

Abstract
Two new peptidic proteasome inhibitors were isolated as trace components from a Curaçao collection of Symploca sp. marine cyanobacteria. Carmaphycin A (1) and carmaphycin B (2) feature a leucine-derived α, β -epoxyketone warhead directly connected to either methionine sulfoxide or methionine sulfone. Their structures were elucidated on the basis of extensive NMR/MS analyses and confirmed by total synthesis, which in turn provided more material for further biological evaluations. Pure carmaphycins A and B were found to inhibit the β5 subunit (chymotrypsin-like activity) of the S. cerevisiae 20S proteasome in the low nanomolar range. Additionally, they exhibited strong cytotoxicity to lung and colon cancer cell lines, as well as exquisite antiproliferative effects in the NCI60 cell line panel. These assay results as well as initial structural biology studies suggest a distinctive binding mode for these new inhibitors.
Keywords: cyanobacteria, anticancer, proteasome inhibitors, total synthesis, structural biology
Introduction
Cyanobacteria (blue-green algae) have shown a remarkable capacity to produce structurally diverse natural products exhibiting a broad spectrum of potent biological activities.[1] Notably, a growing number of cyanobacterial metabolites show high cytotoxicity to a variety of cancer cell lines.[2] For example, the apratoxin family of depsipeptides are highly potent cytotoxins.[1,3] The parent compound, apratoxin A, induces G1-phase cell cycle arrest and apoptosis through interaction with STAT3 and FGFR signaling.[3] Metabolites such as the curacins,[4] the dolastatins and their analogues (e.g. symplostatins, gallinamide A, malevamide D, belamide A),[5] and the cryptophycins[6] have been shown to interfere with the normal function of microtubules. More recently, a Symploca sp. yielded largazole, a new class of HDAC inhibitor with potent cancer cell cytotoxicity.[7] The combination of potent and selective cytotoxic activities, unique mechanisms of action, and novel structural frameworks that characterizes these cyanobacterial natural products, make them important lead compounds in the development of new anticancer chemotherapeutics.
Our ongoing efforts aimed at discovering new marine cyanobacterial metabolites with anticancer potential involve the use of a strategic panel of murine and human cancer cell lines to screen for cytotoxicity.[8] Employing this key resource, we detected potent activity in extracts derived from a Curaçao collection of the cyanobacterium Symploca sp.[9] A combination of bioassay and 1H NMR-guided fractionation led us to isolate the exquisitely active carmaphycins A (1) and B (2),[10] which are cytotoxic and cytostatic through inhibition of the 20S proteasome (Scheme 1). Herein we report the isolation and structure elucidation, total synthesis, biological evaluation, and initial structural biology studies of these new cyanobacterial-derived anticancer agents.
Scheme 1.

Structures of carmaphycins A (1) and B (2).
Results and Discussion
Isolation and Structure Elucidation
Browns tufts of Symploca sp. were collected off an anchor rope by snorkel south of the CARMABI research station in Curaçao. The cyanobacterial tissue was extracted repeatedly with CH2Cl2/MeOH (2:1) and then fractioned by silica gel vacuum column chromatography to produce nine subfractions (A-I). 1H NMR-guided fractionation of the bioactive fraction H involved normal phase column chromatography and reverse phase HPLC, and led to the isolation of carmaphycin A (1) and carmaphycin B (2) as colorless oils [ 1.73 mg (0.02% yield) and 0.26 mg (0.003% yield), respectively ].[11] Their complete structural elucidation was accomplished via spectroscopic analysis and total synthesis, as described below.
HRESIMS of 1 yielded an [ M+Na ] + peak at m/z 538.2918, exhibiting an isotopic pattern comprised of m/z 538/539/540 (100:29:9 ratio), suggesting the presence of at least one sulfur atom. Combined with NMR data (Table 1), this led to the molecular formula C15H45N3O6S (calcd for C15H45N3O6SNa, 538.2921), containing 5 degrees of unsaturation (excluding sulfur). Intense IR absorptions at 3288 (broad) and 1636 cm−1 were consistent with amide functionalities. Analysis of the 1H NMR spectrum of 1 showed resonances characteristic of a peptide, including six NH protons (δ8.05-δ6.13), six α-methines (δ4.85- δ4.27), three deshielded methyl groups (δ2.74, δ2.71 and δ1.52), multiple diastereotopic methylene protons, and a series of shielded methyl doublets and triplets (δ0.95- δ0.86). Intriguingly, the majority of these resonances were arranged in pairs, suggesting this sample was a mixture of two diastereomers. The 13C NMR of 1 confirmed the peptidic nature of the new metabolite (Table 1), and also displayed twinned peaks for some of the resonance bands.
Table 1.
NMR spectroscopic data (CDCl3) for carmaphycin A (1) and carmaphycin B (2)
| N | Carmaphycin A (1) | Carmaphycin B (2) | Type | HMBCc | COSY | ||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Diastereomer 1 | Diastereomer 2 | ||||||||
|
|
|||||||||
| δCa | δH , mult. (J in Hz)b | δCa | δH , mult. (J in Hz)b | δCa | δH , mult. (J in Hz)b | ||||
| 1a | 52.53 | 3.33, d (5.4) | 52.58 | 3.36, d (4.8) | 52.43 | 3.31, d (4.8) | CH2 | 2,3,4 | 1b |
| 1b | 2.90, d (5.4) | 2.91, d (4.8) | 2.91, d (4.8) | 2,3,4 | 1a | ||||
| 2 | 59.35 | 59.34 | 59.23 | C | |||||
| 3 | 16.86 | 1.52, s | 16.84 | 1.53, s | 16.73 | 1.52, s | CH3 | 1,2,4 | |
| 4 | 208.56 | 209.20 | 208.39 | C | |||||
| 5 | 51.36 | 4.45, m | 51.39 | 4.48, m | 51.37 | 4.50, ddd (7.8, 6.7, 1.8) | CH | 4,6,7,10e | 5NH, 6a, 6b |
| 5 | NH | 7.83, d (6.6) | 8.03, d (7.2) | 6.95, d (6.7) | 5e,6e,10 | 5 | |||
| 6a | 39.07 | 1.55, m | 39.25 | 1.53, m | 39.08 | 1.58, m | CH2 | 4,5,7,8,9 | 5,6b,7 |
| 6b | 1.33, m | 1.31, m | 1.31, m | 4,5,7,8,9 | 5,6a,7 | ||||
| 7 | 25.24 | 1.70, m | 25.27 | 1.70, m | 25.21 | 1.68, m | CH | 5e,6e,8e,9e | 6a,6b,8,9 |
| 8 | 21.10 | 0.93, d (6.0) | 21.13 | 0.93, d (6.0) | 21.01 | 0.95, d (5.9) | CH3 | 6,7,9 | 7 |
| 9 | 23.33 | 0.92, d (6.6) | 23.36 | 0.92, d (6.6) | 23.21 | 0.96, d (6.6) | CH3 | 6,7,8 | 7 |
| 10 | 170.29 | 170.26 | 169.79 | C | |||||
| 11 | 50.81 | 4.83, m | 50.63 | 4.86, m | 50.31 | 4.76, dt (7.2, 6.6) | CH | 10,12,13,15e | 11NH,12a,12b |
| 11 | NH | 7.28, d (6.6) | 7.03, d (7.2) | 6.76, d (7.2) | 10e,11e,15 | 11 | |||
| 12a | 28.47 | 2.41, m | 26.14 | 2.50, m | 25.75 | 2.34, m | CH2 | 10,11,13 | 11,12b,13a,13b |
| 12b | 2.10, m | 2.08, m | 2.12, m | 10,11,13 | 11,12a,13a,13b | ||||
| 13a | 48.96 | 3.42, m | 46.67 | 3.44, m | 50.39 | 3.53, m | CH2 | 11,12,14e | 12a,12b,13b |
| 13b | 2.76, m | 2.66, m | 3.21, ddd (14.4, 5.4, 5.4) | 11,12,14e | 12a, 12b, 13a | ||||
| 14 | 38.83 | 2.71, s | 35.95 | 2.74, s | 40.56 | 3.06, s | CH3 | 12f,13 | |
| 15 | 171.41 | 170.95 | 171.10 | C | |||||
| 16 | 58.25 | 4.32, dd (6.0, 8.4) | 58.21 | 4.29, dd (6.6, 8.4) | 58.43 | 4.21, dd (7.7, 7.2) | CH | 15,17,18,19,20 | 16NH,17 |
| 16 | NH | 6.15, d (8.4) | 6.15, d (8.4) | 5.93, d (7.7) | 16e,17e,20 | 16 | |||
| 17 | 31.00 | 2.10, m | 31.21 | 2.06, m | 30.62 | 2.10, m | CH | 15,16,18,19 | 16,18,19 |
| 18 | 19.22 | 0.92, d (6.0) | 19.26 | 0.92, d (6.0) | 19.15 | 0.94, d (7.3) | CH3 | 16,17,19 | 17 |
| 19 | 17.83 | 0.90, d (7.2) | 17.99 | 0.90, d (7.2) | 17.94 | 0.92, d (7.2) | CH3 | 16,17,18 | 17 |
| 20 | 173.42 | 173.30 | 173.43 | C | |||||
| 21 | 36.70 | 2.21, t (7.8) | 36.72 | 2.22, t (7.2) | 36.55 | 2.23, t (7.8) | CH2 | 20,22,23 | 22 |
| 22 | 25.37 | 1.63, m | 25.38 | 1.62, m | 25.23 | 1.63, m | CH2 | 20e,21,23,24f | 21,23 |
| 23 | 31.43 | 1.29, m | 31.43 | 1.29, m | 31.31 | 1.29, m | CH2 | 21,22e,24,25 | 22,24 |
| 24 | 22.37 | 1.31, m | 22.37 | 1.31, m | 22.26 | 1.31, m | CH2 | 22,23,25 | 23,25 |
| 25 | 13.95d | 0.88, t (7.2) | 13.94d | 0.88, t (7.2) | 13.84 | 0.89, t (7.2) | CH3 | 23,24 | 24 |
Recorded at 200 MHz.
Recorded at 600 MHz.
From proton to the indicated carbon.
Assignments may be interchanged.
Observed only for 1.
Observed only for 2.
Comprehensive analysis of 1 by 2D NMR, including HSQC, HMBC and COSY, revealed the sequential combination of a leucine-derived α,β-epoxyketone fragment, a methionine sulfoxide residue, a valine moiety, and a N-hexanoyl appendage (Scheme 1). The presence of methionine sulfoxide, observed as a 1:1 mixture of the R and S sulfoxide diastereomers, was consistent with the occurrence of twinned resonances in the 1H and 13C NMR spectra noted above. A standard interplay of HMBC and ESI-MS2 data were used to sequence these residues and derive the planar structure of 1. As expected, chemical shift differences between the diastereomers (Table 1) are accentuated for those protons (and carbons) neighboring the sulfoxide chiral center and located nearby along the peptidic backbone of carmaphycin A (1).[12]
The second new metabolite, carmaphycin B (2), analyzed for C25H45N3O7S by HRESIMS of the [ M+H ] + peak (m/z 532.3045; calcd for C25H46N3O7S, 532.3051). With the exception of one additional oxygen atom, both 1 and 2 showed very similar isotope patterns by MS as well as comparable IR spectra. Their structural similarity was also apparent by analysis of the 1H and 13C NMR data (Table 1) which further revealed that compound 2 existed as a single stereoisomer. Extensive analysis by 2D NMR localized the main chemical shift differences between 1 and 2 to the methionine-derived residue, and because their molecular formulas differed by a single oxygen atom, it was evident that the sulfoxide functionality in 1 was replaced by a sulfone in 2; this conclusion was supported by MS2 fragmentation analysis (Figures S4-6).
Total Synthesis
Encouraged by preliminary bioactivity results and recognizing that the extremely small amounts of isolated 1 and 2 would preclude their stereochemical elucidation as well as any biological and pharmacological evaluations, we embarked on the total synthesis of both metabolites (Scheme 2).
Scheme 2.
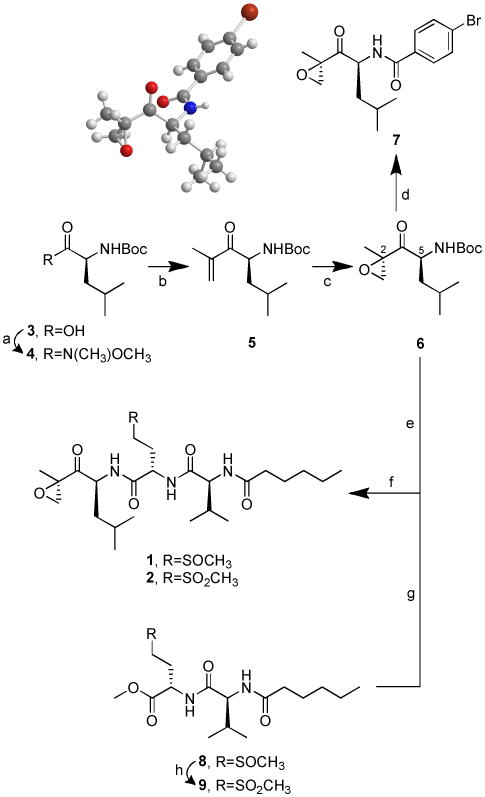
Total synthesis of 1 and 2. (a) Et3N, BOP, CH3O(CH3)NH.HCl, CH2Cl2, 25 °C, 3 h, 97%; (b) isopropenyl magnesium bromide, THF, 0 °C, 6 h, 82%; (c) NaHCO3, trifluoroacetone, oxone, CH3CN/H2O, -10 °C, 1.5 h, 26%; (d) TFA, CH2Cl2, 25 °C, 1.5 h; then 4-bromobenzoyl chloride, DiPEA, CH2Cl2, 25 °C, 16 h, 61% (e) TFA, CH2Cl2, 25 °C, 1 h; (f) PyBOP, DiPEA, CH2Cl2, 25 °C, 15-20 h, 50% of a 1.6:1 diastereomeric mixture from 8, 33% from 9; (g) LiOH.H2O, 1,4-dioxane/H2O, 25 °C, 1 h; (h) NaHCO3, oxone, acetone/H2O, 0 °C, then stirring at 25°C for 1 h, 99%.
A convergent, flexible and scalable approach, in which the readily accessible (2R,5S)-α,β-epoxyketone intermediate 6[13] was attached to either 8 or 9 via PyBOP-mediated coupling, swiftly afforded synthetic carmaphycin A (1) and carmaphycin B (2) with unoptimized overall yields of 10% and 7%, respectively. The absolute configuration at C2 and C5 was determined to be R and S, respectively, via X-ray analysis of derivative 7. Oxone was used to quantitatively oxidize the dipeptide sulfoxide (8) to its corresponding sulfone (9). Both synthetic products were identical by HPLC, MS and NMR comparison with the metabolites isolated from Symploca sp., thus confirming the proposed structures for the natural products 1 and 2. Moreover, each pair showed superimposable CD curves (Figure S9) and shared the same sign of specific rotation, thus allowing the assignment of their absolute configuration as 2R, 5S, 11S, 16S.
Biological Activity
The structures of carmaphycins A (1) and B (2) resemble the potent proteasome inhibitor epoxomicin (10), related α,β-epoxyketone natural products and synthetic analogues.[14] Among these, carfilzomib is currently in phase III clinical trials for treatment of multiple myeloma.[15] These observations prompted us to evaluate both metabolites for their capacity to inhibit the β5 subunit (chymotrypsin-like) of the Saccharomyces cerevisiae 20S proteasome (Table 2).
Table 2.
S. cerevisiae 20S Proteasome Inhibitory and Cytotoxic Activities Determined for Carmaphycins A (1) and B (2), Epoxomicin (10) and Salinosporamide A (11).
| Comp. | Proteasome Inhibition(IC50, nM) | Cytotoxicity | |
|---|---|---|---|
|
| |||
| H-460 (EC50 nM) | HCT-116 (IC50 nM) | ||
| 1 | 2.5 ± 0.3 | 9 ± 2 | 19 ± 1 |
| 2 | 2.6 ± 0.9 | 6 ± 1 | 43 ± 4 |
| 10 | 2.7 ± 0.4 | 4 ± 1 | 23 ± 2 |
| 11 | 1.4 ± 0.2 | 14 ± 6 | 48 ± 3 |
Pure carmaphycin A (1) and carmaphycin B (2) exhibited potent inhibitory activity, comparable to that of epoxomicin (10) and salinosporamide A (11). The latter metabolite is also marine-derived and currently in phase I clinical trials for the treatment of multiple myeloma.[16] Further biological testing of 1 and 2 revealed strong cytotoxicity to human lung adenocarcinoma (H-460) and colon cancer (HCT-116) cell lines. In the NCI 60 cell line panel, both carmaphycins displayed exquisite antiproliferative effects to lung, colon and CNS tumor cell lines; in most of these cell lines, GI50 values were observed between 1 and 50 nM. Interestingly, the most sensitive cell lines to 1 and 2 consistently possess mutations in either the KRAS or tp53 genes, or both. Upon reaching the Total Growth Inhibition line, further concentration increases did not cause enhanced cytotoxicity (e.g. the dose response curves were flat between 100 nM and 10 μM), reminiscent of antitubulin and other cytostatic drugs. Thus, to our knowledge the carmaphycins have unique cellular effects quite different from those of other α,β-epoxyketones.
Structural Biology Studies
Molecular dynamics (MD) simulations based on existing X-ray crystallography data for the epoxomicin:S. cerevisiae 20S proteasome complex (including the known morpholino adduct derived from Thr1 in the catalytic β5 subunit and the α,β-epoxyketone warhead),[17] provided some insight on the binding mode of the carmaphycins (Figures 1a and S10a). Two-dimensional interaction plots displayed a network of hydrogen bonds as well as van der Waals and solvent interactions between both carmaphycins and the surrounding protein residues (Figures 1b and S10b). Of key importance, the sulfoxide/sulfone moieties in the methionine-derived residue are spatially close to the NH group of Gly23, likely enabling a hydrogen bond between these functionalities.[18] Superimposition of bound MD-simulated carmaphycin A (1) with epoxomicin (10), salinosporamide A (11), and the FDA-approved proteasome inhibitor bortezomib (12) (Figure 1c), indicates the above hydrogen bond with Gly23 to be exclusive to 1, thus suggesting a distinctive binding mode for these new inhibitors.
Figure 1.
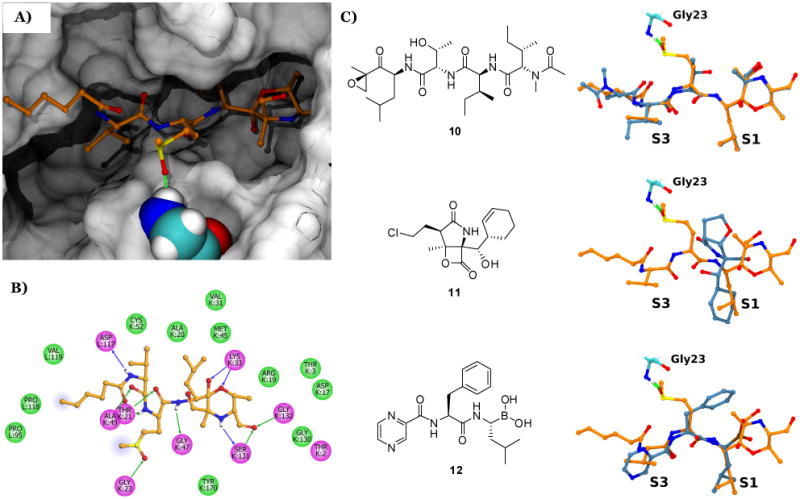
MD modeling of the carmaphycin A (1) bound to the β5 subunit of the S. cerevisiae 20S proteasome. A) Lowest energy structure of carmaphycin A (1). B) Two-dimensional interaction plot consisting of 1 and surrounding protein residues (blue and green arrows represent hydrogen bonds, green colored residue discs refer to van der Waals interactions, magenta discs are either polar/charged or hydrogen bond containing residues, and the light blue airbrush circles on the alkane chain represent solvent interactions. C) Bound structures of different 20S proteasome inhibitors (light blue) relative to carmaphycin A (1) (orange). Pocket locations (S1 and S3) and Gly23 (cyan) of the β5 subunit are shown. For all structures, oxygen, nitrogen, and sulfur atoms are colored red, blue and yellow respectively. See Supporting Information for details.
Conclusion
A combined bioassay/NMR guided approach yielded two new α,β-epoxyketone containing natural products, the first such compounds isolated from the marine environment. In this regard, marine cyanobacteria continue to be exceptionally prolific sources of new drug leads, especially those with anticancer potential. The new carmaphycin natural products have remarkable cellular toxicity, especially to solid tumor cell lines, suggesting that the unique sulfoxide/sulfone functional group is imparting novel properties to these potent proteasome inhibitors. With a chemical synthesis route to the carmaphycins in place, additional features of the SAR of these proteasome inhibitors are under continuing investigation.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a JASCO P-2000 polarimeter, and circular dichroism data were obtained using a JASCO J-815 CD spectrometer. UV and IR spectra were recorded on a Beckman DU800 spectrophotometer and on a Nicolet 100 FT-IR spectrometer, respectively. 1H, 13C, and 2D NMR spectra were collected at a 1H resonance frequency of either 500 MHz (Varian VX500), 600 MHz (Bruker Avance III equipped with 1.7 mm and 5 mm TCI cryoprobes) or 800 MHz (Varian VS800 equipped with a 5 mm cold probe). Chemical shifts were calibrated internally to the residual signal of the solvent in which the sample was dissolved (CDCl3, δH 7.26, δC 77.0). High-resolution mass spectra were obtained on a ThermoFinnigan MAT900XL mass spectrometer. X-ray crystallographic data was acquired in a Bruker APEX-II diffractometer equipped with a CCD detector and low-temperature cryostat. HPLC was carried out using a dual Waters 515 pump system equipped with a Waters 996 photodiode array detector. Vacuum and flash chromatographic separations were performed using type H (10-40 μm, Aldrich) silica and silica gel 60 (40-63 μm, EMD), respectively. Merck TLC sheets (silica gel 60 F254) were used for analytical TLC (aluminum-supported, layer-thickness 200 μm) and preparative TLC (glass-supported, layer-thickness 250 μm). All chemical reagents were obtained from Aldrich in an analytical or higher grade and were used as received unless stated otherwise. Dipeptide 8 was purchased from GenicBio Limited, China. Solvents were acquired as HPLC grade. All reactions were performed under dry nitrogen using glassware previously oven dried (150°C), unless otherwise specified. Glassware was allowed to reach room temperature under a flow of inert gas. Likewise, glass syringes and stainless steel needles, used to handle anhydrous reagents and solvents, were oven dried, cooled in a desiccator, and flushed with inert gas prior to use. THF and CH2Cl2 were distilled from sodium/benzophenone and CaH2, respectively.
Biological Activity
Compounds were assayed for inhibition of the Saccharomyces cerevisiae 20S proteasome β5-subunit as previously described,[19] but using only 0.5 μg/mL of proteasome. Data was plotted on SigmaPlot 12.0 and fit with a “4 parameter logistic” curve to obtain IC50 values with standard errors. Cytotoxicity to H-460 and HCT-116 cells was determined as previously reported.[20]
Cyanobacterial Collection
Samples of cyanobacterial biomass (voucher specimen available from WHG as collection number NAC15/Dec/08-5) were collected by hand at a depth of 0.5-2.5 m south of CARMABI beach, in December 2008. Multiple brown tufts (5-7 cm tall, 2-3 cm across) were collected off an anchor rope used to secure small vessels (Figure S1). They were stored in 70% EtOH at -20 °C prior to extraction.
Extraction, Isolation and Synthesis
Approximately 9.7 g (dry wt) of the cyanobacteria were extracted repeatedly with CH2Cl2/MeOH (2:1) to afford 0.4305 g of extract. A portion of this material (0.3296 g) was fractionated by silica gel vacuum liquid chromatography[21] using a stepwise gradient solvent system of increasing polarity starting from 100% hexanes, then 10% EtOAc in hexanes to 100% MeOH, to produce nine fractions (A-I). The bioactive fraction H (eluting with 25% MeOH/EtOAc, 56.3 mg) was subjected to a 1H NMR-guided fractionation using silica gel column chromatography (isocratic, 10% MeOH in CH2Cl2 + 0.1% HOAc), followed by reverse phase HPLC (Phenomenex Jupiter 10μ C18 300Å, 250 × 10.0 mm, 40% MeCN in H2O at 3.0 mL/min, detection at 211 nm) to yield carmaphycin A (1) as a 1:1 diastereomeric mixture (1.73 mg, 0.02 %), and carmaphycin B (2) (0.26 mg, 0.003 %).
Carmaphycin A (1)
Colorless oil; [ α ] 23D +6.4 (c 0.5, CH3CN); CD (c 0.2, CH3CN) λmax (Δε) 290 (+1.24), 240 (+0.07), 230 (+0.46), 200 (-3.45); IR (neat) 3288, 3060, 2924, 1636, 1452, 1026, 755, 698 cm-1; 1H, 13C, and 2D NMR data, see Table S1; HRESIMS m/z [ M+Na ] + 538.2918 (calcd for C25H45N3O6SNa, 538.2921).
Carmaphycin B (2)
Colorless oil; [ α ] 23D +30 (c 0.04, CH3CN); CD (c 0.2, CH3CN) λmax (Δε) 290 (+8.57), 240 (-0.90), 230 (+0.55), 200 (-16.5); IR (neat) 3282, 3062, 2926, 1640, 1453, 1131, 741, 699 cm-1; 1H, 13C, and 2D NMR data, see Table S1; HRESIMS m/z [ M+H ] + 532.3045 (calcd for C25H46N3O7S, 532.3051).
Boc-L-Leucine N-methoxy-N-methylamide (4)
Triethylamine (1.53 mL, 11.0 mmol) was added to a stirred solution of Boc-Leu (2.54 g, 11.0 mmol) in CH2Cl2 (40 mL). To this solution, benzotriazol-1-yloxytris [ dimethylamino ] phosphonium hexafluorophosphate (BOP, 4.86 g, 11.0 mmol) was added, followed by a solution of O,N-dimethylhydroxylamine hydrochloride (1.18 g, 12.1 mmol) and triethylamine (1.68 mL, 12.1 mmol) in CH2Cl2 (10 mL). The reaction was monitored by TLC. After 3h, the reaction crude was diluted with CH2Cl2 (200 mL) and washed successively with HCl 3M (3 × 30 mL), saturated NaHCO3 (3 × 30 mL), and brine (3 × 30 mL). The organic layer was dried with Na2SO4 and the solvent evaporated. The residual crude was purified by silica gel column chromatography (50% EtOAc/hexanes) to afford 4 as a colorless oil (2.92 g, 97%). [ α ] 24D -11 (c 3.1, CH3CN); 1H NMR (CDCl3, 500 MHz) δ 5.04 (d, J = 9.0 Hz, 1H), 4.70 (m, 1H), 3.76, (s, 3H), 3.17, (s, 3H), 1.69, (m, 1H), 1.45-1.37 (m, 2H), 1.40 (s, 9H), 0.94 (d, J = 6.5 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 173.8 (C), 155.6 (C), 79.4 (C), 61.5 (CH3), 48.9 (CH), 42.0 (CH2), 32.1 (CH3), 28.3 (3CH3), 24.7 (CH3), 23.3 (CH), 21.5 (CH3). HRESIMS m/z [ M+H ] + 275.1966 (calcd for C13H27N2O4, 275.1965).
(S)-t-Butyl-2,6-dimethyl-3-oxohept-1-en-4-ylcarbamate (5)
To a 0 °C solution of the isopropenyl magnesium bromide (43.3 mL, 21.7 mmol, 0.5 M solution in THF) was added a solution of amide 4 (2.83 g, 10.3 mmol) in dry THF (30 mL) dropwise via an addition funnel under an atmosphere of nitrogen. The reaction was stirred at 0 °C for 6h and then slowly poured into a beaker containing 40 mL of saturated NH4Cl solution and approximately 40 mL of ice. The pH of the solution was adjusted to 7 using HCl 6 M when all the ice melted, and the resulting mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with saturated NaHCO3 (3 × 50 mL), brine (3 × 50 mL), and dried over Na2SO4. The Na2SO4 was removed by filtration, and the volatiles were removed under reduced pressure. Purification by silica gel column chromatography (33% EtOAc/hexanes) gave 5 as a colorless oil (2.15 g, 82%). [ α ] 24D +23 (c 1.2, CH3CN); 1H NMR (CDCl3, 500 MHz) δ 6.06 (s, 1H), 5.86 (s, 1H), 5.14 (d, J = 8.7 Hz, 1H), 5.04 (dt, J = 3.8, 8.7 Hz, 1H), 1.88 (s, 3H), 1.71 (m, 1H), 1.46 (ddd, J = 3.8, 9.2, 13.5 Hz, 1H), 1.41 (s, 9H), 1.13 (ddd, J = 3.8, 9.2, 13.5 Hz, 1H), 0.98 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 201.5 (C), 155.5 (C), 142.2 (C), 125.9 (CH2), 79.4 (C), 52.6 (CH), 43.1 (CH2), 28.3 (3CH3), 24.9 (CH), 23.3 (CH3), 21.6 (CH3), 17.8 (CH3). HRESIMS m/z [ M+H ] + 256.1905 (calcd for C14H26NO3, 256.1907).
Boc-L-Leucine epoxyketone (6)
NaHCO3 (23.5 g, 280 mmol) was dissolved in H2O (30 mL) and cooled to -10°C. Trifluoroacetone (27.0 mL, 300 mmol) and intermediate 5 (2.18 g, 8.5 mmol in 37 mL CH3CN) were added followed by the portion wise addition of oxone (43.0 g, 70 mmol). The reaction was diluted with Et2O (200 mL) and partitioned. The aqueous layer was extracted with Et2O (3 × 100 mL) and the combined organic layers were dried with Na2SO4. After solvent removal in vacuo, repetitive silica gel column chromatography (5% and 20% EtOAc/hexanes) provided starting material 5 (0.12 g) and the desired diastereomer 6 (colorless oil, 0.59 g, 26%) as the major reaction product. [ α ] 24D +79 (c 0.7, CH3CN); 1H NMR (CDCl3, 500 MHz) δ 4.84 (d, J = 8.5 Hz, 1H), 4.31 (dt, J = 3.5, 8.5 Hz, 1H), 3.28 (d, J = 5.0 Hz, 1H), 2.88 (d, J = 5.0 Hz, 1H), 1.72 (m, 1H), 1.51 (s, 3H), 1.49 (ddd, J = 3.5, 10.0, 14.0 Hz, 1H), 1.40 (s, 9H), 1.16 (ddd, J = 3.5, 10.0, 14.0 Hz, 1H), 0.96 (d, J = 6.5 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 209.5 (C), 155.6 (C), 79.7 (C), 59.0 (C), 52.3 (CH2), 51.4 (CH), 40.4 (CH2), 28.2 (3CH3), 25.1 (CH), 23.3 (CH3), 21.3 (CH3), 16.7 (CH3). HRESIMS m/z [ M+Na ] + 294.1673 (calcd for C14H25NO4Na, 294.1676).
Boc-L-Leucine epoxyketone derivative (7)
Boc-L-Leu epoxyketone (6) (44.0 μg, 160 μmol) in CH2Cl2 (1.5 mL) was treated with TFA (300 μL, 3.9 mmol) and stirred at 25°C until TLC showed the absence of starting material (1.5 h), whereupon it was concentrated in vacuo to a reddish oil. This oil was dissolved in dry CH2Cl2 (10 mL), and to this solution were sequentially added 4-bromobenzoyl chloride (53.0 μg, 240 μmol) and DiPEA (50 μL, 290 μmol) at 25°C. After stirring 16h at 25°C, the reaction mixture was quenched with saturated NH4Cl, followed by solvent partition and CH2Cl2 (3 × 10 mL) extractions of the aqueous layer. All organic extracts were combined and dried (Na2SO4), to be then concentrated in vacuo. Silica gel column chromatography (20% EtOAc/hexanes) gave 7 as a white solid (35.1 μg, 61%). Additional purification by normal phase HPLC (Phenomenex Luna 5μ Silica 100Å, 250 × 10.0 mm, 1% iso-PrOH/hexanes at 3.0 mL/min, detection at 256 and 280 nm), yielded highly pure 7 (15.2 μg), which was crystallized from EtOAc/iso-octane as colorless tridimensional needles. [ α ] 28D +41 (c 1.5, CH3CN); 1H NMR (CDCl3, 600 MHz) δ 7.61 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 6.52 (d, J = 8.4 Hz, 1H), 4.83 (ddd, J = 3.0, 10.5, 10.8 Hz, 1H), 3.41 (d, J = 5.1 Hz, 1H), 2.94 (d, J = 5.1 Hz, 1H), 1.75 (m, 1H), 1.64 (ddd, J = 3.6, 13.8, 13.8 Hz, 1H), 1.54 (s, 3H), 1.36 (ddd, J = 4.2, 13.8, 13.8, 1H), 1.01 (d, J = 6.6 Hz, 3H), 0.97 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 209.1 (C), 166.4 (C), 132.5 (C), 131.8 (2CH), 128.6 (2CH), 126.5 (C), 59.1 (CH), 52.6 (CH2) 50.8 (CH), 40.4 (CH2), 25.4 (CH), 23.4 (CH3), 21.4 (CH2), 16.7 (CH3). HRESIMS m/z [ M+Na ] + 376.0520 (calcd for C16H20BrNO3Na, 376.0519). Crystallographic data available from CCDC under deposition number 855711.
Dipeptide sulfone (9)
Oxone (270 mg, 420 μmol) was added to a solution of dipeptide 8 (20.0 mg, 53.0 μmol) and NaHCO3 (150 mg, 1.8 mmol) in a 45% acetone/H2O solution (9.6 mL) at 0 °C. The mixture was stirred at 25°C for 1h. After addition of H2O (10 mL), the resulting solution was extracted with CHCl3 (3 × 10 mL). The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to give 9 as a white powder (20.6 mg, 99%). [ α ] 24D -20 (c 0.4, CH3CN); 1H NMR (CDCl3, 600 MHz) δ 7.03 (d, J = 7.8 Hz, 1H), 6.10 (d, J = 7.8 Hz, 1H), 4.69 (m, 1H), 4.24 (dd, J = 7.2, 7.8 Hz, 1H), 3.77 (s, 3H), 3.15 (m, 1H), 3.08 (m, 1H), 2.94 (s, 3H), 2.46 (m, 1H), 2.23 (dd, J = 7.8, 7.8 Hz, 2H), 2.22 (m, 1H), 2.09 (m, 1H), 1.62 (m, 2H), 1.32 (m, 2H), 1.31 (m, 2H), 0.97 (d, J = 6.6 Hz, 3H), 0.96 (d, J = 7.2 Hz, 3H), 0.89 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 173.6 (C), 171.7 (C), 171.0 (C), 58.7 (CH), 52.8 (CH3), 50.7 (CH2), 50.6 (CH), 40.8 (CH3), 36.6 (CH2), 31.4 (CH2), 30.7 (CH), 25.3 (CH2), 24.7 (CH2), 22.3 (CH2), 19.2 (CH3), 18.3 (CH3), 13.9 (CH3). HRESIMS m/z [ M+H ] + 393.2055 (calcd for C17H33N2O6S, 393.2054).
Synthetic Carmaphycin A (1)
A solution of dipeptide sulfoxide 8 (76.2 mg, 200 μmol) and LiOH.H2O (42.1 mg, 1.0 mmol) in 1,4-dioxane/H2O (9 mL, 2:1) was stirred at 25 °C. After 1 h, TLC (80% EtOAc/hexanes) showed absence of starting material. Solvents were evaporated off and the resulting residue was dissolved in H2O (10 mL), acidified and extracted with n-butanol (3 × 15 mL). The combined organic extracts were dried (Na2SO4) and concentrated to obtain the free acid of 8 as a white solid. In a separate reaction, Boc-L-Leu epoxyketone (6) (31.0 mg, 110 μmol) in CH2Cl2 (1 mL) was treated with TFA (200 μL, 2.6 mmol) and stirred at 25°C until TLC showed the absence of starting material (1h), whereupon it was concentrated in vacuo to a reddish oil. This oil was dissolved in CH2Cl2 (2 mL) and added to a solution of the previously prepared free acid (34.0 mg, 93.0 μmol) and PyBOP (50.0 mg, 93.0 μmol) in CH2Cl2 (3 mL) at 25°C; followed by addition of DiPEA (66 μL, 380 μmol). After stirring 20h at 25°C, the reaction mixture was quenched with saturated NH4Cl, followed by solvent partition and CH2Cl2 (3 × 10 mL) extractions of the aqueous layer. All organic extracts were combined and dried (Na2SO4), to be then concentrated in vacuo. Silica gel column chromatography (10% MeOH/EtOAc), followed by normal phase HPLC (Phenomenex Luna 5μ Silica 100Å, 250 × 10.0 mm, 50% iso-PrOH/hexanes at 3.0 mL/min, detection at 211 and 235 nm), yielded synthetic 1 as a colorless oil comprised of a 1.6:1 diastereomeric mixture (24.4 mg, 50%). [ α ] 24D +22 (c 0.5, CH3CN); CD (c 0.2, CH3CN) λmax (Δε) 290 (+3.36), 250 (+0.17), 230 (+1.13), 200 (-6.99); major diastereomer 1H NMR (CDCl3, 600 MHz) δ 7.72 (d, J = 7.2 Hz, 1H), 6.08 (d, J = 8.4 Hz, 1H), 4.82 (m, 1H), 4.48 (m, 1H), 4.29 (dd, J = 6.0, 8.4 Hz, 1H), 3.39 (ddd, J = 4.2, 10.2, 14.4 Hz, 1H), 3.33 (d, J = 5.1 Hz, 1H), 2.90 (d, J = 5.1 Hz, 1H), 2.73 (s, 3H), 2.69 (dt, J = 5.4, 15.0 Hz, 1H), 2.50 (m, 1H), 2.23 (t, J = 7.8 Hz, 2H), 2.14 (m, 1H), 2.12 (m, 1H), 1.70 (m, 1H), 1.64 (m, 2H), 1.55 (m, 1H), 1.52 (s, 3H), 1.34 (m, 1H), 1.31 (m, 2H), 1.29 (m, 2H), 0.94 (d, J = 7.8 Hz, 3H), 0.93 (d, J = 7.2 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 7.2 Hz, 3H), 0.89 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 208.6 (C), 173.5 (C), 171.5 (C), 170.35 (C), 59.3 (C), 58.33 (CH), 52.5 (CH2) 51.3 (CH), 50.9 (CH), 48.9 (CH2), 39.1 (CH2), 38.8 (CH3), 36.7 (CH2), 31.4 (CH2), 30.9 (CH), 28.2 (CH2), 25.4 (CH2), 25.3 (CH), 23.35 (CH3), 22.4 (CH2), 21.1 (CH3), 19.3 (CH3), 17.9 (CH3), 16.8 (CH3), 13.9 (CH3); minor diastereomer 1H NMR (CDCl3, 600 MHz) δ 7.93 (d, J = 7.2 Hz, 1H), 7.00 (d, J = 6.6 Hz, 1H), 6.08 (d, J = 8.4 Hz, 1H), 4.82 (m, 1H), 4.48 (m, 1H), 4.27 (dd, J = 6.6, 8.4 Hz, 1H), 3.39 (ddd, J = 4.2, 10.2, 14.4 Hz, 1H), 3.36 (d, J = 5.1 Hz, 1H), 2.91 (d, J = 5.1 Hz, 1H), 2.77 (dt, J = 5.4, 13.8 Hz, 1H), 2.71 (s, 3H), 2.41 (m, 1H), 2.22 (t, J = 7.2 Hz, 2H), 2.10 (m, 1H), 2.18 (m, 1H), 1.70 (m, 1H), 1.63 (m, 2H), 1.53 (m, 1H), 1.52 (s, 3H), 1.35 (m, 1H), 1.31 (m, 2H), 1.29 (m, 2H), 0.94 (d, J = 7.8 Hz, 3H), 0.93 (d, J = 7.2 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 7.2 Hz, 3H), 0.89 (t, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 209.2 (C), 173.4 (C), 171.1 (C), 170.31 (C), 59.3 (C), 58.28 (CH), 52.6 (CH2) 51.4 (CH), 50.7 (CH), 46.9 (CH2), 39.2 (CH2), 36.7 (CH2), 36.1 (CH3), 31.4 (CH2), 31.1 (CH), 26.0 (CH2), 25.4 (CH2), 25.2 (CH), 23.31 (CH3), 22.4 (CH2), 21.1 (CH3), 19.2 (CH3), 18.0 (CH3), 16.8 (CH3), 13.9 (CH3). HRESIMS m/z [ M+H ] + 516.3100 (calcd for C25H46N3O6S, 516.3102).
Synthetic Carmaphycin B (2)
A solution of dipeptide sulfone (9) (37.6 mg, 96.0 μmol) and LiOH.H2O (21.0 mg, 480 μmol) in 1,4-dioxane/H2O (5 mL, 2:1) was stirred at 25 °C. After 1 h, TLC (80% EtOAc/hexanes) showed absence of starting material. Solvents were evaporated off and the resulting residue was dissolved in H2O (10 mL), acidified and extracted with n-butanol (3 × 15 mL). The combined organic extracts were dried (Na2SO4) and concentrated to obtain the free acid of 9 as a white solid. In a separate reaction, Boc-L-Leu epoxyketone (6) (32.0 mg, 110 μmol) in CH2Cl2 (1 mL) was treated with TFA (200 μL, 2.6 mmol) and stirred at 25°C for 1h, whereupon it was concentrated in vacuo to a reddish oil. This oil was dissolved in CH2Cl2 (2 mL) and added to a solution of the previously prepared free acid and PyBOP (50.0 mg, 96.0 μmol) in CH2Cl2 (3 mL) at 25°C; followed by addition of DiPEA (66 μL, 380 μmol). After stirring 15h at 25°C, the reaction mixture was quenched with saturated NH4Cl, followed by solvent partition and CH2Cl2 (3 × 10 mL) extractions of the aqueous layer. All organic extracts were combined and dried (Na2SO4), to be then concentrated in vacuo. Silica gel column chromatography (100% iso-PrOH), followed by normal phase HPLC (Phenomenex Luna 5μ Silica 100Å, 250 × 10.0 mm, 20% iso-PrOH/hexanes at 3.0 mL/min, detection at 211 and 235 nm), yielded synthetic 2 as a colorless oil (16.7 mg, 33%). [ α ] 24D +8 (c 0.5, CH3CN); CD (c 0.2, CH3CN) λmax (Δε) 290 (+1.09), 240 (-0.017), 230 (+0.21), 200 (-2.76); 1H NMR (CDCl3, 600 MHz) δ 7.11 (broad s, 2H), 6.28 (broad s, 1H), 4.76 (dt, J = 7.2, 7.2 Hz, 1H), 4.51 (ddd, J = 2.4, 7.8, 9.0, 1H), 4.28 (dd, J = 7.2, 7.2 Hz, 1H), 3.39 (ddd, J = 7.2, 7.8, 14.4 Hz, 1H), 3.30 (d, J = 4.8 Hz, 1H), 3.18 (ddd, J = 6.0, 7.2, 14.4 Hz, 1H), 3.02 (s, 3H), 2.91 (d, J = 4.8 Hz, 1H), 2.28 (m, 1H), 2.23 (t, J = 7.8 Hz, 2H), 2.18 (m, 1H), 2.06 (m, 1H), 1.68 (m, 1H), 1.62 (m, 2H), 1.57 (ddd, J = 3.0, 10.2, 13.8 Hz, 1H), 1.51 (s, 3H), 1.31 (m, 1H), 1.31 (m, 2H), 1.29 (m, 2H), 0.94 (d, J = 7.2 Hz, 3H), 0.93 (d, J = 6.6 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 7.8 Hz, 3H), 0.89 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 150 MHz) δ 208.3 (C), 173.5 (C), 171.2 (C), 169.8 (C), 59.1 (C), 58.2 (CH), 52.3 (CH2), 51.1 (CH), 50.4 (CH2), 50.2 (CH), 40.4 (CH3), 38.9 (CH2), 36.3 (CH2), 31.2 (CH2), 30.7 (CH), 25.4 (CH2), 25.2 (CH), 25.0 (CH2), 23.1 (CH3), 22.1 (CH2), 20.9 (CH3), 19.0 (CH3), 17.9 (CH3), 16.6 (CH3), 13.7 (CH3). HRESIMS m/z [ M+Na ] + 554.2867 (calcd for C25H45N3O7SNa, 554.2870).
Computational Methods and Software Packages
X-ray crystal structures for 20S proteasome complexes with epoxomicin (10) (PDB ID 1G65),[17] salinosporamide A (11) (PDB ID 2FAK),[22] and bortezomib (12) (PDB ID 3MG0)[23] were obtained from the literature. Images in Figures 2 and S10 were produced with the VMD 1.9[24] and Discovery Studio 3.1[25] software packages. In silico, the carmaphycin A (1) and B (2) structures were constructed using the crystal structure of epoxomicin (9) (PDBID 1G65)[17] as a guide. Moving left to right with respect to Figure 1, the software package Avogadro[26] was used to convert epoxomicin's threonine side chain into a methionine side chain with either a sulfoxide (R,S) or sulfone moiety. Similarly, we converted the isoleucine side chain to a valine side chain and removed the remaining atoms of epoxomicin (9) leaving the alkane chain of the carmaphycins.
In preparation for molecular dynamics (MD) simulations, we used Gaussian 09[27] with the HF/6-31G* model to calculate the electronic structure around the carmaphycins and Antechamber[28] to assign RESP[29] partial charges to the atoms. For the MD simulations, we excluded most of the 20S proteasome structure except those chains (K and L) that form the β5 chymotrypsin-like site. To prevent instability of the binding pocket, we applied harmonic restraints to these chains during all simulation steps: minimization = 10 kcal/mol/A2, equilibration = 5 kcal/mol/A2, and production = 1 kcal/mol/A2. All simulations were performed with the AMBER 11 simulation package[28] using the Amber ff99SB and GAFF force fields. Water solvation was accounted for via the OBC generalized Born implicit solvation model[30] with a monovalent salt concentration of 0.145 M. During minimization, 1000 steps of steepest descent were done followed by up to 5000 steps of the conjugate gradient method. After minimization, the systems were heated to 300K in 50K increments using a Langevin thermostat over a total time of 75ps followed by 400ps of equilibration at 300K. Langevin dynamics production runs for each system were run for 20ns with 2fs time steps and the SHAKE[31] algorithm applied to the hydrogen bonds.
Supplementary Material
Acknowledgments
We thank J. Nunnery and the CARMABI research station for this cyanobacterial collection. Special thanks to A. Mrse (Chemistry, UCSD) and X. Huang (Biochemistry, UCSD) for assistance with the NMR spectrometers, Y. Su (Mass Spectrometry, UCSD) for HRMS data acquisition, A. L. Rheingold (Crystallography, UCSD) for X-ray data analysis, and N. Engene (SIO, UCSD) for his input in the cyanobacterial identification. We also acknowledge FAPESP (Brazil) for providing a fellowship to H. M. D., as well as NIH/NCI CA100851, CA127622 and GM61300 for funding this research.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Tidgewell K, Clark BT, Gerwick WH. In: Comprehensive Natural Products Chemistry II. Mander LN, Liu HW, editors. Vol. 2. Elsevier; London: 2010. pp. 141–188. [Google Scholar]
- 2.a) Jaspars M, Lawton LA. Curr Opin Drug Discov Devel. 1998;1:77–84. [PubMed] [Google Scholar]; b) Mayer AMS, Gustafson KR. European J Cancer. 2008;44:2387–2387. [Google Scholar]; c) Tan LT. Appl Phycol. 2010;22:659–676. [Google Scholar]
- 3.a) Luesch H, Chanda SK, Raya RM, DeJesus PD, Orth AP, Walker JR, Izpisua Belmonte JC, Schultz PG. Nat Chem Biol. 2006;2:158–167. doi: 10.1038/nchembio769. [DOI] [PubMed] [Google Scholar]; b) Liu Y, Law BK, Luesch H. Mol Pharmacol. 2009;76:91–104. doi: 10.1124/mol.109.056085. [DOI] [PubMed] [Google Scholar]
- 4.a) Gerwick WH, Proteau PJ, Nagle DG, Hamel E, Blokhin A, Slate DL. J Org Chem. 1994;59:1243–1245. [Google Scholar]; b) Luduena RF, Prasad V, Roach MC, Banerjee M, Yoo HD, Gerwick WH. Drug Dev Res. 1997;40:223–229. [Google Scholar]
- 5.a) Harrigan GG, Luesch H, Yoshida WY, Moore RE, Nagle DG, Paul VJ, Mooberry SL, Corbett TH, Valeriote FA. J Nat Prod. 1998;61:1075–1077. doi: 10.1021/np980321c. [DOI] [PubMed] [Google Scholar]; b) Luesch H, Yoshida WY, Moore RE, Paul VJ, Mooberry SL, Corbett TH. J Nat Prod. 2002;65:16–20. doi: 10.1021/np010317s. [DOI] [PubMed] [Google Scholar]
- 6.Al-awar RS, Shin C. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton: 2005. pp. 151–169. [Google Scholar]
- 7.a) Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. J Am Chem Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Y, Salvador LA, Byeon S, Ying Y, Kwan JC, Law BK, Hong J, Luesch H. J Pharmacol Exp Ther. 2010;335:351–361. doi: 10.1124/jpet.110.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Taori K, Paul VJ, Luesch H. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 8.Valeriote FA, Grieshaber CK, Media J, Pietraszkewicz H, Hoffmann J, Pan M, McLaughlin SJ. J Exp Ther Oncol. 2002;2:228–236. doi: 10.1046/j.1359-4117.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- 9.This sample was identified on the basis of morphology and phylogenetic inference of its 16S rRNA gene sequence (see supporting information).
- 10.Metabolites 1 and 2 are named after the CARMABI research station, Curaçao.
- 11.These masses were obtained via NMR quantitation by solvent 13C-satellites. See: Dalisay DS, Molinski TF. J Nat Prod. 2009;72:739–744. doi: 10.1021/np900009b.
- 12.The term “carmaphycin A” refers to a mixture of epimers at the sulfoxide stereocenter.
- 13.This epoxidation is highly diastereoselective. Only the less polar and more abundant diastereomer 6 was purified. See: Witte MD, Florea BI, Verdoes M, Adeyanju O, Van der Marel GA, Overkleeft HS. J Am Chem Soc. 2009;131:12064–12065. doi: 10.1021/ja901231w.Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BHB, Crews CM. Bioorg Med Chem Lett. 1999;9:2283–2288. doi: 10.1016/s0960-894x(99)00376-5.Zhou H, Aujay MA, Bennett MK, Dajee M, Demo SD, Fang Y, Ho MN, Jiang J, Kirk CJ, Laidig GJ, Lewis ER, Lu Y, Muchamuel T, Parlati F, Ring E, Shenk KD, Shields J, Shwonek PJ, Stanton T, Sun CM, Sylvain C, Woo TM, Yang J. J Med Chem. 2009;52:3028–3038. doi: 10.1021/jm801329v.
- 14.a) Tsuchiya K, Kobayashi S, Nishikiori T, Nakagawa T, Tatsuta K. J Antibiot. 1997;50:261–263. [PubMed] [Google Scholar]; b) Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Chem Biol. 1999;6:811–822. doi: 10.1016/s1074-5521(99)80128-8. [DOI] [PubMed] [Google Scholar]; c) Meng L, Mohan R, Kwok BHB, Eloffsson M, Sin N, Crews CM. Proc Natl Acad Sci USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sugawara K, Hatori M, Nishiyama Y, Tomita K, Kamei H, Konishi M, Oki T. J Antibiot. 1990;43:8–18. doi: 10.7164/antibiotics.43.8. [DOI] [PubMed] [Google Scholar]; e) Hanada M, Sugawara K, Kaneta K, Toda S, Nishiyama Y, Tomita K, Yamamoto H, Konishi M, Oki T. J Antibiot. 1992;45:1746–1752. doi: 10.7164/antibiotics.45.1746. [DOI] [PubMed] [Google Scholar]
- 15.a) Kisselev AF, van der Linden WA, Overkleeft HS. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk FKD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK. Cancer Res. 2007;67:6383–6391. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]; c) Sterz J, Von Metzler I, Hahne JC, Lamottke B, Rademacher J, Heider U, Terpos E, Sezer O. Expert Opin Investig Drugs. 2008;17:879–895. doi: 10.1517/13543784.17.6.879. [DOI] [PubMed] [Google Scholar]
- 16.a) Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew Chem Int Ed. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]; b) Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, Chandra J, Chauhan D, Cusack JC, Jr, Fenical W, Ghobrial B, Groll M, Jensen PR, Lam KS, Lloyd GK, McBride W, McConkey DJ, Miller CP, Neuteboom STC, Oki Y, Ovaa H, Pajonk F, Richardson PG, Roccaro AM, A M, Sloss CM, Spear MA, Valashi E, Younes A, Palladino MA. Curr Cancer Drug Targets. 201111:254–284. doi: 10.2174/156800911794519716. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gulder TAM, Moore BS. Angew Chem Int Ed. 2010;49:9346–9367. doi: 10.1002/anie.201000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groll M, Kim KB, Kairies N, Huber R, Crews CM. J Am Chem Soc. 2000;122:1237–1238. [Google Scholar]
- 18.A similar interaction was observed in the crystal structure of the 20S proteasome β2 subunit with TMC-95A, a non-covalent cyclic peptide inhibitor isolated from the fungus Agiospora montagnei. See Groll M, Koguchi Y, Huber R, Kohno J. J Mol Biol. 2001;311:543–548. doi: 10.1006/jmbi.2001.4869.
- 19.Nett M, Gulder TAM, Kale AJ, Hughes CC, Moore BS. J Med Chem. 2009;52:6163–6167. doi: 10.1021/jm901098m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Tidgewell K, Engene N, Byrum T, Media J, Doi T, Valeriote FA, Gerwick WH. ChemBioChem. 2010;11:1458–1466. doi: 10.1002/cbic.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mevers E, Liu WT, Engene N, Mohimani H, Byrum T, Pevzner PA, Dorrestein PC, Spadafora C, Gerwick WH. J Nat Prod. 2011;74:928–936. doi: 10.1021/np200077f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gross H, McPhail KL, Goeger DE, Valeriote FA, Gerwick WH. Phytochem. 2010;71:1729–1735. doi: 10.1016/j.phytochem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coll JC, Bowden BF. J Nat Prod. 1986;49:934–936. [Google Scholar]
- 22.Groll M, Huber R, Potts BC. J Am Chem Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 23.Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barret C, Liu JX, Soucy TA, Sappal DS, Bump N, Olhava EJ, Fleming P, Dick LR, Tsu C, Sintchak MD, Blank JL. Biochem J. 2010;430:461–476. doi: 10.1042/BJ20100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1996;14.1:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Discovery Studio 3.1. Accelrys; San Diego, CA: 2011. [Google Scholar]
- 26.Avogadro: an open-source molecular builder and visualization tool. Version 1.03. http://avogadro.openmolecules.net/
- 27.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.01. Gaussian; Wallingford CT: 2009. [Google Scholar]
- 28.Case DA, Darden TA, CheathamIII TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossvai I, Wong KF, Paesani F, Vanicek J, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews H, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 11. University of California; San Francisco, CA: 2010. [Google Scholar]
- 29.Cornell WD, Cieplak P, Bayly CI, Kollmann PA. J Am Chem Soc. 1993;115:9620–9631. [Google Scholar]
- 30.Onufriev A, Bashford D, Case DA. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 31.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.