

Abstract
Nicotine intake affects CNS responses to stressors. We reported that nicotine self-administration (SA) augmented the hypothalamo-pituitary-adrenal (HPA) stress response, in part due to altered neurotransmission and neuropeptide expression within hypothalamic paraventricular nucleus (PVN). Limbic-PVN interactions involving medial prefrontal cortex, amygdala, bed nucleus of the stria terminalis (BST) greatly impact the HPA stress response. Therefore, we investigated the effects of nicotine SA on stress-induced neuronal activation in limbic-PVN network, using c-Fos protein immunohistochemistry and retrograde tracing. Nicotine decreased stress-induced c-Fos in prelimbic cortex (PrL), anteroventral BST (avBST), and peri-PVN; but increased c-Fos induction in medial amygdala (MeA), locus coeruleus, and PVN. Fluoro-gold (FG) was injected into avBST or PVN, since GABAergic neurons in avBST projecting to PVN corticotrophin-releasing factor (CRF) neurons relay information from both PrL glutamatergic and MeA GABAergic neurons. The stress-induced c-Fos expression in retrograde-labeled FG+ neurons was decreased in PrL by nicotine, but increased in MeA, and also reduced in avBST. Therefore, within limbic-PVN network, nicotine SA exerts selective regional effects on neuronal activation by stress. These findings expand the mechanistic framework by demonstrating altered limbic-BST-PVN interactions underlying the disinhibition of PVN CRF neurons, an essential component of the amplified HPA response to stress by nicotine.
Keywords: nicotine, stress, medial prefrontal cortex, bed nucleus of the stria terminalis, amygdale, corticotrophin-releasing factor
Introduction
There are complex bidirectional interactions between stress and smoking (Richards et al. 2011). Stress is related to initiation (Finkelstein et al. 2006), maintenance (Payne et al. 1991) and relapse to smoking (Siahpush & Carlin 2006). However, the effect of smoking itself on CNS stress responses is complex. Habitual smokers tend to exhibit greater and prolonged emotional reactions to stressful problem solving tasks (Tsuda et al. 1996), while smoking appears to reduce emotional discomfort after a stressful task (Perkins et al. 1992). The hypothalamic-pituitary-adrenal (HPA) response to psychological stress (i.e. public speaking and mental arithmetic) can be blunted in smokers (Kirschbaum et al. 1993, Childs & de Wit 2009, Rohleder & Kirschbaum 2006); on the other hand, during psychological stress, ad libitum smokers also show greater and prolonged cortisol secretion (Pomerleau & Pomerleau 1990, al’Absi et al. 2003, McKee et al. 2011). In rats, chronic forced administration of nicotine prevented the elevation of plasma corticosterone after acute restraint stress, an emotive stressor (Cheng et al. 2005); Yet, HPA responses to acute mild footshock stress, an emotive stressor involving limbic pathways (Li & Sawchenko 1998, Herman & Cullinan 1997), were augmented during chronic nicotine self-administration (SA) (Chen et al. 2008). As we have previously reported (Chen et al. 2008), the effects of nicotine SA are stressor-specific. This emphasizes the importance of studies focused on the specific CNS pathways mediating the effects of a single well-defined stressor.
The mechanisms of the enhanced HPA responsiveness to mild footshock stress during nicotine SA are only partially understood. First, nicotine SA increases the expression of arginine vasopressin (AVP) in PVN corticotrophin-releasing factor (CRF) neurons, the critical neuropeptides regulating adrenocorticotropin secretion during stress (Antoni 1986), and facilitates the activation of these CRF+/AVP+ neurons in response to stress (Yu et al. 2008). The CRF neurons in PVN are innervated by glutamatergic, and GABAergic inputs (Cummings & Seybold 1988, Herman et al. 2000). Nicotine SA augments PVN glutamate release, but diminishes GABA release induced by stress; this exaggerated reduction in GABA release and enhanced glutamate release contribute to the enhanced HPA response to stress by nicotine (Yu et al. 2010). Therefore, both altered neuropeptide expression and modulation of neurotransmission within PVN are pivotal local factors that account for the enhanced HPA response to stress by nicotine. However, the effects of nicotine SA on the limbic network that modulates local PVN neurotransmission are still unknown.
Multiple regions of the limbic system, such as medial prefrontal cortex (mPFC), amygdala, and hippocampus, influence the HPA response to a variety of stressors (Herman et al. 2005). HPA regulation by limbic structures is region-specific: dorsal mPFC (prelimbic and anterior cingulate cortex) and ventral subiculum of hippocampus inhibit the HPA response to emotional stressors, whereas ventral mPFC (infralimbic cortex) and medial amygdala (MeA) enhance this response (Dayas et al. 1999, Radley et al. 2006, Herman et al. 1995). However, limbic structures do not regulate PVN directly; instead, their efferents innervate subcortical relay sites such as bed nucleus of the stria terminalis (BST), peri-PVN area and so on, which in turn innervate PVN CRF neurons. Various limbic structures may affect a particular relay site. For example, projections from prelimbic cortex (PrL) and ventral subiculum converge on a common relay, the GABAergic neurons in anteroventral BST (avBST), including the fusiform and dorsomedial subdivisions of BST (Ju & Swanson 1989). These neurons regulate PVN CRF neurons by inhibiting the HPA responses to acute emotional stress (Radley et al. 2009, Radley & Sawchenko 2011). Lastly, limbic structures are regulated by other regions also involved in the integrated response to stress. For example, noradrenergic innervation of PrL from locus coeruleus modulates HPA responses to acute restraint stress (Radley et al. 2008).
Based on the critical role of the limbic system in providing the emotive component of the HPA response to stress (Herman et al. 2005) and the acute effects of nicotine on multiple limbic regions (Matta et al. 1993, Seppa et al. 2001), we hypothesize that chronic nicotine SA alters stress-induced neuronal activation of critical pathways in the limbic-PVN network. To evaluate this, we determined the effects of nicotine on: (i) induction of c-Fos by mild footshock stress in the brain (ii) stress-induced c-Fos within the Fluoro-gold (FG) labeled neurons in limbic-BST-PVN projection pathway. These experiments found that nicotine diminished stress-induced activation in neuronal circuits projecting from PrL to avBST and from avBST to PVN. Increased activity in this pathway is known to inhibit PVN CRF neurons (Radley et al. 2009). In contrast, nicotine augmented the stress-induced activation of neurons projecting from MeA that inhibit avBST (Herman et al. 2003). The results reported herein expand the mechanistic framework by demonstrating that chronic nicotine SA modulates stress-induced neuronal activation in the limbic-PVN network, leading to a reduction in the activity of inhibitory neuronal projections from avBST to PVN and an increase in the stress-responsiveness of PVN CRF neurons.
Materials and methods
Materials
(−)-Nicotine hydrogen tartrate was purchased from Sigma-Aldrich (St. Louis, MO). Dual channel swivels, polyethylene buttons and metal springs for intravenous infusion were purchased from Instech Laboratories (Plymouth Meeting, PA). Operant chambers, circuit boards, interface modules and L2T2 software for nicotine SA were purchased from Coulbourn Instruments (Allentown, PA). Fluoro-gold (FG) was purchased from Fluorochrome (Denver, CO). Midgard ™ precision current source for iontophoretic injection was purchased from Stoeling Co. (Wood Dale, IL).
Animals and surgeries
Adult male Sprague-Dawley rats (280–300 g, Harlan, Madison, WI) were given 7 d to recovery from shipping and acclimated to a reversed 12:12 h light:dark cycle. Standard rat chow and water were provided ad libitum throughout all experiments. All procedures conformed to NIH guidelines and were approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center. Rats received jugular catheters under ketamine/xylazine anesthesia (13:87 mg/kg body weight, respectively, i.p.). For retrograde labeling of avBST or PVN-projection neurons, 2% FG solution in saline (Schmued & Fallon 1986) was iontophoretically injected (0.5 A, 7 s on/off) into avBST or PVN by using glass micropipettes (15–20 μm tip diameter) for 5 min and 2.5 min respectively. The stereotaxic coordinates for avBST were: anteroposterior (AP) = −0.3 mm; mediolateral (ML) = +1.4 mm; dorsoventral (DV) = −7.2 mm (Paxinos & Watson 1997). The coordinates for PVN were: AP = −1.8; ML = +0.3; DV = −7.4. The micropipette was kept in place for 10 min after injection to reduce back-diffusion of the FG along the injection track.
Nicotine self-administration
The nicotine SA procedure was conducted using our published protocol (Valentine et al. 1997). After recovery (3 d) from jugular surgery in operant chambers, rats were given access to nicotine or saline 23 h/d for 12 d, without prior training, priming or food deprivation. The final hour of the 12 h lights-on cycle (i.e., 9:00–10:00 AM) was reserved for housekeeping tasks. The operant chamber contained two horizontal levers, and a green cue light above each lever was illuminated when nicotine was available. Lever presses were recorded and syringe pumps were controlled by computers and interfaces. Pressing the active lever, randomly designated, elicited a computer-driven injection (i.v., 50 μl/0.81 s) of nicotine (0.03 mg/kg) or saline via jugular vein. Each injection was followed by a 7-s period during which the green cue light was extinguished but nicotine was unavailable. Pressing the alternate (inactive) lever had no programmed consequence.
Mild footshock stress
Electric footshock or sham-shock was delivered 4 h after the beginning of the SA session on the final day of SA, allowing animals ad libitum access to nicotine prior to, during, and after the shock session. A total of five shocks (0.6 mA, 0.5 s per shock) were randomly delivered over 5 min through the grid floor. During the hour immediately after the brief episode of footshock, lever press behavior was unchanged (Chen et al. 2008).
Experiment protocols
In the first experiment, the effects of nicotine or saline SA (referred to as nicotine or saline, respectively) on c-Fos induction throughout whole brain regions by mild footshock stress were identified. On d 13 of SA, 2 h after shock (maximal c-Fos induction occurred 2 h after footshock stress) (Li & Sawchenko 1998), nicotine or saline (n=8 in each group, 3 for sham-shock, and 5 for shock treatment) rats were deeply anesthetized with sodium pentobarbital (80mg/Kg, i.p.) and perfused transcardially with 100 ml of 0.9% saline followed by 500 ml of 4% paraformaldehyde in 0.1 M phosphate buffer. After perfusion, brains were immediately removed, post-fixed overnight at 4 °C, cryoprotected in 30% sucrose in 0.1 M phosphate buffer for 2 or 3 d at 4 °C, and then fast frozen and stored at −80 °C until processing. In the second experiment, the effects of nicotine on stress-induced c-Fos expression in limbic-BST-PVN projection pathway were determined. In a separate group of animals, FG was deposited into avBST or PVN on the day of jugular surgery. On d 13 of SA (i.e., 15 d after FG injection), nicotine (n = 6 for avBST and PVN) or saline (n = 6 for avBST and PVN) rats were sacrificed 2 h after footshock and perfused with 4% paraformaldehyde. Brains were post-fixed, cryoprotected in 30% sucrose, and stored at −80 °C until processing as described above. Animals were excluded due to inaccurate deposition of FG: 1 in the nicotine and saline avBST groups and 3 in each of the respective PVN groups.
Immunohistochemistry of c-Fos protein
Coronal brain sections (30 μm) from medial frontal cortex to brainstem were obtained by cryosection (CM1850, Leica, Nussloch, Germany) and mounted onto pre-cleaned slides. Every fifth section was saved for staining. Immunoperoxidase labeling of c-Fos was performed, as described previously (Valentine et al. 1996). Briefly, tissue sections were blocked with 2% normal goat serum containing 0.03% triton-X100 in 0.01 M phosphate buffered saline for 30 min, and then incubated in rabbit anti-c-Fos antibody (detects acid residues 4–17 of rat c-Fos; 1:10000; Chemicon, Billerica, MA) at 4 °C for 3 d. Then slides were sequentially incubated in biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA) for 60 min, and in VECTASTAIN™ ABC reagent (Vector Laboratories) for 30 min at 20–25 °C, and visualized by a 5 min exposure to 3,3′-diaminobenzidine with metal (nickel) enhancer (Vector Laboratories). Finally, the slides were dehydrated, cleared, and cover-slipped.
Dual immunohistochemistry of FG and c-Fos
Coronal sections (30 μm) of medial prefrontal cortex, MeA (regions projecting to avBST) or avBST (projects to PVN) were obtained by cryosection and mounted onto pre-cleaned slides. Every fifth section was saved for staining. Dual immunoperoxidase labeling for FG and c-Fos was performed in two separate stages. First, slides were blocked with 2% normal goat serum for 30 min, and then sequentially incubated in rabbit anti-FG antibody (1:200, Fluorochrome, Denver, CO) at 20–25 °C overnight, and in biotinylated goat anti-rabbit IgG (1:200) for 60 min, followed by application of the VECTASTAIN™ ABC reagent for 30 min. FG was then visualized with diaminobenzidine to produce brown cytoplasmic staining. In the second stage, after rinsing, slides were sequentially incubated in rabbit anti-c-Fos antibody (1:10000, Chemicon) at 4 °C for 3 d, biotinylated goat anti-rabbit IgG and the ABC reagent. Diaminobenzidine with nickel was then used to produce gray-black nuclear staining. Finally, the slides were dehydrated, cleared, and cover-slipped.
Microscopy and data analysis
The expression of c-Fos in serial sections from mPFC to brainstem was analyzed using an Olympus BX60 light microscope (Olympus America Inc., Center Valley, PA). A single investigator, blind to the assignment of treatment groups, determined the level of c-Fos expression. The expression of c-Fos in a wide range of brain regions was first classified by four levels according to the following criteria: (+++), represents c-Fos expression in the majority of cells in a given brain region; (++), a moderate number of cells expressing c-Fos; (+), a low level of c-Fos expression in widely scattered cells; (−), absence of detectable c-Fos, or < 3 positive cells in a brain region. Based on the c-Fos expression and their relevance to the stress response, the following brain regions were selected for detailed counting of c-Fos+ neurons: PrL (4.2, 3.7, 3.2 mm), infralimbic cortex (3.2, 2.7, 2.2 mm), avBST (−0.2, −0.3, −0.4) hypophysiotrophic (medial parvocellular) region of PVN and peri-PVN (−1.8, −2.0, −2.2), MeA (−2.6, −2.8, −3.0), and locus coeruleus (−9.6, −9.8, −10.0). For each brain region, three sections, corresponding to its rostral, middle, and caudal aspects, were selected for analysis, with their bregma based coordinates (Paxinos and Watson 1997) provided above in the parenthesis after each brain region. Images of these brain regions were captured using an Olympus DP21 digital camera combined with either 10x or 20x objectives. The c-Fos positive cells were counted unilaterally using NIH ImageJ software (http://rsb.indo.nih.gov/ij). The sum of the c-Fos positive cells from 3 representative brain sections was used for data analysis. The c-Fos and FG double positive (c-Fos+-FG+) neurons were identified under 40x and 100x magnification and counted in the same sections as c-Fos+ neurons in PrL, avBST and MeA. The DAB reaction product indicative of FG+ neuron is a brown, punctate cytoplasmic pigment; while the nickel enhanced reaction product indicative of c-Fos+ neuron is a gray/blue nuclear pigment. Although an occasional c-Fos+-FG+ neuron showed a low level of bluish cytoplasmic discoloration, reflecting some leaching of nickel enhanced product outside of the nucleus, as infrequently observed in c-Fos single positive neurons, these neurons were readily identified by their cellular profiles of homogenous blue nuclear staining surrounded by brown cytoplasmic pigment.
Statistics
The number of lever presses during SA was analyzed by two-way ANOVA with repeated measures. The data of c-Fos induction by footshock or sham-shock in nicotine and saline groups were subjected to two-way ANOVA, and further analyzed by post hoc tests (Scheffe) for multiple comparisons between individual groups. The data of stress-induced c-Fos expression in FG-labeled neurons between the nicotine and saline shock groups were analyzed by t-test. Data were expressed as mean ± SEM. Statistical significance was assigned at p < 0.05.
Results
Nicotine self-administration
Adult male Sprague-Dawley rats acquired and maintained self-administering of nicotine or saline for 12 d. Fig. 1 represents the collective lever press record of the rats in all experiments. In the nicotine group, active lever presses were 3 to 4-fold greater than inactive presses (F1, 30 = 105.2, p < 0.001), whereas presses on both levers were similar in the saline group (F1, 30 = 1.69, p > 0.05). Additionally, active lever presses were greater in the nicotine than saline group (F1, 30 = 97.1, p < 0.001), but inactive lever presses was not different (F1, 30 = 0.7, p > 0.05). The average daily nicotine intake during last 3 d of SA was: 1.14 ± 0.01 mg/kg/day, attributable to approximately 38 infusions per d.
Fig. 1.

The acquisition and maintenance of chronic nicotine self-administration. Nicotine (30 μg/kg, i.v.) or saline was available 23 h/d for 12 d. Nicotine rats had significantly more active than inactive lever presses (repeated measure ANOVA, F1, 30 = 105.2, p < 0.001), and more active presses than saline rats (F1, 30 = 97.1, p < 0.001). Active and inactive presses were similar in saline rats (F1, 30 = 1.69, p > 0.05). All data are expressed as mean ± SEM.
Relative levels of basal c-Fos expression and induction by mild footshock stress in whole brain of nicotine rats
The results of rostro-caudal assessment of c-Fos immunoreactivity throughout the brain are reported in Table 1. In the saline and nicotine sham-shock groups, the levels of c-Fos expression were very low to undetectable in most brain regions, with a few exceptions of comparable and low expression (i.e. +) in specific regions of cortex, olfactory region, amygdala, thalamus, hypothalamus, and superior colliculus. Mild footshock stress increased c-Fos expression in multiple brain regions, including cortex, olfactory region, lateral septum, bed nucleus of the stria terminalis (BST), amygdala, thalamus, hypothalamus and brainstem. Compared to saline rats receiving footshock, nicotine altered stress-induced c-Fos expression in prelimbic cortex (PrL), anteroventral BST (avBST), medial amygdala (MeA), hypothalamic paraventricular nucleus (PVN), peri-PVN region, locus coeruleus (LC), nucleus of solitary tract (NTS)-A2, and -C2 region.
Table 1.
Relative levels of c-Fos expression induced by mild footshock stress or sham-shock in the brain of rats self-administering nicotine vs. saline.
| Brain regions | Sham-shock
|
Shock
|
||
|---|---|---|---|---|
| Saline | Nicotine | Saline | Nicotine | |
| I. Forebrain | ||||
| A. Cortex | ||||
| 1. Prelimbic | − | − | +++ | + |
| 2. Infralimbic | − | − | ++ | ++ |
| 3. Anterior cingulate | + | + | +++ | +++ |
| 4. Motor | + | + | ++ | ++ |
| 5. Somatosensory | − | − | + | + |
| 6. Insular | − | − | + | + |
| 7. Claustrum | − | − | + | + |
| B. Olfactory region | ||||
| 1. Anterior olfactory nucleus | + | + | +++ | +++ |
| 2. Piriform cortex | + | + | ++ | ++ |
| 3. Olfactory tubercle | − | − | + | + |
| C. Hippocampus | ||||
| 1. CA1 | − | − | − | − |
| 2. CA3 | − | − | − | − |
| 3. Dentate gyrus | − | − | − | − |
| 4. Dorsal subiculum | − | − | − | − |
| 5. Ventral subiculum | − | − | + | + |
| D. Lateral septum nucleus | ||||
| 1. Dorsal part | − | − | − | − |
| 2. Ventral part | − | − | + | + |
| E. Accumbens nucleus | − | − | − | − |
| F. Bed nucleus of stria terminalis | ||||
| 1. Anterodorsal region | − | − | + | + |
| 2. Anteroventral region | − | − | ++ | + |
| 3. Posterior region | − | − | + | + |
| G. Amygdala | ||||
| 1. Medial nucleus | + | + | ++ | +++ |
| 2. Central nucleus | − | − | + | + |
| 3. basolateral nucleus | − | − | + | + |
| 4. Basomedial nucleus | − | − | + | + |
| 5. Cortical nucleus | + | + | ++ | ++ |
| H. Thalamus | ||||
| 1. Habenula nucleus | − | − | + | + |
| 2. Paraventricular nucleus | + | + | +++ | +++ |
| 3. Centromedial nucleus | − | − | + | + |
| 4. Reuniens nucleus | − | − | + | + |
| 5. Mediodorsal nucleus | − | − | − | − |
| 6. Anterior thalamic area | − | − | − | − |
| 7. Lateral thalamic area | − | − | − | − |
| 8. Ventral thalamic area | − | − | − | − |
| 9. Posterior thalamic area | − | − | − | − |
| 10. Reticular nucleus | − | − | − | − |
| 11. Zona incerta | − | − | + | + |
| I. Hypothalamus | ||||
| 1. Paraventricular nucleus | ||||
| Dorsal parvocellular part | − | − | + | + |
| Medial Parvocellular part | − | − | + | +++ |
| Magnocellular part | − | − | − | − |
| 2. Peri-PVN region | − | − | ++ | + |
| 3. Medial preoptic area | + | + | ++ | ++ |
| 4. Anterior hypothalamic area | + | + | ++ | ++ |
| 5. Lateral hypothalamic area | + | + | ++ | ++ |
| 6. Dorsal hypothalamic area | + | + | ++ | ++ |
| 7. Dorsomedial nucleus | − | − | + | + |
| 8. Ventromedial nucleus | − | − | + | + |
| 9. Posterior hypothalamic area | − | − | + | + |
| 10. Arcuate nucleus | + | + | ++ | ++ |
| 11. Supraoptic nucleus | − | − | + | + |
| II. Brainstem | ||||
| 1. Ventral tegmental area | − | − | − | − |
| 2. Superior colliculus | + | + | ++ | ++ |
| 3. Dorsal raphe | − | − | + | + |
| 4. Periaqueductal gray | − | − | + | + |
| 5. Cuneiform nucleus | − | − | + | + |
| 6. Dorsal tegmental nucleus | − | − | + | + |
| 7. Barrington’s nucleus | − | − | + | + |
| 8. Locus coeruleus | − | − | + | ++ |
| 9. Lateral parabrachial nucleus | − | − | − | − |
| 10. A2/C2 region | − | − | ++ | + |
| 11. Inferior olive | − | − | + | + |
| 12. Lateral reticular nucleus | − | − | + | + |
| 13. A1/C1 region | − | − | − | − |
Level of c-Fos immunoreactivity in selected brain regions was evaluated 2 h after footshock or sham-shock in 3–5 rats from each group. A 4-point scale was used for comparison: + + +, represents c-Fos expression in the majority of cells in a given brain region; ++, a moderate number of cells expressing c-Fos; +, a low level of c-Fos expression in widely scattered cells; −, absence of detectable c-Fos, or < 3 positive cells in a brain region.
Semiquantitative effects of nicotine on stress-induced c-Fos expression
Photomicrographic images and semi-quantitative analyses of c-Fos expression are shown in Figs. 2–5. In PrL, there were very few c-Fos+ neurons in the saline (Fig. 2a, e) and nicotine (Fig. 2b, f) groups after sham-shock. Mild footshock stress greatly increased c-Fos expression in the saline group (Fig. 2c, g), but to a much less degree in the nicotine group (Fig. 2d, h); the number of stress-induced c-Fos+ neurons decreased by 49% (Fig. 2j; saline vs. nicotine/shock: 268.2 ± 23.3 vs. 136.2 ± 15.0, p < 0.01; effect of shock, F1,12 = 115.0, p < 0.001; effect of nicotine, F1,12 = 12.8, p < 0.01; nicotine × shock interaction, F1,12 = 12.7, p < 0.01).
Fig. 2.

Effect of nicotine self-administration on stress-induced neuronal activation in prelimbic cortex. Photomicrographs (a–d) illustrate immunohistochemical labeling of c-Fos in coronal tissue sections of prelimbic cortex (PrL). Boxes indicate the locations of areas used for counting c-Fos+ neurons; these areas are shown in panels e-h at higher-magnification. Schematic representations of PrL (i) are adapted from Paxions and Watson 1997, with the position of the counting square (380 × 380 μm2) shown. Both the saline (a, e) and nicotine (b, f) rats expressed c-Fos at very low levels after sham-shock. Mild footshock stress greatly increased c-Fos expression in the saline group (c, g), but to a lesser degree in the nicotine group (d, h). Mean ± SEM number of stress-induced c-Fos+ neurons is shown in panel j. *: p < 0.01, compared to the corresponding sham-shock controls. #: p < 0.01, compared to saline rats receiving footshock. n=3 for sham-shock, and n=5 for shock groups in both the saline and nicotine rats in Figs. 2–5. Scale bars: panels a–d, 100 μm; panels e–h, 50 μm.
Fig. 5.

Effects of nicotine self-administration on stress-induced neuronal activation in amygdala and hypothalamic paraventricular nucleus. Photomicrographs illustrate immunohistochemical labeling of c-Fos in coronal sections of medial amygdala (MeA; a–c) and hypothalamic paraventricular nucleus (PVN) and peri-PVN region (peri-PVN: defined as the subparaventricular zone (Spa) and the region immediately adjacent the lateral and dorsal PVN; d–f). The number of c-Fos+ neurons was counted in the area enclosed by dashed lines. In MeA, after sham-shock, both the saline (a) and nicotine rats (images not shown) exhibit scattered c-Fos+ neurons; footshock increased the expression of c-Fos in the saline group (b), and to a greater degree in the nicotine group (c). In PVN and peri-PVN, unstressed saline (a) and nicotine (images not shown) rats expressed minimal c-Fos. In the saline group (e), footshock induced c-Fos in peri-PVN, dorsal (dp) and medial parvocellular PVN (mp), but not in posterior magnocellular PVN (pm); in the nicotine group (f), foot shock induced more c-Fos in mpPVN, but less c-Fos in peri-PVN compared to the saline group. The mean ± SEM number of stress-induced c-Fos+ neurons is shown in panel g. *: p < 0.01, compared to the corresponding sham-shock controls. #: p < 0.01, compared to saline rats receiving footshock. Scale bar: panels a–c and d–f, 50 μm. opt: optic tract; AH: anterior hypothalamic area.
In infralimbic cortex (IL), there also were very few c-Fos+ neurons in the saline (Fig. 3a, d) and nicotine (images not shown) groups after sham-shock. In contrast to PrL, footshock-induced c-Fos+ neurons was similar in IL of the saline (Fig. 3b, e) and nicotine (Fig. 3c, f) groups (Fig. 3h; nicotine vs. saline/shock: 76.1 ± 6.0 vs. 82.4 ± 7.1, p > 0.05; effect of shock, F1,12 = 149.0, p < 0.001; effect of nicotine, F1,12 = 0.3, p > 0.05).
Fig. 3.

Effect of nicotine self-administration on stress-induced neuronal activation in infralimbic cortex. Photomicrographs (a–c) illustrate immunohistochemical labeling of c-Fos in coronal sections of infralimbic cortex (IL). Boxes indicate the positions of areas used for counting c-Fos+ neurons; these areas are shown in panels d-f at higher-magnification. A schematic representation of IL is shown in panel g, with the position of the counting square (380 × 380 μm2) shown. Both saline (a, d) and nicotine (images not shown) rats expressed c-Fos at very low levels after sham-shock. Mild footshock stress increased c-Fos expression to comparable levels in both the saline (b, e) and nicotine (c, f) groups. The mean ± SEM number of stress-induced c-Fos+ neurons is shown in panel h. *: p < 0.01, compared to the corresponding sham-shock controls. Scale bars: panels a–c, 100 μm; panels d–f, 50 μm. fmi: forceps minor of the corpus callosum.
In avBST, footshock induced less c-Fos in the nicotine (Fig. 4b) than saline (Fig. 4a) groups; the number of stress-induced c-Fos+ neurons decreased by 48% in avBST (Fig. 4c; saline vs. nicotine/shock: 234.4 ± 12.5 vs. 121.6 ± 17.2, p < 0.01; effect of shock, F1,12 = 155.0, p < 0.001; effect of nicotine, F1,12 = 15.8, p < 0.01; nicotine × shock interaction, F1,12 = 15.8, p < 0.01).
Fig. 4.

Effects of nicotine self-administration on stress-induced neuronal activation in bed nucleus of the stria terminals and locus coeruleus. Photomicrographs illustrate immunohistochemical labeling of c-Fos in coronal sections of the anteroventral bed nucleus of the stria terminals (avBST; a, b) and locus coeruleus (LC; d, e). The number of c-Fos+ neurons was counted in the area enclosed by dashed lines. Footshock induced less c-Fos in the nicotine (b) than saline (a) groups in avBST; but more c-Fos in the nicotine (e) than saline groups (d) in LC. The mean ± SEM number of stress-induced c-Fos+ neurons in avBST and LC are shown in panel c and f, respectively. *: p < 0.01, compared to the corresponding sham-shock controls. #: p < 0.01, compared to saline rats receiving footshock. Scale bars: panels a, b and d, e, 100 μm. ac: anterior commissure; 4V: fourth ventricle; Bar: Barrington’s nucleus; Me5: mesencephalic trigeminal nucleus.
In contrast to our previous report showing that nicotine decreased stress-induced c-Fos expression in catecholaminergic neurons of NTS (Yu & Sharp 2010), in LC, the number of stress-induced c-Fos+ neurons increased by 88% in the nicotine (Fig. 4d) compared to saline (Fig. 4e) groups (Fig. 4f; saline vs. nicotine/shock: 62.4 ± 9.2 vs. 117.6 ± 17.0, p < 0.01; effect of shock, F1,12 = 56.5, p < 0.001; effect of nicotine, F1,12 = 6.4, p < 0.05; nicotine × shock interaction, F1,12 = 6.1, p < 0.05).
In MeA, scattered c-Fos+ neurons were present after sham-shock in the saline (Fig. 5a) and nicotine groups (image not shown). Footshock increased the expression of c-Fos in the saline group (Fig. 5b), and to a greater degree in the nicotine group (Fig. 5c); the number of stress-induced c-Fos+ neurons increased by 92% (Fig. 5g; saline vs. nicotine/shock: 168.6 ± 6.3 vs. 323.2 ± 26.9, p < 0.01; effect of shock, F1,12 = 105.5, p < 0.001; effect of nicotine, F1,12 = 16.3, p < 0.01; nicotine × shock interaction, F1,12 = 17.0, p < 0.01).
In PVN and the peri-PVN region, minimal c-Fos+ neurons were observed after sham-shock in the saline (Fig. 5d) and nicotine (image not shown) groups. Footshock increased c-Fos expression in the dorsal (dp) and medial parvocellular (mp) PVN, and peri-PVN of the saline group, but not in the posterior magnocellular (pm) PVN (Fig. 5e). In the nicotine group, footshock induced less c-Fos in peri-PVN, but much greater c-Fos in the medial parvocellular PVN compared to the saline group (Fig. 5f). The number of stress-induced c-Fos+ neurons in peri-PVN decreased by 52% (Fig. 5g; saline vs. nicotine/shock: 136.4 ± 6.1 vs. 66.2 ± 3.2, p < 0.01; effect of shock, F1,12 = 418.1, p < 0.001; effect of nicotine, F1,12 = 57.9, p < 0.001; nicotine × shock interaction, F1,12 = 56.8, p < 0.001), whereas it increased by 172% in PVN (Fig. 5g; saline vs. nicotine/shock: 121.8 ± 6.6 vs. 331.8 ± 29.3, p < 0.01; effect of shock, F1,12 = 120.0, p < 0.001; effect of nicotine, F1,12 = 27.3, p < 0.001; nicotine × shock interaction, F1,12 = 27.6, p < 0.001).
In summary, nicotine decreased stress-induced neuronal activation in PrL (not IL), avBST, and peri-PVN region, but increased the activity in MeA, LC and PVN.
Semiquantitative effect of nicotine on stress-induced c-Fos expression within neurons projecting to avBST or PVN
Fluoro-gold (FG) was injected into avBST (Fig. 6i). After 2 weeks, FG-labeled neurons were primarily found in PrL and IL, nucleus accumbens shell, medial preoptic nucleus, medial and central amygdala, dorsomedial hypothalamic nucleus, arcuate nucleus, and NTS-A2 and -C2 regions. Dual immunocytochemical detection of FG and c-Fos was performed on PrL and MeA tissue sections, which had shown differences in stress-induced c-Fos expression between the nicotine and saline groups. As shown in Fig 6, c-Fos+-FG+ neurons consistently demonstrated blue pigment usually exclusively in nuclei and brown staining restricted to cytoplasm (see inserts in panels e, f, and h), while FG single positive neurons only showed brown punctate cytoplasmic staining (see insert in panels f and g). In PrL (Fig. 6a, b), FG+ neurons were located primarily in cortical layer V, and a similar number of FG+ neurons were present in the saline and nicotine groups (Fig. 6j). In the saline group, most FG+ neurons were also labeled with c-Fos induced by footshock (Fig. 6a, e). In contrast, there were fewer c-Fos+-FG+ neurons in the nicotine group (Fig. 6b, f). The number of c-Fos+-FG+ neurons decreased by 50% (Fig. 6j; saline vs. nicotine/shock: 56.2 ± 2.5 vs. 28.6 ± 1.2, p < 0.01) and the percentage of FG+ neurons that were double-positive (i.e., c-Fos+-FG+) also decreased (Fig. 6j; saline vs. nicotine/shock: 85.8% ± 1.4 vs. 49.0% ± 2.4, p < 0.01).
Fig. 6.

Effects of nicotine self-administration on stress-induced activation of neurons in prelimbic cortex and medial amygdala that project to anteroventral bed nucleus of the stria terminals. Dark-field photomicrography (i) showed that Fluoro-gold (FG) was deposited in anteroventral bed nucleus of the stria terminals (avBST; enclosed by dashed lines). Bright-field photomicrographs (a–d, 40x) illustrate dual immunohistochemical labeling of c-Fos (gray/blue nucleus) and FG (brown cytoplasm) in coronal sections of prelimbic cortex (PrL) and medial amygdala (MeA). The boxes indicate the position of the areas shown in panels e–h at higher-magnification (100x). The inserts in panels e–h are high power views of representative neurons. The c-Fos+-FG+ neurons (indicated by solid arrows) consistently demonstrated blue pigmented nuclei and brown staining restricted to cytoplasm (see inserts in panels e, f, and h). FG single positive neurons (indicated by open arrows) showed brown punctate cytoplasmic staining (see inserts in panels f and g). In PrL, footshock induced c-Fos expression in the majority of FG+ neurons in the saline group (a, e); in contrast, footshock induced c-Fos in fewer FG+ neurons in the nicotine group (b, f). In MeA, footshock induced c-Fos within more FG+ neurons in the nicotine (d, h) than saline (c, g) groups. The number of Fos+-FG+ neurons and the percentage of FG+ neurons that were double positive (mean ± SEM) were both reduced by nicotine in PrL (j) (within a 380 × 380 μm2 area, see Fig. 2i), but were increased by nicotine in MeA (k). * p < 0.001, compared to the saline group. n=5 in each group in both PrL and MeA. Scale bar: panels a–d, 50 μm; panels e-h, 20 μm, panel I, 100 μm.
In MeA, some FG+ neurons were double-labeled with c-Fos induced by footshock in the saline group (Fig. 6c, g). However, the nicotine group expressed a greater number of c-Fos+-FG+ neurons (Fig. 6d, h); the number of double-positive neurons increased by 36% (Fig. 6k; saline vs. nicotine/shock: 118.2 ± 5.7 vs. 160.5 ± 6.4, p < 0.01) and the percentage of FG+ neurons that were double-positive also increased (Fig. 6k; saline vs. nicotine/shock: 67.1% ± 2.1 vs. 91.7% ± 1.8, p < 0.01).
In a separate experiment, FG was injected into the medial parvocellular PVN (Fig. 7e), wherein CRF neurons receive axonal projections from avBST (Radley et al. 2009). Retrogradely labeled FG+ neurons were primarily found in the avBST, medial preoptic area, dorsomedial hypothalamic nucleus, and NTS-A2, and -C2. Dual immunocytochemical detection of FG and c-Fos, performed in avBST, showed differences in stress-induced c-Fos expression between the nicotine and saline groups. In the saline group, most FG+ neurons were double-labeled with c-Fos induced by footshock (Fig. 7a, c). In contrast, fewer c-Fos+-FG+ neurons were observed in the nicotine group (Fig. 7b, d); the number decreased by 35% (Fig. 7f; saline vs. nicotine/shock: 84.7 ± 10.4 vs. 56.7 ± 9.0, p < 0.01) and the percentage of FG+ neurons that were double-positive also decreased (Fig. 7f; saline vs. nicotine/shock: 77.5% ± 1.6 vs. 47.3% ± 1.7, p < 0.01).
Fig. 7.

Effect of nicotine self-administration on stress-induced activation of neurons in anteroventral bed nucleus of the stria terminals that project to hypothalamic paraventricular nucleus. Dark-field photomicrograph (e) showed Fluoro-gold (FG) was primarily deposited in the medial parvocellular PVN (mp). Bright-field photomicrographs illustrate dual immunohistochemical labeling of c-Fos (gray/blue nucleus) and FG (brown cytoplasm) in coronal sections of avBST (a, b). The boxes indicate the positions of the areas shown in panels c and d at higher magnification (100x). In the saline group (a, c), footshock induced c-Fos in the majority of FG+ neurons, as indicated by solid arrows, while open arrows indicate FG single positive neurons (d). In contrast, footshock induced c-Fos in fewer FG+ neurons in the nicotine group (b, d). The number of Fos+-FG+ cells and the percentage of FG+ neurons that were double positive in avBST (mean ± SEM) were reduced by nicotine (f). * p < 0.01, compared to the saline group. n=3 in each group. Scale bars: panels a–b, and e, 100 μm; c–d, 20 μm.
In summary, nicotine diminished the stress-induced activation of neurons projecting from PrL to avBST, but augmented the activity of neurons projecting from MeA to avBST. Moreover, nicotine reduced the activity of neurons projecting from avBST to PVN.
Discussion
As we previously reported, nicotine augments HPA responses to acute mild footshock stress, and this effect depends on changes in neurotransmission and neuropeptide expression within PVN CRF neurons (Chen et al. 2008, Yu et al. 2008, Yu & Sharp 2010, Yu et al. 2010). The current study expands this mechanistic framework by demonstrating that chronic nicotine SA modulates stress-induced neuronal activation in the limbic-PVN network, which in turn regulates local PVN neurotransmission and function (Herman et al. 2005). Nicotine diminished the stress-induced activation of pyramidal neurons projecting from PrL to avBST, and from avBST to PVN. The glutamatergic neurons in PrL innervate GABAergic neurons in avBST, and then inhibit PVN CRF neurons and the HPA stress response (Radley et al. 2009). In contrast, nicotine increased the activity of neurons innervating avBST from MeA. The majority of neurons in MeA that project to BST are GABAergic (Herman et al. 2005). Therefore, in response to stress, the reduced activity of avBST neurons by nicotine was associated with increased activity of neurons in the medial parvocellular PVN. We have previously shown that these correspond to CRF neurons (Yu et al. 2008). Fig. 8 is a schematic representation of the modulation of stress-induced neuroactivity by nicotine in the limbic-PVN network.
Fig. 8.
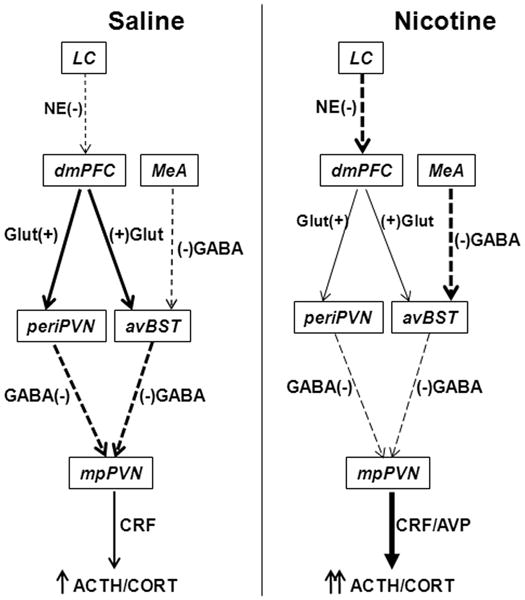
Schematic diagram of the effects of chronic nicotine self-administration on stress-induced neuroactivity in the limbic-PVN network. Nicotine diminished stress-induced activity of glutamatergic (Glut) excitatory (+) neurons (designated by thinner solid arrows compared to saline control) that project from prelimbic cortex (PrL) to anteroventral bed nucleus of the stria terminalis (avBST) and to peri-PVN. The reduced activity of PrL neurons may result from the enhanced activity of norepinephrinergic (NE) inhibitory (−) neurons (thicker dashed arrow compared to saline) projecting from locus coeruleus (LC). In contrast, nicotine increased the activity of GABAergic inhibitory (−) neurons that project from medial amygdala (MeA) to avBST. Therefore, the activities of GABAergic inhibitory (−) neurons projecting from avBST and peri-PVN to medial parvocellular PVN (mpPVN) were diminished. These effects of chronic nicotine on the limbic-PVN network result in the disinhibition of corticotrophin-releasing factor (CRF) neurons by reduced stress-induced GABA release. This disinhibition facilitates the stimulation of CRF neurons and the release of CRF and arginine vasopressin (AVP) by the enhanced stress-induced PVN glutamate release, which we have previously reported (Yu et al. 2008, Yu et al. 2010).
In addition to avBST, GABAergic neurons in peri-PVN also relay the output signals from both PrL and MeA to CRF neurons in the medial parvocellular PVN (Herman et al. 2003, Herman et al. 2005). Thus, the diminished activity of peri-PVN neurons by nicotine was also associated with reduced activity of PrL excitatory neurons and increased activity of MeA inhibitory neurons. The reduced activity of peri-PVN inhibitory neurons induced by nicotine also contributes the increased responsiveness of CRF neurons in the medial parvocellular PVN.
The PrL is known to inhibit the activity of hypophysiotropic CRF neurons in the medial parvocellular PVN, whereas IL activates autonomic neurons in the dorsal parvocellular PVN in response to emotional stress (Radley et al. 2006). Nicotine SA selectively reduced stress-induced activation of neurons in PrL, without affecting IL. Reflecting this difference, neuronal activity increased in the medial but not dorsal parvocellular PVN. Although chronic treatment with nicotine has been shown to increase the activity of autonomic nervous system (i.e. plasma epinephrine) induced by restraint stress (Morse 1989), the current study found no evidence of an effect of nicotine SA on the autonomic neurons of dorsal parvocellular PVN.
Nicotine SA reduced the stress-induced activity of layer V neurons in PrL that project to avBST. In mPFC, nicotinic cholinergic receptors (nAChRs) have not been detected on layer V pyramidal neurons, although GABAergic interneurons in this layer do express nAChRs. By binding to α4β2 nAChRs, nicotine induced these GABAergic neurons to express higher levels of glutamic acid decarboxylase 67 (GAD67), a GABA neuron-specific protein (Satta et al. 2008, Maloku et al. 2011). Additionally, acute treatment with nicotine may increase the inhibitory input to layer 5 pyramidal neurons by exciting GABA interneurons through α4β2 nAChRs (Mansvelder et al. 2009). We postulate that chronic exposure to nicotine in vivo may reduce the activity of PrL pyramidal neurons in layer V by exciting local GABA interneurons.
Pyramidal neurons in PrL layer V are innervated by noradrenergic projections from LC (Lee et al. 2005, Wang et al. 2010). These noradrenergic projections from LC inhibit stress-induced neuronal activation of PrL, thereby facilitating the activity of PVN CRF neurons and the HPA response to stress (Radley et al. 2008). Therefore, the increased activation of LC neurons by stress during nicotine SA may contribute to the reduced activity of layer V pyramidal neurons and indirectly to the increased activity of CRF neurons in the medial parvocellular PVN. Acute treatment with nicotine stimulates the expression of c-Fos in both NTS and LC (Matta et al. 1993). Chronic exposure to nicotine induced sustained transcription of tyrosine hydroxylase in LC (Sun et al. 2004). In contrast to increased c-Fos in LC, shown herein, and to the effects on tyrosine hydroxylase, we previously showed that expression of c-Fos in catecholaminergic neurons of NTS-A2 (70% of saline) and -C2 (50% of saline) was reduced by nicotine SA (Yu & Sharp 2010). Therefore, nicotine SA differentially affects stress-induced c-Fos expression in primary noradrenergic areas of the brain.
The mechanism underlying the enhanced activation of MeA neurons by stress during nicotine SA is unknown. The GABAergic neurons in MeA are innervated by glutamatergic neurons projecting from the ventral subiculum of hippocampus (Herman & Mueller 2006). Nicotine is known to increase glutamate release via presynaptic a7 nAChRs on glutamatergic terminals in various brain sites, including the ventral tegmental area, nucleus accumbens, medial prefrontal cortex and hippocampus (Mansvelder & McGehee 2002, Nomikos et al. 2000). Chronic exposure to nicotine may potentiate excitatory inputs to MeA GABAergic neurons by stimulating presynaptic α7 nAChRs on glutamate terminals within MeA.
Chronic nicotine SA did not affect basal expression of c-Fos in sham-shock groups in limbic-PVN network. This coheres with previous reports that basal adrenocorticotropin, corticosterone, and PVN glutamate and GABA levels were unaffected by chronic nicotine SA (Chen et al. 2008, Yu et al. 2010). In contrast, nicotine acutely stimulated c-Fos expression in hypothalamus (e.g. PVN), brainstem (e.g. NTS-A2/C2), and limbic system (e.g. cingulate cortex, and central amygdala) (Matta et al. 1993). Thus, desensitization of nicotine-induced c-Fos expression also occurs during chronic nicotine SA, similar to other chronic stressors (e.g. footshock) (Li & Sawchenko 1998). In a model of nicotine SA, using limited nicotine access and food deprivation, c-Fos expression increased in sensory areas (e.g. superior colliculus), as well as limbic structures involved in natural reward pathways (e.g. medial frontal cortex and nucleus accumbens shell). However, in the current study of nicotine self-administration, with 23 h access to nicotine in the absence of food deprivation, basal c-Fos expression was unchanged in the foregoing regions. This may reflect model-dependent effects.
Sensitization of the HPA response to a novel stressor that is preceded by exposure to a chronic stressor, i.e. repeated restraint, chronic intermittent cold or hypoxia exposure, has been postulated to arise from altered regulation within several brain regions, i.e. limbic structures, thalamus, hypothalamus and brainstem, that modulate the HPA stress response (Pardon et al. 2003, Ma et al. 2008, Ma & Morilak 2005, Bhatnagar & Dallman 1998, Jedema et al. 2001). For example, chronic intermittent cold stress selectively increased restraint-induced activation of neurons in parabrachial nucleus, posterior paraventricular nucleus of thalamus, central, basolateral and basomedial amygdala, and parvocellular PVN, which underlie sensitization of the HPA response to a novel stressor (Bhatnagar & Dallman 1998, Bhatnagar et al. 2000). In contrast, the stress of chronic intermittent hypoxia increased the activity of excitatory neurons in LC induced by immobilization stress, but decreased the activity in lateral hypothalamus, an HPA-inhibitory region (Ma et al. 2008). As shown herein, nicotine SA modulated the stress-induced activity of neural circuits involving the LC-PrL-avBST and MeA-avBST that project to PVN. Thus, it appears that nicotine facilitates the HPA response to a novel stressor based on mechanisms of limbic modulation that differ from those of other chronic stressors.
The effects of nicotine on the limbic-PVN network are manifest by region-specific changes in the stress-induced activity of limbic neuronal pathways. Nicotine SA diminished the activity of excitatory neurons projecting from PrL to avBST and peri-PVN. The reduced activity of PrL neurons may due to the enhanced excitation of inhibitory LC neurons and, perhaps, increasing activity of GABAergic interneurons within PrL. In contrast, nicotine augmented the activity of inhibitory neurons projecting from MeA to avBST. Therefore, the activity of inhibitory neurons, projecting from avBST and peri-PVN to medial parvocellular PVN, was reduced. These effects of chronic nicotine on the limbic-PVN network result in the disinhibition of CRF neurons by reducing stress-induced GABA release, this disinhibition facilitates the stimulation of CRF neurons to enhanced release of glutamate or even diminished norepinephrine release within PVN in response to stress, which we have shown previously (Yu & Sharp 2010, Yu et al. 2010). From this expanded perspective, nicotine SA alters neural interactions controlling the limbic-BST-PVN network, thereby modulating the responsiveness of PVN CRF neurons and the HPA to stress.
Acknowledgments
This research was supported by DA-03977 (B.M.S.) from NIDA. We thank Hao Chen, Ph.D. for his assistance in preparation of this manuscript.
Abbreviations used
- AVP
arginine vasopressin
- avBST
anteroventral bed nucleus of the stria terminalis
- CRF
corticotrophin-releasing factor
- FG
Fluoro-gold
- HPA
hypothalamo-pituitary-adrenal
- IL
infralimbic cortex
- LC
locus coeruleus
- MeA
medial amygdala
- mPFC
medial prefrontal cortex
- NTS
nucleus of solitary tract
- PrL
prelimbic cortex
- PVN
hypothalamic paraventricular nucleus
- SA
self-administration
Footnotes
No financial or other conflict of interest was involved.
References
- al’Absi M, Wittmers LE, Erickson J, Hatsukami D, Crouse B. Attenuated adrenocortical and blood pressure responses to psychological stress in ad libitum and abstinent smokers. Pharmacol Biochem Behav. 2003;74:401–410. doi: 10.1016/s0091-3057(02)01011-0. [DOI] [PubMed] [Google Scholar]
- Antoni FA. Hypothalamic control of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin-releasing factor. Endocr Rev. 1986;7:351–378. doi: 10.1210/edrv-7-4-351. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Dallman M. Neuroanatomical basis for facilitation of hypothalamic-pituitary-adrenal responses to a novel stressor after chronic stress. Neuroscience. 1998;84:1025–1039. doi: 10.1016/s0306-4522(97)00577-0. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Viau V, Chu A, Soriano L, Meijer OC, Dallman MF. A cholecystokinin-mediated pathway to the paraventricular thalamus is recruited in chronically stressed rats and regulates hypothalamic-pituitary-adrenal function. J Neurosci. 2000;20:5564–5573. doi: 10.1523/JNEUROSCI.20-14-05564.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fu Y, Sharp BM. Chronic nicotine self-administration augments hypothalamic-pituitary-adrenal responses to mild acute stress. Neuropsychopharmacology. 2008;33:721–730. doi: 10.1038/sj.npp.1301466. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Glazkova D, Serova L, Sabban EL. Effect of prolonged nicotine infusion on response of rat catecholamine biosynthetic enzymes to restraint and cold stress. Pharmacol Biochem Behav. 2005;82:559–568. doi: 10.1016/j.pbb.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Childs E, de Wit H. Hormonal, cardiovascular, and subjective responses to acute stress in smokers. Psychopharmacology (Berl) 2009;203:1–12. doi: 10.1007/s00213-008-1359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings S, Seybold V. Relationship of alpha-1- and alpha-2-adrenergic-binding sites to regions of the paraventricular nucleus of the hypothalamus containing corticotropin-releasing factor and vasopressin neurons. Neuroendocrinology. 1988;47:523–532. doi: 10.1159/000124965. [DOI] [PubMed] [Google Scholar]
- Dayas CV, Buller KM, Day TA. Neuroendocrine responses to an emotional stressor: evidence for involvement of the medial but not the central amygdala. Eur J Neurosci. 1999;11:2312–2322. doi: 10.1046/j.1460-9568.1999.00645.x. [DOI] [PubMed] [Google Scholar]
- Finkelstein DM, Kubzansky LD, Goodman E. Social status, stress, and adolescent smoking. J Adolesc Health. 2006;39:678–685. doi: 10.1016/j.jadohealth.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Morano MI, Akil H, Watson SJ. Contribution of the ventral subiculum to inhibitory regulation of the hypothalamo-pituitary-adrenocortical axis. J Neuroendocrinol. 1995;7:475–482. doi: 10.1111/j.1365-2826.1995.tb00784.x. [DOI] [PubMed] [Google Scholar]
- Herman JP, Eyigor O, Ziegler DR, Jennes L. Expression of ionotropic glutamate receptor subunit mRNAs in the hypothalamic paraventricular nucleus of the rat. J Comp Neurol. 2000;422:352–362. [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Herman JP, Mueller NK. Role of the ventral subiculum in stress integration. Behav Brain Res. 2006;174:215–224. doi: 10.1016/j.bbr.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Jedema HP, Finlay JM, Sved AF, Grace AA. Chronic cold exposure potentiates CRH-evoked increases in electrophysiologic activity of locus coeruleus neurons. Biol Psychiatry. 2001;49:351–359. doi: 10.1016/s0006-3223(00)01057-x. [DOI] [PubMed] [Google Scholar]
- Ju G, Swanson LW. Studies on the cellular architecture of the bed nuclei of the stria terminalis in the rat: I. Cytoarchitecture. J Comp Neurol. 1989;280:587–602. doi: 10.1002/cne.902800409. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Strasburger CJ, Langkrar J. Attenuated cortisol response to psychological stress but not to CRH or ergometry in young habitual smokers. Pharmacol Biochem Behav. 1993;44:527–531. doi: 10.1016/0091-3057(93)90162-m. [DOI] [PubMed] [Google Scholar]
- Lee HS, Kim MA, Waterhouse BD. Retrograde double-labeling study of common afferent projections to the dorsal raphe and the nuclear core of the locus coeruleus in the rat. J Comp Neurol. 2005;481:179–193. doi: 10.1002/cne.20365. [DOI] [PubMed] [Google Scholar]
- Li HY, Sawchenko PE. Hypothalamic effector neurons and extended circuitries activated in “neurogenic” stress: a comparison of footshock effects exerted acutely, chronically, and in animals with controlled glucocorticoid levels. J Comp Neurol. 1998;393:244–266. [PubMed] [Google Scholar]
- Ma S, Mifflin SW, Cunningham JT, Morilak DA. Chronic intermittent hypoxia sensitizes acute hypothalamic-pituitary-adrenal stress reactivity and Fos induction in the rat locus coeruleus in response to subsequent immobilization stress. Neuroscience. 2008;154:1639–1647. doi: 10.1016/j.neuroscience.2008.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Morilak DA. Chronic intermittent cold stress sensitises the hypothalamic-pituitary-adrenal response to a novel acute stress by enhancing noradrenergic influence in the rat paraventricular nucleus. J Neuroendocrinol. 2005;17:761–769. doi: 10.1111/j.1365-2826.2005.01372.x. [DOI] [PubMed] [Google Scholar]
- Maloku E, Kadriu B, Zhubi A, Dong E, Pibiri F, Satta R, Guidotti A. Selective alpha4beta2 nicotinic acetylcholine receptor agonists target epigenetic mechanisms in cortical GABAergic neurons. Neuropsychopharmacology. 2011;36:1366–1374. doi: 10.1038/npp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Mertz M, Role LW. Nicotinic modulation of synaptic transmission and plasticity in cortico-limbic circuits. Semin Cell Dev Biol. 2009;20:432–440. doi: 10.1016/j.semcdb.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Foster CA, Sharp BM. Nicotine stimulates the expression of cFos protein in the parvocellular paraventricular nucleus and brainstem catecholaminergic regions. Endocrinology. 1993;132:2149–2156. doi: 10.1210/endo.132.5.8386611. [DOI] [PubMed] [Google Scholar]
- McKee SA, Sinha R, Weinberger AH, Sofuoglu M, Harrison EL, Lavery M, Wanzer J. Stress decreases the ability to resist smoking and potentiates smoking intensity and reward. J Psychopharmacol. 2011;25:490–502. doi: 10.1177/0269881110376694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse DE. Neuroendocrine responses to nicotine and stress: enhancement of peripheral stress responses by the administration of nicotine. Psychopharmacology (Berl) 1989;98:539–543. doi: 10.1007/BF00441956. [DOI] [PubMed] [Google Scholar]
- Nomikos GG, Schilstrom B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH. Role of alpha7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behav Brain Res. 2000;113:97–103. doi: 10.1016/s0166-4328(00)00204-7. [DOI] [PubMed] [Google Scholar]
- Pardon MC, Ma S, Morilak DA. Chronic cold stress sensitizes brain noradrenergic reactivity and noradrenergic facilitation of the HPA stress response in Wistar Kyoto rats. Brain Res. 2003;971:55–65. doi: 10.1016/s0006-8993(03)02355-2. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coorinates. 3. Academic Press; New York: 1997. [Google Scholar]
- Payne TJ, Schare ML, Levis DJ, Colletti G. Exposure to smoking-relevant cues: effects on desire to smoke and topographical components of smoking behavior. Addict Behav. 1991;16:467–479. doi: 10.1016/0306-4603(91)90054-l. [DOI] [PubMed] [Google Scholar]
- Perkins KA, Grobe JE, Fonte C, Breus M. “Paradoxical” effects of smoking on subjective stress versus cardiovascular arousal in males and females. Pharmacol Biochem Behav. 1992;42:301–311. doi: 10.1016/0091-3057(92)90531-j. [DOI] [PubMed] [Google Scholar]
- Pomerleau OF, Pomerleau CS. Cortisol response to a psychological stressor and/or nicotine. Pharmacol Biochem Behav. 1990;36:211–213. doi: 10.1016/0091-3057(90)90153-9. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Arias CM, Sawchenko PE. Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci. 2006;26:12967–12976. doi: 10.1523/JNEUROSCI.4297-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Gosselink KL, Sawchenko PE. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J Neurosci. 2009;29:7330–7340. doi: 10.1523/JNEUROSCI.5924-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Sawchenko PE. A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J Neurosci. 2011;31:9683–9695. doi: 10.1523/JNEUROSCI.6040-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Williams B, Sawchenko PE. Noradrenergic innervation of the dorsal medial prefrontal cortex modulates hypothalamo-pituitary-adrenal responses to acute emotional stress. J Neurosci. 2008;28:5806–5816. doi: 10.1523/JNEUROSCI.0552-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JM, Stipelman BA, Bornovalova MA, Daughters SB, Sinha R, Lejuez CW. Biological mechanisms underlying the relationship between stress and smoking: state of the science and directions for future work. Biol Psychol. 2011;88:1–12. doi: 10.1016/j.biopsycho.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohleder N, Kirschbaum C. The hypothalamic-pituitary-adrenal (HPA) axis in habitual smokers. Int J Psychophysiol. 2006;59:236–243. doi: 10.1016/j.ijpsycho.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, Guidotti A. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci U S A. 2008;105:16356–16361. doi: 10.1073/pnas.0808699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Fallon JH. Fluoro-Gold: a new fluorescent retrograde axonal tracer with numerous unique properties. Brain Res. 1986;377:147–154. doi: 10.1016/0006-8993(86)91199-6. [DOI] [PubMed] [Google Scholar]
- Seppa T, Salminen O, Moed M, Ahtee L. Induction of Fos-immunostaining by nicotine and nicotinic receptor antagonists in rat brain. Neuropharmacology. 2001;41:486–495. doi: 10.1016/s0028-3908(01)00093-4. [DOI] [PubMed] [Google Scholar]
- Siahpush M, Carlin JB. Financial stress, smoking cessation and relapse: results from a prospective study of an Australian national sample. Addiction. 2006;101:121–127. doi: 10.1111/j.1360-0443.2005.01292.x. [DOI] [PubMed] [Google Scholar]
- Sun B, Chen X, Xu L, Sterling C, Tank AW. Chronic nicotine treatment leads to induction of tyrosine hydroxylase in locus ceruleus neurons: the role of transcriptional activation. Mol Pharmacol. 2004;66:1011–1021. doi: 10.1124/mol.104.001974. [DOI] [PubMed] [Google Scholar]
- Tsuda A, Steptoe A, West R, Fieldman G, Kirschbaum C. Cigarette smoking and psychophysiological stress responsiveness: effects of recent smoking and temporary abstinence. Psychopharmacology (Berl) 1996;126:226–233. doi: 10.1007/BF02246452. [DOI] [PubMed] [Google Scholar]
- Valentine JD, Hokanson JS, Matta SG, Sharp BM. Self-administration in rats allowed unlimited access to nicotine. Psychopharmacology (Berl) 1997;133:300–304. doi: 10.1007/s002130050405. [DOI] [PubMed] [Google Scholar]
- Valentine JD, Matta SG, Sharp BM. Nicotine-induced cFos expression in the hypothalamic paraventricular nucleus is dependent on brainstem effects: correlations with cFos in catecholaminergic and noncatecholaminergic neurons in the nucleus tractus solitarius. Endocrinology. 1996;137:622–630. doi: 10.1210/endo.137.2.8593811. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang QJ, Liu J, Ali U, Gui ZH, Hui YP, Wang T, Chen L, Li Q. Noradrenergic lesion of the locus coeruleus increases the firing activity of the medial prefrontal cortex pyramidal neurons and the role of alpha2-adrenoceptors in normal and medial forebrain bundle lesioned rats. Brain Res. 2010;1324:64–74. doi: 10.1016/j.brainres.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Yu G, Chen H, Wu X, Matta SG, Sharp BM. Nicotine self-administration differentially modulates glutamate and GABA transmission in hypothalamic paraventricular nucleus to enhance the hypothalamic-pituitary-adrenal response to stress. J Neurochem. 2010;113:919–929. doi: 10.1111/j.1471-4159.2010.06654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Chen H, Zhao W, Matta SG, Sharp BM. Nicotine self-administration differentially regulates hypothalamic corticotropin-releasing factor and arginine vasopressin mRNAs and facilitates stress-induced neuronal activation. J Neurosci. 2008;28:2773–2782. doi: 10.1523/JNEUROSCI.3837-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Sharp BM. Nicotine self-administration diminishes stress-induced norepinephrine secretion but augments adrenergic-responsiveness in the hypothalamic paraventricular nucleus and enhances adrenocorticotropic hormone and corticosterone release. J Neurochem. 2010;112:1327–1337. doi: 10.1111/j.1471-4159.2009.06551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]