

Abstract
Bluetongue virus (BTV) is transmitted by biting midges (Culicoides spp.). It causes disease mainly in sheep and occasionally in cattle and other species. BTV has spread into northern Europe, causing disease in sheep and cattle. The introduction of new serotypes, changes in vector species, and climate change have contributed to these changes. Ten BTV serotypes have been isolated in Australia without apparent associated disease. Simplified methods for preferential isolation of double-stranded RNA (dsRNA) and template preparation enabled high-throughput sequencing of the 10 genome segments of all Australian BTV prototype serotypes. Phylogenetic analysis reinforced the Western and Eastern topotypes previously characterized but revealed unique features of several Australian BTVs. Many of the Australian BTV genome segments (Seg-) were closely related, clustering together within the Eastern topotypes. A novel Australian topotype for Seg-5 (NS1) was identified, with taxa spread across several serotypes and over time. Seg-1, -2, -3, -4, -6, -7, -9, and -10 of BTV_2_AUS_2008 were most closely related to the cognate segments of viruses from Taiwan and Asia and not other Australian viruses, supporting the conclusion that BTV_2 entered Australia recently. The Australian BTV_15_AUS_1982 prototype was revealed to be unusual among the Australian BTV isolates, with Seg-3 and -8 distantly related to other BTV sequences from all serotypes.
INTRODUCTION
Bluetongue virus (BTV; type 20) was first detected in Australia when isolated from Culicoides (Diptera: Ceratopogonidae) collected in northern Australia in 1975. Ten BTV serotypes (1, 2, 3, 7, 9, 15, 16, 20, 21, and 23) of the described 26 serotypes have been detected in Australia through national surveillance programs (1, 9–11, 15, 17, 27, 28). Regular occurrence of BTV 1 and 21 has been documented in northern Western Australia, the Northern Territory, Queensland, and the northeastern areas of New South Wales. Long-term monitoring has shown that, in spite of the geographic distribution of BTV across northern and eastern Australia, disease has remained absent for the past 30 years. The pattern of sporadic detection of some serotypes and the occasional appearance of new serotypes suggests regular introductions of BTV into Australia from southeast Asia via windborne Culicoides (7). BTV 2 (1) has recently (2008) been detected in northern and eastern Australia, including regions in which only BTV 1 and 21 had been previously recorded.
In Europe, virus and disease have spread into the northern parts of the continent where they were previously absent. This suggests that the apparent stable pattern of the absence of bluetongue disease in Australia could be fragile. Factors that have influenced the changes observed in Europe are the appearance of previously exotic serotypes (BTV 8 in 2006, BTV 6 in 2008, and BTV 11 in 2009), changes in the distribution and competence of Culicoides species, and climate change. BTV 8 in Europe is unusual in that cattle have developed clinical disease (6, 8). The spread of several serotypes into Europe may also favor the emergence of new variants by genome segment reassortment. The routes of entry of these BTV serotypes into northern and western Europe have not been defined, even though detailed tracing of animal movements, analysis of virus genome sequences, and modeling of meteorological conditions suitable for potential vector movement have been undertaken (30).
BTV is classified in the genus Orbivirus in the family Reoviridae. It has a 10-segment double-stranded RNA (dsRNA) genome. Genetic diversity is generated by genome segment reassortment and mutation and selection leading to genetic shift and genetic drift, respectively (5). Intrasegment recombination has been described as an additional mechanism for the generation of genetic diversity in BTV (14). Genotype/topotype analysis of Australian BTV isolates has relied upon the comparison of sequences from Seg-10 (690-bp region of the NS3 coding region) and Seg-3 (540-bp region of the VP3 coding region) and conventional serotyping (13, 24). While those analyses have provided some insights into the relationships and likely origins of BTVs in Australia, the lack of a comprehensive strategy for determining the sequence of all genomic segments of isolates means that genetic flows associated with reassortment would be undetected. This paper describes simple, reliable, and high-throughput methods for determining genome sequences from multiple BTV isolates and their application to the first comprehensive analysis of Australian BTV prototype serotypes. This analysis has generated novel observations on the relationship of Australian BTVs to those in Asia and the rest of the world and provides evidence for entry of BTV serotypes into Australia in recent times. Notably, this publication almost doubles the number of BTV isolates for which there is sequence determination for each of the 10 genome segments.
MATERIALS AND METHODS
Cell line and viruses.
BTVs were cultured using BHK-21 BSR cells and Eagle's basal medium (BME) supplemented with 2.5% or 5% fetal bovine serum at 37°C or using C6/36 (Aedes albopictus) cells in M199 medium supplemented with 10% fetal bovine serum at 28°C. Virus stocks were stored at 4°C or −80°C. The origins and passage histories of BTV isolates are listed in Table 1. The earliest available isolate for each BTV serotype with a documented passage history was used to ensure that it was as close as possible to the original field material. Nonetheless, many of the viruses had undergone extensive passage.
Table 1.
Australian BTV prototype serotypes subjected to genome sequencinga
| Serotype | Isolate (reference)b | Source and location where isolate was collected | Longitude | Latitude | Isolation method | Passage history | Sample | Yr of collection |
|---|---|---|---|---|---|---|---|---|
| BTV 1c | CSIRO156 (27) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Cell culture | 1 × BHK/3 × PPVero/3 × BHK/4 × Vero/2 × BHK/1 × BHKBSR | Cattle blood | 1979 |
| BTV 2 | DPP7291 (1) | Douglas Daly, NT | 131o11′E | 13o49′S | Cell culture | At least 3 × BHKBSR | Cattle blood | 2008 |
| BTV 3 | DPP973 (11) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Egg embryo | Egg/2 × sheep/Egg/4 × BHK | Cattle blood | 1986 |
| BTV 7 | DPP6963 (15) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Egg embryo | 1 × Egg/3 × BHK/BSR | Cattle blood | 2007 |
| BTV 9 | DPP0837 (10) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Cell culture | 3 × BHK21/2 × BHKBSR | Cattle blood | 1985 |
| BTV 15 | DPP192 (9) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Cell culture | 6 × BHK21/4 × BHK/BSR | Cattle blood | 1982 |
| BTV 16 | DPP96 (11) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Egg embryo | 2 × Egg/2 × SV/1 × BHK/BSR | Cattle blood | 1986 |
| BTV 20 | CSIRO19 (28) | Beatrice Hill/CPRS, NT | 131o20′E | 12o39′S | Cell culture | 1 × mouse/17 × BHK | Culicoides mixed species | 1977-1978 |
| BTV 21 | DPP86 (27) | VRRS, NT | 131o17′E | 17o2′S | Cell culture | 3 × BHK21/2 × BHK/BSR | Cattle blood | 1979 |
| BTV 23 | DPP90 (9) | TFRF, NT | 131o08′E | 13o13′S | Cell culture | 6 × BHK/2 × Vero/3 × BHK/3 × mouse/2 × BHKBSR | Cattle blood | 1982 |
NT, Northern Territory, Australia. BHK, baby hamster kidney cell culture; BHK/BSR, BSR line of BHK cells; Vero, Vero cell culture; CPRS, Coastal Plains Research Station; VRRS, Victoria River Research Station; TFRF, Tortilla Flats Research Farm.
GenBank and Sequence Read Archive accession numbers are listed in Table S1 in the supplemental material.
The cDNA template for sequencing was prepared using the FLAC technique (21).
Purification of BTV dsRNA.
Two 150-cm2 cultures of BHK-21 BSR or C6/36 cells at 70% to 80% cell confluence were infected at a multiplicity of infection of 0.1 50% tissue culture infective dose (TCID50) per cell. The infected cells were harvested 4 to 5 days postinfection at a cytopathic effect level of 50% to 70%. The cell pellet was dissolved in TRIzol LS reagent (Invitrogen) and total RNA extracted. dsRNA was prepared using differential precipitation of ssRNA and 2 M LiCl and recovery of the dsRNA from the supernatant by precipitation with isopropanol (23). The integrity of the dsRNA was evaluated after separation on a 1% agarose gel (Tris-acetate-EDTA [TAE]) containing SYBR Safe (Invitrogen) (1:10,000).
Preparation of BTV ds cDNA.
dsRNA was DNase I digested, heat denatured in the presence of 15% dimethyl sulfoxide (DMSO), and snap cooled. cDNA was reverse transcribed using the Superscript III system (Invitrogen) in the presence of 50 ng of random hexamers and 0.5 ng of primers specific to the ends of the BTV genome segments (5′ GTTAAAN 3′ and 5′′ GTAAGTN 3′) (22). cDNA was purified after digestion with RNase H and RNase I. ds cDNA was prepared by treatment of the cDNA with the NEB Next End Repair Module. The concentration of dsDNA was measured using a Quant-IT dsDNA HS assay kit (Invitrogen) and measured on a Qubit fluorometer (Invitrogen).
High-throughput genomic sequencing by multiplexed Illumina.
The cDNA samples of BTV genomic material were prepared for sequencing using the TruSeq (Illumina) protocols and standard multiplex adaptors available in March 2011. A paired-end, 100- or 150-base-read protocol was used for sequencing on an Illumina GAIIx instrument.
Sequence assembly.
Low-quality reads were removed prior to assembly; filtering excluded reads with one or more Ns and those reads with regions of quality of less than Q20. De novo assemblies were performed using Velvet (31). Read mapping was performed using SHRiMP (25) as implemented in NESONI (http://bioinformatics.net.au/software.nesoni.shtml). A combination of read mapping and de novo assembly methods was used to construct a consensus sequence for each genome segment of each of the 10 BTV isolates. Read sets were deposited in the Sequence Read Archive and final consensus sequences submitted to GenBank (see Table S1 in the supplemental material).
Phylogenetic and bioinformatics analysis.
BTV sequences containing at least the coding region of a segment were downloaded from GenBank on 7 December 2011. The following convention was used to identify sequences—BTV_serotype number_three-letter country code_year of isolation_numerical identifier of sequence as listed in Table S1 in the supplemental material. ISO 3166-1 alpha-3 was used for the three letter country code. Where a country or date of isolation was not available, XXX or 9999, respectively, was used. Isolates and associated GenBank entries are listed in Table S1 in the supplemental material. Alignments and phylogenetic analyses were performed using the MEGA5 suite of programs (29). MEGA was used to find the best-fit substitution model for each genome segment. The model with the lowest Bayesian Information Criterion value was used to undertake a maximum-likelihood analysis with 1,000 bootstraps. Where appropriate, the nomenclature for the geographically based phylogenetic groups was assigned as described by Maan et al. (18, 20).
Nucleotide sequence accession numbers.
The nucleotide sequences determined in this work are available under GenBank accession numbers JN881985 to JN881994, JQ086221 to JQ086310, and JQ240321 to JQ240330. The Sequence Read Archive accession number is SRA030511.
RESULTS
BTV genomic cDNA preparation and sequencing.
The method produced BTV cDNA with a quantity (from 250 ng to 2,000 ng per 2 × 150-cm2 flasks, depending upon the extent of growth of the strain) and quality suitable for input into the Illumina sequencing platform. Inclusion of primers targeting the conserved 5′ and 3′ ends of the BTV genome segments (22) improved the lower coverage routinely observed toward the ends of the genomic segments (data not shown). Near-full-length sequence was generated for all of the Australian BTV prototype serotype genome segments (see Table S1 in the supplemental material). The sequence for BTV_1_AUS_1979_23 (CSIRO 156) reported here was generated from cDNA template prepared using the FLAC technique (21) and sequenced on the Illumina platform.
BTV outer capsid protein genes: Seg-2 (VP2) and Seg-6 (VP5).
VP2 (950 to 961 amino acids [aa], depending upon serotype) (19) and VP5 (526 aa), comprising the outer capsid proteins of the mature virion, are encoded by Seg-2 and Seg-6, respectively. Together, Seg-2 and Seg-6 encode the proteins that define the BTV serotype, with the major contribution coming from VP2. Analysis of the 10 nucleotide sequences encoding VP2 of the Australian prototype serotypes confirmed their identity by correlation with existing serotype lineages. Figure S2 in the supplemental material shows the 10 Australian prototype sequences within the 10 nucleotypes (A to J). Previously, nucleotypes were defined as having <35% difference in the Seg-2 nucleotide sequence (19). It was possible to define Western and Eastern topotypes within Seg-2 for some serotypes.
The sequences of Seg-6 have been shown to form a number of distinct nucleotypes that have been designated A to F (16, 26) and have been associated with particular serotypes, although this is not an absolute relationship (18, 19, 26) (see Fig. S6 in the supplemental material). There is evidence for Western and Eastern topotypes (e.g., BTV 1, 2, 9, and 23) within Seg-6 nucleotypes, but these relationships are based upon small numbers of sequences and the relationships are complicated by the association of Seg-6 nucleotypes with more than one serotype. The Seg-6 sequences from the 10 Australian prototype serotypes fell into the already defined nucleotypes (see Fig. S6 in the supplemental material).
BTV core protein genes: core surface Seg-7 (VP7).
Seg-7 encodes the serogroup antigen VP7, which is a 349-amino-acid protein located on the surface of the core particle. The addition of the 10 Australian BTV VP7 sequences confirmed and strengthened the identification of the six major topotypes previously assigned—Western 1, 2, and 3 and Eastern 1, 2, and 3 (20). The Australian VP7 sequences clustered in Eastern topotypes 1, 2, and 3 (Fig. 1; see also Fig. S3 in the supplemental material). Six Australian VP7 sequences were located in Eastern topotype 2; of these, five (BTV 1, 9, 20, 21, and 23) clustered closely together. In contrast, BTV_2_AUS_2008_24 clustered with viruses from Japan, Taiwan, and China. Eastern topotype 3 contained viruses from China, India, and Italy along with BTV_16_AUS_1986_29 and BTV_3_AUS_1986_25. Eastern topotype 1 was distantly related to all of the other groups. This topotype contained a disparate group of sequences: those of BTV_15_AUS_1982_28, BTV_7_AUS_2007_26, and vaccine strains for BTV 7, 15, and 19 from South Africa.
Fig 1.

Phylogenetic tree showing relationship between the nucleotide coding regions for Seg-7 (VP7) for all serotypes. Australian sequences: ●, prototype; ○, nonprototype.
BTV core protein genes: subcore Seg-1 (VP1), Seg-3 (VP3), Seg-4 (VP4), and Seg-9 (VP6).
The BTV subcore protein genes are the most conserved of those encoding BTV structural proteins. They provide essential structural or replication functions for the virus—VP1 polymerase (1,302 aa), VP3 structural component (901 aa), VP4 capping enzyme (644 aa), and VP6 helicase (329 aa). A new nonstructural protein, NS4, that is encoded by Seg-9 has recently been identified (3). Nonetheless, it was possible to identify Western and Eastern topotypes aligned with the geographic origins of the virus isolates (20). The addition of 10 full coding sequences for each of these genes to those already in the databases confirmed the Western and Eastern topotypes (Fig. 2; see also Fig. S1, S3, S4, and S9 in the supplemental material). All of the Australian prototype serotypes (with the exception of Seg-3 BTV_15_AUS_1982_28) fell within the Eastern topotype (Fig. 2). BTV-15_AUS_1982_28 Seg-3 was a single member of a topotype distinct from the Western and Eastern topotypes. BTV-2_AUS_2008_24 Seg-1, -3, -4, and -9 (Fig. 2A, b, c, and d, respectively) were more closely allied to sequences from BTV_2_TWN_2003_4, BTV_12_TWN_2003_17, and other Asian viruses than to the Australian viruses. The other Australian BTV genome segments clustered together within the Eastern topotypes.
Fig 2.

Phylogenetic trees showing relationships between the nucleotide coding regions for (a) Seg-1 (VP1), (b) Seg-3 (VP3), (c) Seg-4 (VP4), and (d) Seg-9 (VP6) for all serotypes. Australian sequences: ●, prototype; ○, nonprototype.
BTV nonstructural protein genes: Seg-5 (NS1), Seg-8 (NS2), and Seg-10 (NS3).
Seg-5 encodes the NS1 protein (553 aa), which accumulates in the cytoplasm of infected cells as tubules. NS1 function is unknown. Previous phylogenetic analysis of Seg-5 identified distinct Western and Eastern topotypes (18). Addition of sequences from the 10 Australian prototype serotypes identified a third distinctly Australian topotype (including BTV 1, 7, 9, 20, and 21) (Fig. 3; see also Fig. S5 in the supplemental material). The other five Australian BTV NS1 sequences (BTV 2, 3, 15, 16, and 23) clustered within the previously identified Eastern topotype, which includes viruses from Taiwan and the Middle East through to Italy and from South Africa. Within this topotype, three of the Australian NS1 sequences (BTV 3, 15, and 23) clustered very closely together, with BTV 2 and 16 more distantly related. Although BTV_2_AUS_2008_24 was an outlier in this topotype, it did not cluster with BTV_2 and _12_TWN_2003 isolates, in contrast to the apparent relationships of BTV_2_AUS_2008_24 to BTV_2 and BTV_12_TWN_2003 revealed by the analyses of Seg-1, -3, -4, and -9. BTV_16_AUS_1986_29 clustered with viruses from Taiwan and the Middle East through to Italy and from South Africa.
Fig 3.

Phylogenetic tree showing relationship between the nucleotide coding regions for Seg-5 (NS1) for all serotypes. Australian sequences: ●, prototype; ○, nonprototype.
Seg-8 encodes the highly conserved viral inclusion body matrix protein NS2 (354 aa). Inclusion of the 10 Australian NS2 sequences reinforced the division into Western and Eastern topotypes (Fig. 4; see also Fig. S8 in the supplemental material). Nine of the 10 Australian BTV NS2 sequences aligned in the Eastern topotype, which contained two possible clades—an Australian clade containing BTV 1, 2, 3, 7, 9, 16, 20, 21, and 23 and a Middle East-to-Asian clade. BTV_15_AUS_1982_28 was an exception with respect to the Eastern topotype (Fig. 4). BTV_15_AUS_28 Seg-8 was as a deep-branching member of the Western topotype.
Fig 4.

Phylogenetic tree showing relationship between the nucleotide coding regions for Seg-8 (NS2) for all serotypes. Australian sequences: ●, prototype; ○, nonprototype.
Seg-10 encodes the NS3 (229 aa) and NS3a (216 aa) proteins. This segment is highly conserved, with nucleotide sequence identity > 80% across all of the isolates. Analysis of the 10 Australian sequences confirmed the existence of three major topotypes—Western 1 and 2 and Eastern 1 (Fig. 5; see also Fig. S10 in the supplemental material). A second Eastern topotype was proposed previously but to date contains only a single virus sequence—that of BTV_15_CHN_1996_304 (18). All of the Australian Seg-10 sequences fell into the Eastern 1 topotype. BTV 1, 3, 9, and 23 aligned closely along with BTV_21 in a clade encompassing predominantly Australian viruses. BTV_2_AUS_2008_24 once again aligned more closely with viruses from Taiwan and Asia. BTV_15_AUS_1982_28 and BTV_16_AUS_1986_29 formed a clade with several sequences from Asia but were distant from the clade in which BTV_2_AUS_2008_24 aligned.
Fig 5.
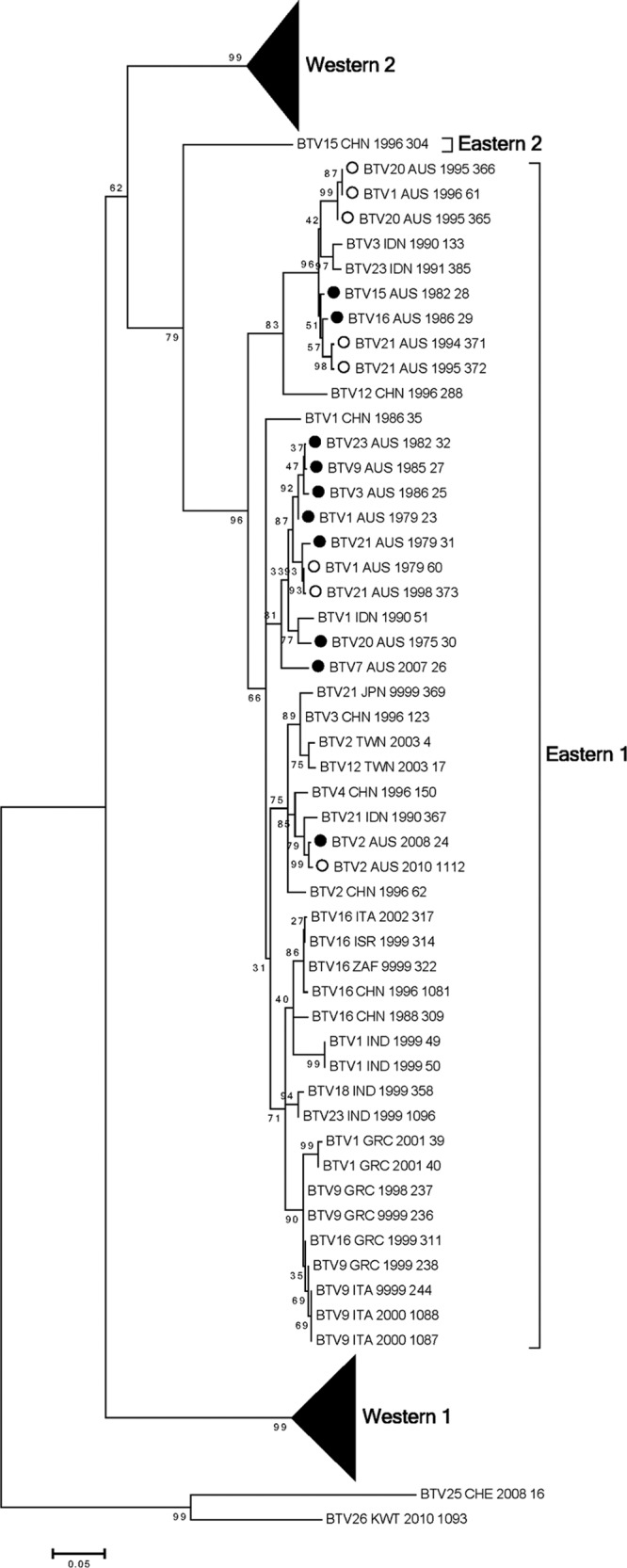
Phylogenetic tree showing relationship between the nucleotide coding regions for Seg-10 (NS3) for all serotypes. Australian sequences: ●, prototype; ○, nonprototype.
BTV2_AUS_2008.
The first isolation of BTV 2 in Australia occurred in 2008 from a healthy sentinel bovine in the Northern Territory of Australia (Table 1). Analysis of the 10 genome segments of this virus in comparison with the other Australian prototype serotypes supported the hypothesis of entry into Australia in the recent past. For Seg-1, -2, -3, -4, -6, -7, -9, and -10, there was strong support for the idea of BTV_2_AUS_2008_24 being closely related to viruses in Taiwan and elsewhere in Asia (Fig. 1, 2, and 5; see also Fig. S1, S2, S3, S4, S6, S7, S9, and S10 in the supplemental material). Seg-5 (NS1) and -8 (NS2) of BTV_2_AUS_2008_24 clustered with sequences from the other Australian prototype serotypes, although they tended to be deep-branching members of these groups (Fig. 3 and 4; see also Fig. S5 and S8 in the supplemental material).
BTV_15_AUS_1982.
BTV 15 has been isolated infrequently in Australia. Analysis of the genome segments of BTV_15_AUS_1982_28 provided some insights into its possible origins but also raised some significant questions. Analysis of Seg-1, -4, -5, -7, -9, and -10 placed BTV_15_AUS_1982_28 in the Eastern topotypes defined previously (18) (Fig. 1, 2, 3, and 5; see also Fig. S1, S4, S5, S7, S9, and S10 in the supplemental material). Seg-3 (VP3) and Seg-8 (NS2) placed BTV_15_AUS_1982_28 in distinctly different groups (Fig. 2B and 4; see also Fig. S3 and S8 in the supplemental material). For Seg-3, BTV_15_AUS_1982_28 was an isolated sequence distantly related to the Western and Eastern topotypes. Surprisingly, Seg-8 (NS2) of BTV_15_AUS_1982_28 aligned within the Western topotype, in marked contrast to all of the other Australian prototype serotypes. It was notable that BTV_15_CHN_1996_304 Seg-10 did not fall into the readily defined Western 1 and 2 and Eastern topotypes (Fig. 5; see also Fig. S10 in the supplemental material). BTV_15_AUS_1982_28 Seg-10 was not related to Seg-10 from BTV_15_CHN_1996_304.
DISCUSSION
Elucidation of all coding sequences for all of the genomic segments of Australian prototype serotypes of BTV has allowed a detailed analysis of their relationships to viruses from other areas of the world. Notably, this report almost doubles the number of BTV isolates for which there is a genome sequence determined for each of the 10 segments. Australia lies at a geographic extremity of BTV distribution, with BTV serotypes regularly detected in northern Australia and BTV 1, 21, and now 2 occurring in eastern Australia (9–11, 15, 27, 28); so far, disease has not been detected in commercial livestock.
Genome segments of Australian viruses largely fitted into the previously defined Eastern topotype(s) (18). The relationships for each of Seg-1, -3, -4, -8, and -9 are remarkably similar, each with two topotypes—Eastern and Western. The evolutionary distance between the Eastern and Western topotypes inferred for each of these segments indicates significant geographically independent evolution of the segments. The notable exception is BTV_15_AUS_1982, for which Seg-3 falls outside the Eastern and Western topotypes and Seg-8 aligns within the Western topotype. Within these broad Western and Eastern topotypes, it was possible to identify clades which may have associations with smaller geographic regions, e.g., Seg-1, -3, -4, -5, -7, -8, -9, and -10. Confirmation of the geographic associations observed in these clades within the topotypes would require the acquisition of sequences from more BTV isolates across the full geographic distribution of BTV. Genotype/topotype analysis of Australian BTV isolates has been based on comparisons of short regions of highly conserved genome segments (Seg-10 NS3 [690 bp] and Seg-3 VP3 [540 bp]) (13, 24). An ability to analyze BTVs at the whole genome level should shed new light on the origins and movement of these important viruses.
The analysis of the 10 genome segments of BTV_2_AUS_2008_24 supports the hypothesis of entry into Australia in the recent past. Eight of 10 genome segments provided strong support for the idea of this virus having its origins in Asia. Since no live ruminants can be imported, BTV vaccines (live or killed) are not permitted for use in Australia and as northern Australia is separated from southeast Asia by water, the movement of BTVs into Australia would be by way of wind-borne dispersal of infected insect vectors. Recent modeling of possible routes of entry into Australia support the likely entry from Timor and other near neighbors via wind-borne Culicoides (7). The movement of Australian Culicoides in the opposite direction must also be considered plausible. Introduction of exotic species of Culicoides and exotic BTVs, along with changes in landscape utilization, climate change, and climate variability, has the potential to lead to the emergence of bluetongue disease in Australia in a manner similar to what has occurred in Europe (30).
The potential for reassortment of genome segments at high frequency in the vertebrate or invertebrate host confounds the ability to assign origins to the viruses with any degree of certainty (5, 12). The phylogenetic analyses of the prototype Australian serotypes revealed instances where viruses share identical or near-identical genome segments, suggesting a common origin through reassortment. For example, BTV 1, 3, and 23 share common or very closely related Seg-1, -3, and -9 segments; BTV 3 and 23 share Seg-4, -5, and -8; and BTV 1 and 20 share Seg-4, Seg-5, and Seg-8. The significance and frequency of such reassortment events would be revealed only by sequencing of a larger number of BTV isolates collected at different times and locations in Australia. The experience in Europe has been for reassortment to take place rapidly, including that seen with live attenuated vaccine strains used for disease control (2).
The methodology we have developed provided full coding sequences of multiple BTV isolates. It has application to any dsRNA virus or samples, including clinical or vector samples from which dsRNA can be preferentially extracted. Analysis of viruses at extensive landscape and temporal scales as well as metagenomic analysis of viruses in insect vectors is amenable to the approach developed here. Currently, we lack genetic determinants for BTV virulence (4). This is, in part, due to the difficulty of maintaining virulence during passage in cell culture, necessitating animal-to-animal transmission for virulence studies. The technique for dsRNA isolation should be applicable to blood, clinical samples, and samples of Culicoides vectors, facilitating an exploration of genetic determinants of virulence by direct sequencing of such samples and by appropriate reverse genetic studies. Only with complete genome sequences from BTVs across the entire geographic spread of the virus will it be possible to assign origins of viruses and genome segments with a high degree of confidence.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the funding contributed by the Australian Biosecurity CRC for Emerging Infectious Diseases in support of this work.
Ross Lunt and Debbie Eagles provided valuable discussion and knowledge in the selection of isolates for sequence determination.
Footnotes
Published ahead of print 18 April 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Anonymous 2009. National arbovirus monitoring program report. Anim. Health Surveill. Q. Rep. 13:5–7 [Google Scholar]
- 2. Batten CA, Maan S, Shaw AE, Maan NS, Mertens PP. 2008. A European field strain of bluetongue virus derived from two parental vaccine strains by genome segment reassortment. Virus Res. 137:56–63 [DOI] [PubMed] [Google Scholar]
- 3. Belhouchet M, et al. 2011. Detection of a fourth orbivirus non-structural protein. PLoS One 6:e25697 doi:10.1371/journal.pone.0025697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caporale M, et al. 2011. Determinants of bluetongue virus virulence in murine models of disease. J. Virol. 85:11479–11489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carpi G, Holmes EC, Kitchen A. 2010. The evolutionary dynamics of bluetongue virus. J. Mol. Evol. 70:583–592 [DOI] [PubMed] [Google Scholar]
- 6. Dal Pozzo F, Saegerman C, Thiry E. 2009. Bovine infection with bluetongue virus with special emphasis on European serotype 8. Vet. J. 182:142–151 [DOI] [PubMed] [Google Scholar]
- 7. Eagles D, Deveson T, Walker PJ, Zalucki M, Durr P. J. 29 December 2011. Evaluation of long-distance dispersal of Culicoides midges into northern Australia using a migration model. Med. Vet. Entomol. [Epub ahead of print.] doi:10.1111/j.1365-2915.2011.01005.x [DOI] [PubMed] [Google Scholar]
- 8. Elbers AR, et al. 2008. Field observations during the bluetongue serotype 8 epidemic in 2006. I. Detection of first outbreaks and clinical signs in sheep and cattle in Belgium, France and the Netherlands. Prev. Vet. Med. 87(1–2):21–30 [DOI] [PubMed] [Google Scholar]
- 9. Gard GP, Shorthose JE, Cybinski DH, Zakrzewski H. 1985. The isolation from cattle of 2 bluetongue viruses new to Australia. Aust. Vet. J. 62:203. [DOI] [PubMed] [Google Scholar]
- 10. Gard GP, Shorthose JE, Weir RP, Erasmus BJ. 1987. The isolation of a bluetongue serotype new to Australia. Aust. Vet. J. 64:87–88 [DOI] [PubMed] [Google Scholar]
- 11. Gard GP, Weir RP, Melville LF, Lunt RA. 1987. The isolation of bluetongue virus types 3 and 16 from northern Australia. Aust. Vet. J. 64:388. [DOI] [PubMed] [Google Scholar]
- 12. Gibbs EP, Greiner EC. 1994. The epidemiology of bluetongue. Comp. Immunol. Microbiol. Infect. Dis. 17:207–220 [DOI] [PubMed] [Google Scholar]
- 13. Gould AR, Pritchard LI. 1990. Relationships amongst bluetongue viruses revealed by comparisons of capsid and outer coat protein nucleotide sequences. Virus Res. 17:31–52 [DOI] [PubMed] [Google Scholar]
- 14. He CQ, et al. 2010. Intragenic recombination as a mechanism of genetic diversity in bluetongue virus. J. Virol. 84:11487–11495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lunt R, et al. 2009. Laboratory characterization of BTV-7, a serotype previously not isolated in Australia, p 93–95 In Ryan P, Aaskov J, Russell R. (ed), Arbovirus research in Australia. Proceedings of Arbovirus Research in Australia 10, Brisbane, Australia The Queensland Institute of Medical Research, Brisbane, Australia [Google Scholar]
- 16. Maan S, et al. 2009. Molecular epidemiology studies of bluetongue virus, p 135–166 In Mellor PS, Baylis M, Mertens PC. (ed), Bluetongue. Academic Press, London, United Kingdom [Google Scholar]
- 17. Maan S, et al. 2011. Complete genome characterisation of a novel 26th bluetongue virus serotype from Kuwait. PLoS One 6:e26147 doi:10.1371/journal.pone.0026147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maan S, et al. 2008. Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virology 377:308–318 [DOI] [PubMed] [Google Scholar]
- 19. Maan S, et al. 2007. Analysis and phylogenetic comparisons of full-length VP2 genes of the 24 bluetongue virus serotypes. J. Gen. Virol. 88:621–630 [DOI] [PubMed] [Google Scholar]
- 20. Maan S, et al. 2010. Full genome characterisation of bluetongue virus serotype 6 from the Netherlands 2008 and comparison to other field and vaccine strains. PLoS One 5:e10323 doi:10.1371/journal.pone.0010323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maan S, et al. 2007. Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J. Virol. Methods 143:132–139 [DOI] [PubMed] [Google Scholar]
- 22. Mertens PP, Sangar DV. 1985. Analysis of the terminal sequences of the genome segments of four orbiviruses. Virology 140:55–67 [DOI] [PubMed] [Google Scholar]
- 23. Potgieter AC, et al. 2009. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 90:1423–1432 [DOI] [PubMed] [Google Scholar]
- 24. Pritchard LI, et al. 2004. Genetic diversity of bluetongue viruses in south east Asia. Virus Res. 101:193–201 [DOI] [PubMed] [Google Scholar]
- 25. Rumble SM, et al. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 5:e1000386 doi:10.1371/journal.pcbi.1000386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh KP, et al. 2004. Phylogenetic analysis of bluetongue virus genome segment 6 (encoding VP5) from different serotypes. Vet. Ital. 40:479–483 [PubMed] [Google Scholar]
- 27. St George TD, et al. 1980. The isolation of two bluetongue viruses from healthy cattle in Australia. Aust. Vet. J. 56:562–563 [DOI] [PubMed] [Google Scholar]
- 28. St George TD, et al. 1978. The isolation of a bluetongue virus from Culicoides collected in the Northern Territory of Australia. Aust. Vet. J. 54:153–154 [DOI] [PubMed] [Google Scholar]
- 29. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilson AJ, Mellor PS. 2009. Bluetongue in Europe: past, present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364:2669–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.