaging by the bacteriophage SPP1: insights into the headful packaging mechanism. J. Mol. Biol. 353:529–539 [DOI] [PubMed] [Google Scholar]
- 40. Orlov IM, Morgan DG, Cheng RH. 2006. Efficient implementation of a filtered back-projection algorithm using a voxel-by-voxel approach. J. Struct. Biol. 154:287–296 [DOI] [PubMed] [Google Scholar]
- 41. Orlova EV, et al. 2003. Structure of a viral DNA gatekeeper at 10 A resolution by cryo-electron microscopy. EMBO J. 22:1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ouali M, King RD. 2000. Cascaded multiple classifiers for secondary structure prediction. Protein Sci. 9:1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parent KN, et al. 2010. P22 coat protein structures reveal a novel mechanism for capsid maturation: stability without auxiliary proteins or chemical crosslinks. Structure 18:390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pettersen EF, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 45. Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. 2010. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 170:427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333:721–745 [DOI] [PubMed] [Google Scholar]
- 47. Sao-Jose C, de Frutos M, Raspaud E, Santos MA, Tavares P. 2007. Pressure built by DNA packing inside virions: enough to drive DNA ejection in vitro, largely insufficient for delivery into the bacterial cytoplasm. J. Mol. Biol. 374:346–355 [DOI] [PubMed] [Google Scholar]
- 48. Sherman MB, Weaver SC. 2010. Structure of the recombinant alphavirus Western equine encephalitis virus revealed by cryoelectron microscopy. J. Virol. 84:9775–9782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith DE, et al. 2001. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature 413:748–752 [DOI] [PubMed] [Google Scholar]
- 50. Tang L, Gilcrease EB, Casjens SR, Johnson JE. 2006. Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure 14:837–845 [DOI] [PubMed] [Google Scholar]
- 51. Tavares P, Lurz R, Stiege A, Ruckert B, Trautner TA. 1996. Sequential headful packaging and fate of the cleaved DNA ends in bacteriophage SPP1. J. Mol. Biol. 264:954–967 [DOI] [PubMed] [Google Scholar]
- 52. Thuman-Commike PA, et al. 1999. Mechanism of scaffolding-directed virus assembly suggested by comparison of scaffolding-containing and scaffolding-lacking P22 procapsids. Biophys. J. 76:3267–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Topf M, et al. 2008. Protein structure fitting and refinement guided by cryo-EM density. Structure 16:295–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. 1996. A new generation of the IMAGIC image processing system. J. Struct. Biol. 116:17–24 [DOI] [PubMed] [Google Scholar]
- 55. Vinga I, et al. 2012. Role of bacteriophage SPP1 tail spike protein gp21 on host cell receptor binding and trigger of phage DNA ejection. Mol. Microbiol. 83:289–303 [DOI] [PubMed] [Google Scholar]
- 56. Vinga I, et al. 2006. The minor capsid protein gp7 of bacteriophage SPP1 is required for efficient infection of Bacillus subtilis. Mol. Microbiol. 61:1609–1621 [DOI] [PubMed] [Google Scholar]
- 57. Wikoff WR, et al. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129–2133 [DOI] [PubMed] [Google Scholar]
- 58. Wommack KE, Colwell RR. 2000. Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64:69–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.