

Abstract
Mitochondria are compartmentalized organelles essential for numerous cellular functions including ATP generation, iron-sulfur cluster biogenesis, nucleotide and amino acid metabolism as well as apoptosis. To promote biogenesis and proper function, mitochondria have a dedicated repertoire of molecular chaperones to facilitate protein folding and quality control proteases to degrade those proteins that fail to fold correctly. Mitochondrial protein folding is challenged by the complex organelle architecture, the deleterious effects of electron transport chain-generated reactive oxygen species and the mitochondrial genome’s susceptibility to acquiring mutations. In response to the accumulation of unfolded or misfolded proteins beyond the organelle’s chaperone capacity, cells mount a mitochondrial unfolded protein response (UPRmt). The UPRmt is a mitochondria-to-nuclear signal transduction pathway resulting in the induction of mitochondrial protective genes including mitochondrial molecular chaperones and proteases to re-establish protein homeostasis within the mitochondrial protein-folding environment. Here, we review the current understanding of UPRmt signal transduction and the impact of the UPRmt on diseased cells.
1. Introduction
Mitochondrial protein homeostasis is maintained through proper folding and assembly of newly translated polypeptides, as well as efficient trafficking and turnover of those proteins that fail to fold correctly [1–3]. The load of unfolded proteins in mitochondria must precisely match the chaperone protein-folding capacity. If the chaperone capacity is exceeded, each organelle becomes susceptible to the deleterious effects of protein misfolding and aggregation. However, during stress cells employ strategies to protect the protein-folding environment including organelle-specific quality control proteases to degrade the unfolded or misfolded proteins [4] and mitochondrial unfolded protein responses to increase chaperone capacity and re-establish homeostasis within the mitochondrial protein-folding environment [5]. Several factors challenge the mitochondrial protein-folding environment including complexities in mitochondrial biogenesis, DNA and protein damaging reactive oxygen species (ROS) that are generated within mitochondria, as well as environmental factors such as changes in temperature and exposure to toxins [6].
1.1 Complexities of mitochondrial biogenesis that threaten protein homeostasis
Mitochondria are double-membrane bound organelles composed of four compartments: the outer and inner membranes, the intermembrane space (IMS) and the matrix. Each compartment is a separate protein-folding environment, which must be maintained for efficient mitochondrial biogenesis and proper function. The mitochondrial proteome is composed of approximately 1200 proteins encoded by two separate genomes [7]. The mitochondrial genomes (mtDNA) are localized within the matrix and encode 13 essential components of the electron transport chain (ETC) and the ATP synthase as well as a number of mitochondrial-specific tRNAs. The remainder of the mitochondrial proteome is encoded by the nuclear genome, translated in the cytosol and imported into each mitochondrion [8, 9]. Because ETC complexes I, III, IV and the ATP synthase are composed of components encoded by both genomes, it is imperative that expression from both genomes be coordinated to prevent the accumulation of orphaned subunits.
The mitochondrial protein-folding environment can be disturbed by excessive ROS generated from the ETC primarily via the NADH-ubiquinone oxidoreductase (complex I) and the ubiquinol cytochrome c oxidoreductase (complex III) [10, 11], which directly perturb protein folding and structure. Additionally, mtDNA is prone to the accumulation of mutations, presumably because of its exposure to ROS and that it is not protected by histones [12, 13]. Mutations that reduce expression of ETC components or perturb their ability to fold, compromise assembly of the individual complexes putting stress on the protein-folding environment. Additionally, numerous toxins impair protein homeostasis such as paraquat, which causes high levels of ROS accumulation [14], and rotenone, which impairs complex I assembly and function [15].
1.2 Mitochondrial protein quality control: chaperones and proteases
To promote efficient mitochondrial protein folding and complex assembly, mitochondria have a dedicated repertoire of localized molecular chaperones located in both the IMS and matrix [1, 9]. The Hsp60 chaperonin is in the matrix and consists of both Hsp60 and Hsp10 subunits which form a barrel-shaped complex. Hsp60 primarily facilitates the folding of relatively small, soluble monomeric proteins [16–18]. mtHsp70 also resides in the matrix where it performs multiple functions. At the translocase of the inner membrane (TIM23) channel, mtHsp70 functions in the multi-subunit PAM (Presequence Translocase-Associated Motor) where it interacts with the translocating polypeptides to drive their movement through the import channel into the matrix [9, 19]. In a separate complex, mtHsp70 promotes protein folding and complex assembly of imported polypeptides while preventing aggregation [20–22]. Additionally, mtHsp70 is required for the biogenesis of iron-sulfur clusters within the matrix [23]. Mitochondria also contain an Hsp90 isoform known as TRAP-1 (TNF receptor-associated protein 1) that is thought to promote protein folding in a manner similar to the cytosolic isoforms of Hsp90 [24]. At present, no member of the Hsp60 or Hsp70 family of molecular chaperones has been observed in the IMS of mitochondria, however the Tim9-Tim10 complex promotes the import of highly hydrophobic membrane spanning proteins by preventing non-productive protein-protein interactions as the polypeptides traverse the IMS [25].
In addition to molecular chaperones, mitochondria house several quality control proteases that recognize and degrade those proteins that fail to fold or assemble correctly. Both ClpXP and Lon are AAA proteases (ATPase Associated with diverse cellular Activities) located within the matrix that primarily degrade misfolded soluble proteins [3, 4]. Interestingly, the Lon protease has been shown to preferentially degrade oxidatively-damaged proteins including aconitase [26]. Both Paraplegin (encoded by the SPG7 gene) and YME1L are AAA proteases that are anchored within the inner membrane with their active sites facing the matrix and IMS, respectively. The primary role of YME1L and Paraplegin is to degrade misfolded or misassembled subunits of the ETC [27], although a recent role has been described for Paraplegin in mitochondrial ribosome biogenesis [28, 29]. OMI/Htra2 resides in the IMS where it has been suggested to recognize and degrade soluble proteins that fail to fold correctly [30].
Numerous disease scenarios in which mitochondrial protein homeostasis is compromised emphasize the importance of maintaining the mitochondrial protein-folding environment [13, 31, 32]. Both paraquat and rotenone, as well as the Htra2-deletion cause Parkinson’s-like symptoms in mice [15, 33]. Mutations in the mitochondrial chaperonin Hsp60 and mitochondrial quality control protease Paraplegin cause the neurodegenerative disease spastic paraplegia [34]. In addition to a variety of diseases, loss of mitochondrial protein homeostasis has been closely associated with the aging process [6, 13].
2. Compartment-specific unfolded protein response pathways
The cytosol, endoplasmic reticulum and mitochondria are all exposed to nascent polypeptides, thus each compartment requires dedicated protein-folding machinery, which constitutes each organelle’s protein-folding capacity. Stress occurs when the quantity of unfolded or misfolded proteins exceeds a compartment’s protein-folding capacity, rendering the organelle susceptible to catastrophic damage. To adjust folding capacity, eukaryotic cells have evolved organelle-specific signaling pathways known as unfolded protein responses (UPRs).
The heat shock response protects the cytosolic protein-folding environment and is regulated by the transcription factor Heat Shock Factor 1 (HSF1) [35]. In the absence of stress, HSF1 associates with the cytosolic chaperones Hsp70 and Hsp90. However, when unfolded proteins accumulate beyond the cytosolic chaperone capacity, HSF1 dissociates allowing it to trimerize and interact with the promoters of genes that constitute the heat shock response [36]. HSF1 mediates the expression of a number of chaperone genes that localize to the cytosol and nucleus including Hsp70 and Hsp90. Additionally, HSF1 induces the expression of a number of components of the ubiquitin-proteasome system to degrade terminally misfolded proteins and reduce the burden on the cytosolic protein-folding machinery [36]. Conditions that activate the heat shock response include increased temperatures and exposure to toxins such as arsenite that perturb protein folding in the cytosol [37, 38].
The protein-folding environment of the ER is protected by a separate unfolded protein response (UPRER) [39]. The most conserved branch of the UPRER consists of the ER membrane spanning kinase Ire1 and the bZip transcription factor Xbp1. Ire1 monitors the protein-folding environment of the ER lumen and initiates URPER signaling by directly recognizing unfolded proteins [40]. If stress occurs in the ER, Ire1 oligomerizes [41] activating its cytosolic kinase domain. Once activated, the cytosolic domain of Ire1 splices an intron from the Xbp1 transcript [42] allowing translation of a functional bZip protein which traffics to the nucleus to induce the UPRER. The UPRER includes a number of ER-targeted protein-folding machineries including BiP, an ER-targeted Hsp70, protein disulfide isomerase and the glycosylation machinery [43]. Additionally, the UPRER activates expression of ERAD (ER-associated degradation) components which serve to recognize misfolded proteins, retrotranslocate them across the ER membrane to the cytosol where they are ubiquitylated and degraded by the proteasome [44].
In addition to increasing ER-specific protein folding and quality control machinery, a branch of the UPRER also briefly attenuates protein translation to reduce the load on ER folding capacity [39]. In response to ER stress, the ER membrane spanning kinase PERK (Pancreatic enriched ER kinase) dimerizes and phosphorylates the alpha subunit of eIF2 (eukaryotic initiation factor), which serves to attenuate protein translation [45].
Conceptually similar to the UPRER and heat shock response, accumulating evidence supports the existence of a UPRmt; a mitochondria-to-nucleus signal transduction pathway that senses unfolded protein stress within the organelle and transmits a signal out of mitochondria, through the cytosol to the nucleus where the up-regulation of genes encoding mitochondrial chaperones and quality control proteases takes place to re-establish mitochondrial protein homeostasis [5].
3. Retrograde transcriptional responses from mitochondria suggested the presence of a UPRmt
The survival of cells devoid of mtDNA (ρ0 cells) suggested the activation of nuclear responses as compensation for severe mitochondrial dysfunction. Indeed, ρ0 cells undergo a number of changes in nuclear gene expression including the induction of mitochondrial molecular chaperone and protease genes [46–49]. The absence of mtDNA places a considerable amount of stress on the mitochondrial protein-folding environment as those ETC components encoded by the nucleus are still imported into mitochondria but unable to assemble into stoichiometric complexes in the absence of their mtDNA-encoded binding partners. In addition to increasing the mitochondrial protein homeostasis machinery, cells down-regulate the expression of the ETC components in complex I, III and IV to protect the protein-folding environment but not the ATP synthase as it is required to maintain the membrane potential and cell viability [46]. ρ0 cells also down-regulate numerous components required for protein translation [49], consistent with slowed import reducing the load on the mitochondrial protein-folding environment [50]. Interestingly, the lack of mtDNA also affects the expression of a number of cell cycle regulators such as p19, a cyclin dependent kinase inhibitor involved in cell cycle arrest at G1 [46], suggesting that the cell cycle is slowed during periods of mitochondrial dysfunction to prevent replication of cells with defective mitochondria.
Other forms of mitochondrial stress also elicit transcriptional programs consistent with the presence of a UPRmt. For example, deletion of the yeast Paraplegin homolog Yta12, which encodes a mitochondrial quality control protease [4], results in mitochondrial protein aggregation and widespread changes in nuclear-encoded genes [51, 52]. Similarly, inhibition of mtHsp90 results in the up-regulation molecular chaperone genes as well as a number of genes involved in metabolism [53]. Together, these data suggest the presence of specific stress response pathways that protect against mitochondrial dysfunction.
Perhaps the best-characterized mitochondria-to-nuclear signal transduction pathway is the retrograde response (RTG), thoroughly characterized in yeast. The RTG pathway is activated during mitochondrial dysfunction including that caused by mtDNA depletion to increase activity of numerous metabolic pathways that compensate for the lack of mitochondrial activity. For example, a peroxisomal isoform of citrate synthase (CIT2) is induced to increase activity of the glyoxylate cycle, a variant of the tricarboxlic acid cycle induced in ρ0 cells [54]. The RTG response is mediated by RTG1 and RTG3, two basic helix-loop-helix leucine zipper (bHLH/Zip) transcription factors [55, 56]. When mitochondria are functional, RTG3 is hyperphosphorylated, sequestering both itself and RTG1 in the cytoplasm [56]. However, mitochondrial dysfunction causes RTG3 to be partially dephosphorylated allowing nuclear translocation of the RTG1/3 complex and the induction of the compensatory response.
While the RTG response is required for the induction of many genes required for metabolic adaptations, it does not regulate the expression of mitochondrial chaperone or protease genes during mitochondrial stress, suggesting an independent mechanism for UPRmt activation. Here, we review the emerging data regarding the mechanisms of stress sensing and mitochondria-to-nucleus signal transduction.
4. Mitochondrial-to-nuclear UPRmt signal transduction in mammalian cell culture
To demonstrate the presence of a UPRmt, a terminally misfolded, mutant form of ornithine transcarbamylase (OTC) was targeted to the mitochondrial matrix. The presence of unfolded protein in the matrix resulted in the increased expression of several genes that promote mitochondrial protein homeostasis including Hsp60, Hsp10, mtDnaJ and ClpP (Figure 1) [57]. Importantly, expression of cytosolic and ER chaperone genes were unaffected, indicating specificity of the UPRmt. The promoters of the Hsp60, ClpP and mtDnaJ genes were shown to contain a mitochondrial stress responsive element that corresponds to the CHOP (CCAAT/enhancer-binding protein (C/EBP)-homologous protein) transcription factor consensus binding site [57, 58]. Additionally, the CHOP gene itself is up-regulated during mitochondrial stress, further supporting a central role for CHOP in UPRmt signaling [57].
Figure 1. Model of the mammalian UPRmt signaling pathway.

Activation of the UPRmt in mammalian cells occurs in response to stress originating from the mitochondrial matrix or the intermembrane space (IMS), each having distinct signal transduction pathways and transcriptional responses. Accumulation of unfolded proteins within the matrix stimulates the transcriptional up-regulation of the transcription factor CHOP via JNK2 and c-Jun [57, 62]. CHOP subsequently activates the transcription of genes including the quality control protease ClpP and the chaperonin Hsp60 [58]. Alternatively, accumulating unfolded proteins in the IMS causes activation of the kinase AKT and phosphorylation of the estrogen receptor (ERα) leading to the transcriptional up-regulation of the IMS protease Htra2 and the transcription factor NRF1 [64].
However, it was somewhat surprising that CHOP was shown to induce a mitochondrial specific response considering CHOP is also known to be activated by ER stress, genotoxic stress, as well as arsenite exposure [59–61]. Interestingly, an AP-1 (activator protein-1) element within the CHOP promoter is necessary for mitochondrial stress-induced expression [62]. The transcription factor c-Jun is known to bind to the AP-1 consensus sequence upon activation by JNK (c-Jun N-terminal kinase) [63], suggesting that c-jun and JNK play a role upstream of CHOP in UPRmt signaling. Indeed, increased mitochondrial unfolded protein stress stimulates the phosphorylation of JNK2, providing further support for a role of this kinase in CHOP activation [62] (Figure 1). Numerous downstream regulatory events in UPRmt signaling have been documented including transcription factor activation and the resulting transcriptional outputs. However, several questions still remain including the mechanism cells use to sense unfolded proteins within the matrix and how the signal is transmitted across both mitochondrial membranes. Additionally, does the response, which has been studied in cell lines, occur in vivo and does CHOP inhibition impair mitochondrial chaperone induction?
Recent findings have indicated the presence of a separate UPRmt signaling pathway that specifically responds to unfolded protein stress within the IMS (Figure 1) [30, 64]. Expression of mutant EndoG that localizes to the IMS causes AKT phosphorylation and activation of the nuclear hormone receptor estrogen receptor alpha (ERα) [64], consequently resulting in the increased expression of the IMS-localized quality control protease HtrA2 and the transcription factor NRF1, which is involved in mitochondrial biogenesis [64]. UPRmt regulation by the estrogen receptor is consistent with its documented role in promoting mitochondrial fitness [65]. In addition to HtrA2, activity of the proteasome is also increased in response to IMS stress which is hypothesized to prevent the accumulation of misfolded IMS proteins by ubiquitylating them prior to import [64]. The early studies on the IMS-specific UPRmt provide a framework for signal transduction but several similar questions to those of the matrix-specific response remain to be addressed.
5. Mitochondrial-to-nuclear UPRmt signal transduction in C. elegans
To identify additional UPRmt signaling components and dissect the signaling mechanisms we established a genetically tractable model system using C. elegans. Mitochondrial chaperone gene expression is monitored in vivo using UPRmt reporter worms which harbor promoters of the mitochondrial chaperone genes Hsp60 or mtHsp70 driving expression of GFP [52]. Similar to mammalian cells, treatment of C. elegans with ethidium bromide, a chemical reagent known to reduce mtDNA transcription and replication [66], causes increased expression of the mitochondrial chaperone reporters as well as endogenous mitochondrial chaperone genes [52]. Additionally, knockdown of the mitochondrial quality control protease SPG-7/Paraplegin and mitochondrial chaperone genes activates mitochondrial chaperone gene expression, indicating that perturbations in the mitochondrial protein-folding environment activate this transcriptional response.
Using the UPRmt reporter worms, we performed a genome-wide RNAi-based screen to identify a number of components required for UPRmt signal transduction that constitute a signaling pathway connecting the mitochondrial matrix to the nucleus [67–69]. The current data suggest the following model for how stress is sensed in the mitochondrial matrix and transmitted to the nucleus. As unfolded proteins exceed the matrix chaperone capacity, they are degraded by a quality control protease into peptides, which are pumped across the inner membrane by a peptide transporter. Peptide efflux leads to the activation of a bZip transcription factor, which accumulates in the nucleus to activate mitochondrial chaperone gene induction (Figure 2) [5, 69]. The data that support this model are described in the following sections, however it should be noted that perturbations that affect the mitochondrial protein-folding environment are likely to also affect diverse aspects of mitochondrial biology and potentially unidentified signaling mechanisms.
Figure 2. Model of the C. elegans UPRmt signaling pathway.

UPRmt signaling is initiated as unfolded proteins accumulate beyond the resident chaperone folding capacity. To prevent protein aggregation, unfolded or misfolded proteins are degraded to peptides by the quality control protease ClpP in the mitochondrial matrix [68]. HAF-1-mediated peptide efflux leads to activation of the bZip transcription factor ATFS-1 that accumulates in the nucleus. HAF-1-mediated peptide efflux is also required for the accumulation of the ubiquitin-like protein UBL-5 which complexes with the transcription factor DVE-1 [69]. ATFS-1 and DVE-1/UBL-5 then cooperatively induce the expression of mitochondrial chaperone genes including Hsp60 and mtHsp70 in order to restore protein homeostasis.
5.1 Initiation of UPRmt signaling
Knockdown of the mitochondrial quality control protease ClpP abolishes induction of mitochondrial chaperone genes during mitochondrial stress [68], which has more recently been observed in mammalian cells [70]. Consistent with a role in mitochondrial protection, worms with reduced ClpP activity develop much slower in the presence of mitochondrial stress [68]. The localization of ClpP within the mitochondrial matrix, the same compartment where the stress originates, suggests an upstream function for the quality control protease in UPRmt signaling.
ClpP recognizes and degrades misfolded proteins into peptides of approximately 8–20 residues [71]. Interestingly, peptides that accumulate in the mitochondrial matrix are extruded into the IMS via an ABC (ATP Binding Cassette) transporter; Mdl1 in yeast [72] and HAF-1 in C. elegans [69] suggesting ClpP-derived peptides may act as signaling components in the UPRmt. Indeed, deletion of the ATP-dependent peptide transporter HAF-1 attenuates UPRmt activation during mitochondrial stress [69]. HAF-1 is localized within the mitochondrial inner membrane and is essential for survival under conditions of protein misfolding [69] similar to ClpP. Additionally, ATP-dependent efflux of peptides from mitochondria is reduced following loss of clpp-1 and haf-1, suggesting a role of ClpP upstream of HAF-1 [69].
5.2 UPRmt signaling requires the transcription factor ATFS-1
The requirement for ClpP and HAF-1 suggest a means to sense unfolded protein stress and transmit the signal to the cytoplasm, however the downstream transcription factor was unknown. We recently identified the bZip transcription factor ATFS-1 (Activating Transcription Factor associated with Stress, previously known as ZC376.7) as being required for UPRmt signaling and acting downstream of HAF-1 [69]. Similar to inhibition of ClpP and HAF-1, worms lacking ATFS-1 develop much more slowly in the presence of mitochondrial stress consistent with a role in mitochondrial protection. Furthermore, during mitochondrial stress, ATFS-1 accumulates in the nucleus in a HAF-1-dependent manner, demonstrating that matrix protein degradation and peptide efflux act upstream of ATFS-1 [69]. The mechanism by which the efflux of mitochondrial-derived peptides influences ATFS-1 remains to be determined. However, it is conceivable that the released peptides are recognized by specialized receptors or perhaps the rate of peptide efflux determines downstream activation of ATFS-1.
5.3 The UPRmt requires a second transcriptional complex consisting of DVE-1 and UBL-5
The homeobox transcription factor DVE-1 and the ubiquitin-like protein UBL-5 are also required for transcriptional up-regulation of mitochondrial molecular chaperone genes [68]. DVE-1 is localized within nuclei of all cells but undergoes a nuclear re-distribution when the mitochondrial protein-folding environment is perturbed, at which time it binds to the promoters of mitochondrial molecular chaperone genes [68]. ClpP functions upstream of DVE-1 as ClpP inhibition impairs the nuclear redistribution of DVE-1 during stress [68]. Interestingly, the transporter HAF-1 is unnecessary for DVE-1 nuclear redistribution [69], suggesting a separate means of activation. During mitochondrial stress, ubl-5 expression is up-regulated in a DVE-1 and HAF-1-dependent manner and forms a complex with DVE-1 that is required for UPRmt activation [68]. Interestingly, we demonstrated that the mammalian orthologues of DVE-1 and UBL-5 (SatB2 and Ubl5) are also able to form a complex suggesting a similar role in mammalian cells [68]. However, it has yet to be determined if SatB2 is required for UPRmt signaling in mammalian systems.
The current model of UPRmt signal transduction suggests similarities and differences with the well-characterized UPRER (Section 2). While both culminate in the transcriptional induction of compartment-specific protein folding machinery by organelle-responsive transcription factors, the means by which unfolded proteins are detected and the signal transmitted to the respective transcription factors are different. The different signaling mechanisms appear to stem from differences in ER and mitochondrial architecture. The luminal domain of the ER-localized membrane spanning kinase, Ire1, directly senses unfolded proteins within the ER lumen [40]) and transmits the signal to the cytosolic domain of Ire1, which directly activates the transcription factor Xbp1 [39]. Mitochondria have separate stress responses that respond to perturbations in the matrix [52, 57] or IMS [64]. The current model for UPRmt signaling suggests that unfolded or misfolded proteins are detected in the matrix by the quality control protease ClpP, which degrades them to peptides. The peptides are then pumped across the inner membrane leading to the activation of ATFS-1 through an unknown mechanism [69]. As data emerges on the variety of UPRmt signaling mechanisms it will be interesting to compare and contrast them to the UPRER. For example, a separate branch of the UPRER attenuates protein synthesis to protect the protein-folding environment within the ER lumen when the load of unfolded proteins exceeds the capacity of ER chaperones. A conceptually similar response has not been identified in the UPRmt despite pharmacological inhibition of translation being protective against mitochondrial dysfunction [50].
6. The relationship between the UPRmt and other mitochondrial stress response pathways
Recent studies have indicated that severely damaged or energetically dead mitochondria are cleared from the cell via autophagic degradation through a pathway known as mitophagy [73–75]. The kinase PINK1 accumulates specifically on the outer membrane of mitochondria in which the membrane potential has been completely dissipated [76]. PINK1 then recruits Parkin and the downstream autophagy machinery that directs the defective organelles to lysosomes for degradation [77]. Interestingly, similar stresses that ultimately result in mitophagy also activate the UPRmt. For example, depletion of mitochondrial DNA, expression of mutant components of the ETC [78] as well as exposure to paraquat strongly activate both the UPRmt as well as the mitophagy pathway [74]. Because the UPRmt promotes total cellular mitochondrial function by up-regulating protective components to re-establish organellar homeostasis while mitophagy eliminates severely defective or dead organelles, we hypothesize that the UPRmt is activated prior to the mitophagy pathway. If the organelle cannot maintain a membrane potential despite UPRmt activation the defective organelle enters the mitophagy pathway (Figure 3). Ultimately, if mitochondrial damage becomes too pervasive, the cell undergoes apoptosis. As data emerge on all three pathways, it will be of interest to determine how the pathways integrate to protect mitochondrial, cellular, tissue and ultimately organismal health.
Figure 3. Proposed relationship between the UPRmt, mitophagy and apoptosis during mitochondrial stress.
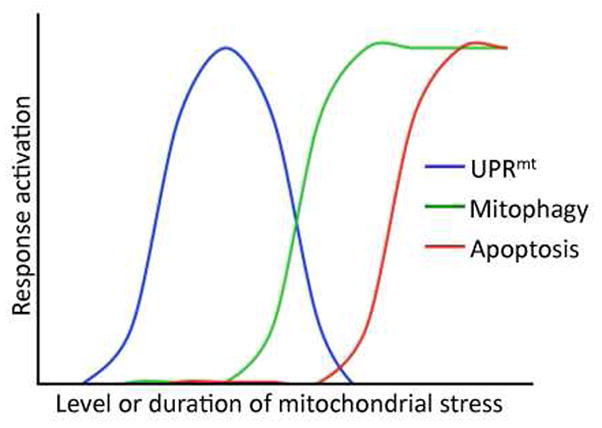
Mitochondrial stress and dysfunction result in the activation of at least three cellular responses including the UPRmt, mitophagy and apoptosis. We propose the depicted relationship between the three pathways as a function of mitochondrial stress level or the duration of mitochondrial stress. Because the UPRmt is a mitochondrial protective response activated to re-establish homeostasis within stressed organelles and mitophagy removes severely defective mitochondria with a completely dissipated inner membrane potential, we propose the UPRmt is activated at lower levels of stress or prior to the induction of mitophagy. As stress in individual mitochondrion exceeds the cytoprotective capacity of the UPRmt, mitophagy eliminates the dead organelles. However, if the total cellular mitochondrial damage becomes too great the cell may undergo apoptosis.
7. Perspective: the UPRmt and the Regulation of Lifespan
The relationship between mitochondrial health and organismal life span has been the focus of much attention in recent years with numerous studies reporting a decline in mitochondrial function with age [12]. Therefore it is somewhat surprising that in worms, flies and mice, mutations that cause ETC dysfunction extend lifespan as much as 50% [79–82]. Many groups had hypothesized that the extension in lifespan was due to the activation of a cyto-protective compensatory response. Recently, it has been shown that ETC mutations that extend lifespan also activate the UPRmt [83]. Impressively, the UPRmt was required for the lifespan extension observed in the ETC mutants [83], supporting a role for the maintenance of the mitochondrial protein-folding environment in lifespan determination [6]. However, it has yet to be determined if HAF-1 or ATFS-1 are required for lifespan extension in the ETC mutants.
While the UPRmt is necessary for lifespan extension, it has yet to be determined if it is sufficient. It will be interesting to determine if UPRmt activation, perhaps by over-expression of ATFS-1, is capable of extending lifespan independent of mitochondrial dysfunction. Additional signaling pathways also contribute to the lifespan extension of ETC mutants including CEH-23 [84] and Hif-1 [85] indicating that multiple compensatory responses contribute to lifespan extension in the ETC mutants.
8. Perspective: the UPRmt in cancer therapeutics
Mounting evidence suggests that cancer cells are exposed to higher levels of mitochondrial stress than normal cells, suggesting a dependence on cellular pathways and components that protect the mitochondrial protein-folding environment [86]. Cells within the tumor interior are exposed to hypoxic conditions, which inhibit protein folding in the IMS and cause remodeling of mitochondrial structure and function [87]. Additionally, cancer cells accumulate mtDNA mutations at relatively high rates [88] and mtDNA depletion has been associated with cancer progression [89]. Similarly, reduced expression of mitochondrial ETC components, including those of complexes I and III, have been observed in tumors [90–92]. While there is little doubt that cancer cells incur greater mitochondrial damage than normal cells, it is unclear whether the damage-associated alterations in mitochondrial function provides an advantage or disadvantage. Regardless, these observations suggest a role for the UPRmt to protect cancer cell mitochondrial function.
For example, Hsp60 expression is increased in a number of cancers including tumors of the digestive, reproductive, nervous systems [93–96]. Elevated levels of Hsp60 likely promote efficient protein folding within the stressed folding environment, although alternative functions of Hsp60 have also been suggested consistent with forced overexpression of Hsp60 causing transformation of embryonic fibroblasts [96]. Hsp60 has been shown to prevent apoptosis by stabilizing the anti-apoptotic protein Survivin [95] as well as by inhibiting the pro-apoptotic proteins Bax and Bak [97], although it is unclear where in the cell this interaction occurs. Consistent with a prominent role for Hsp60 in cancer cell survival, reducing Hsp60 levels by RNAi reduces the oncogenic properties of multiple tumor cell lines while having minimal effects on normal cells [53, 95], demonstrating the attractiveness of this chaperone in cancer target drug development.
Similarly, the Hsp90-related, mitochondrial localized TNF receptor-associated protein-1 (TRAP-1) chaperone is also highly expressed in a number of cancerous tissues [98]. Similar to Hsp60, TRAP-1 prevents apoptotic cell death via an inhibitory interaction with the immunophilin chaperone Cyclophilin D [99]. Recently, the compound Gamitrinib was shown to specifically inhibit the mitochondrial pool of Hsp90 chaperones and activate a UPRmt [53, 100]. Interestingly, Gamitrinib treatment results in the specific death of tumor cells, suggesting a role for TRAP-1 in the function of cancer cell mitochondria. Gamitrinib-activated treatment also caused increased sensitivity to apoptosis [53] opening future possibilities for combined cancer therapies that target the UPRmt and apoptotic pathways simultaneously as a means to enhance treatment.
Summary and future directions
The UPRmt is the collective cellular response to increased levels of mitochondrial unfolded or misfolded proteins through the increased transcription of nuclear-encoded genes that act to promote mitochondrial protein folding. While models detailing the UPRmt signal transduction pathways have been proposed (Figures 1 & 2), many unresolved questions remain. Of particular interest is how extruded peptides from mitochondria activate the transcription factor ATFS-1. In the yeast retrograde pathway, the phosphorylation state of the transcription factor RTG3 determines its activity [56]. ATFS-1 has a serine-rich domain suggesting it may be regulated in a similar manner where mitochondrial peptide efflux may affect phosphorylation of ATFS-1 to impact its nuclear localization, although a putative kinase or phosphatase has not been identified. A second unresolved issue is the relationship between the transcription factors DVE-1 and ATFS-1. While both transcription factors are required for stress-induced expression of mtHsp70 and Hsp60, the contribution of each is unclear. Conceivably, each regulates a subset of genes or alternatively, both are required for the induction of every UPRmt gene. It will be important to identify the entire transcriptional output of each transcription factor and the promoter elements with which ATFS-1 and DVE-1 interact. Evidence from mammalian systems indicates that the DVE-1 orthologue, SatB2, acts as a more general regulator of transcription as it binds throughout the genome to AT rich sequences functioning as a nuclear scaffolding to affect global chromatin organization [101]. Potentially, DVE-1 is required for nuclear remodeling allowing ATFS-1 to directly interact with the promoters of the UPRmt genes.
Many components involved in UPRmt signaling have been identified in mammalian systems as well. It will be important to understand the degree of conservation between the two systems. ClpP is required for both the worm and mammalian UPRmt but the component(s) that connect ClpP to the downstream transcription factors are currently unclear. The mammalian homologues of HAF-1 and ATFS-1 have yet to be identified although CHOP is a potential orthologue of ATFS-1 as both are bZip proteins. Additionally, the role of the mammalian UPRmt has primarily been explored in cell culture. It will be interesting to examine its role in vivo to address the role of mitochondrial protective mechanisms in diseases associated with mitochondrial dysfunction including neurodegeneration and cancer [31, 32, 53]. Further dissection of the UPRmt pathway will yield a better understanding of how cells cope with mitochondrial dysfunction and allow for the discovery of new targets to modulate mitochondrial protein-folding capacity to promote cell survival or death.
Highlights.
Mitochondrial protein homeostasis is maintained by localized chaperones and proteases
Mitochondrial protein folding capacity can be adjusted by the mitochondrial UPRs
Mitochondrial UPRs are mitochondrial-to-nuclear stress signaling pathways
Mitochondrial dysfunction and stress responses are linked to aging and cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 2.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 3.Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. Embo J. 2008;27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tatsuta T, Langer T. AAA proteases in mitochondria: diverse functions of membrane-bound proteolytic machines. Res Microbiol. 2009;160:711–717. doi: 10.1016/j.resmic.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 6.Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- 9.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, Chandel NS. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011;14:537–544. doi: 10.1016/j.cmet.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 14.Castello PR, Drechsel DA, Patel M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem. 2007;282:14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nistico R, Mehdawy B, Piccirilli S, Mercuri N. Paraquat- and rotenone-induced models of Parkinson’s disease. Int J Immunopathol Pharmacol. 2011;24:313–322. doi: 10.1177/039463201102400205. [DOI] [PubMed] [Google Scholar]
- 16.Chakraborty K, Chatila M, Sinha J, Shi Q, Poschner BC, Sikor M, Jiang G, Lamb DC, Hartl FU, Hayer-Hartl M. Chaperonin-catalyzed rescue of kinetically trapped states in protein folding. Cell. 2010;142:112–122. doi: 10.1016/j.cell.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 17.Cheng MY, Hartl FU, Martin J, Pollock RA, Kalousek F, Neupert W, Hallberg EM, Hallberg RL, Horwich AL. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]
- 18.Yamano K, Kuroyanagi-Hasegawa M, Esaki M, Yokota M, Endo T. Step-size analyses of the mitochondrial Hsp70 import motor reveal the Brownian ratchet in operation. J Biol Chem. 2008;283:27325–27332. doi: 10.1074/jbc.M805249200. [DOI] [PubMed] [Google Scholar]
- 19.Kang PJ, Ostermann J, Shilling J, Neupert W, Craig EA, Pfanner N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 1990;348:137–143. doi: 10.1038/348137a0. [DOI] [PubMed] [Google Scholar]
- 20.Scherer PE, Krieg UC, Hwang ST, Vestweber D, Schatz G. A precursor protein partly translocated into yeast mitochondria is bound to a 70 kd mitochondrial stress protein. Embo J. 1990;9:4315–4322. doi: 10.1002/j.1460-2075.1990.tb07880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ostermann J, Voos W, Kang PJ, Craig EA, Neupert W, Pfanner N. Precursor proteins in transit through mitochondrial contact sites interact with hsp70 in the matrix. FEBS Lett. 1990;277:281–284. doi: 10.1016/0014-5793(90)80865-g. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Krzewska J, Liberek K, Craig EA. Mitochondrial Hsp70 Ssc1: role in protein folding. J Biol Chem. 2001;276:6112–6118. doi: 10.1074/jbc.M009519200. [DOI] [PubMed] [Google Scholar]
- 23.Dutkiewicz R, Schilke B, Knieszner H, Walter W, Craig EA, Marszalek J. Ssq1, a mitochondrial Hsp70 involved in iron-sulfur (Fe/S) center biogenesis. Similarities to and differences from its bacterial counterpart. J Biol Chem. 2003;278:29719–29727. doi: 10.1074/jbc.M303527200. [DOI] [PubMed] [Google Scholar]
- 24.Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbamcr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koehler CM. The small Tim proteins and the twin Cx3C motif. Trends Biochem Sci. 2004;29:1–4. doi: 10.1016/j.tibs.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol. 2002;4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 27.Leonhard K, Herrmann JM, Stuart RA, Mannhaupt G, Neupert W, Langer T. AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. Embo J. 1996;15:4218–4229. [PMC free article] [PubMed] [Google Scholar]
- 28.Koppen M, Langer T. Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit Rev Biochem Mol Biol. 2007;42:221–242. doi: 10.1080/10409230701380452. [DOI] [PubMed] [Google Scholar]
- 29.Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m- AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Radke S, Chander H, Schafer P, Meiss G, Kruger R, Schulz JB, Germain D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–12685. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulteau AL, Bayot A. Mitochondrial proteases and cancer. Biochim Biophys Acta. 2011;1807:595–601. doi: 10.1016/j.bbabio.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Martinelli P, Rugarli EI. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim Biophys Acta. 2010;1797:1–10. doi: 10.1016/j.bbabio.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 33.Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Durr A, Fontaine B, Ballabio A. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 35.Sorger PK. Heat shock factor and the heat shock response. Cell. 1991;65:363–366. doi: 10.1016/0092-8674(91)90452-5. [DOI] [PubMed] [Google Scholar]
- 36.Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujimoto M, Nakai A. The heat shock factor family and adaptation to proteotoxic stress. Febs J. 2010;277:4112–4125. doi: 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- 38.Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. Faseb J. 2001;15:1118–1131. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- 39.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 40.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 43.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 46.Delsite R, Kachhap S, Anbazhagan R, Gabrielson E, Singh KK. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol Cancer. 2002;1:6. doi: 10.1186/1476-4598-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Magda D, Lecane P, Prescott J, Thiemann P, Ma X, Dranchak PK, Toleno DM, Ramaswamy K, Siegmund KD, Hacia JG. mtDNA depletion confers specific gene expression profiles in human cells grown in culture and in xenograft. BMC Genomics. 2008;9:521. doi: 10.1186/1471-2164-9-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Hoj PB, Hoogenraad NJ. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240:98–103. doi: 10.1111/j.1432-1033.1996.0098h.x. [DOI] [PubMed] [Google Scholar]
- 49.Mineri R, Pavelka N, Fernandez-Vizarra E, Ricciardi-Castagnoli P, Zeviani M, Tiranti V. How do human cells react to the absence of mitochondrial DNA? PLoS One. 2009;4:e5713. doi: 10.1371/journal.pone.0005713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Zuo X, Kucejova B, Chen XJ. Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat Cell Biol. 2008;10:1090–1097. doi: 10.1038/ncb1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guha S, Lopez-Maury L, Shaw M, Bahler J, Norbury CJ, Agashe VR. Transcriptional and cellular responses to defective mitochondrial proteolysis in fission yeast. J Mol Biol. 2011;408:222–237. doi: 10.1016/j.jmb.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 52.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 53.Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, Ross AH, Plescia J, Altieri DC. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest. 2011;121:1349–1360. doi: 10.1172/JCI44855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet. 2006;40:159–185. doi: 10.1146/annurev.genet.40.110405.090613. [DOI] [PubMed] [Google Scholar]
- 55.Rothermel BA, Thornton JL, Butow RA. Rtg3p, a basic helix-loop-helix/leucine zipper protein that functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J Biol Chem. 1997;272:19801–19807. doi: 10.1074/jbc.272.32.19801. [DOI] [PubMed] [Google Scholar]
- 56.Sekito T, Thornton J, Butow RA. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol Biol Cell. 2000;11:2103–2115. doi: 10.1091/mbc.11.6.2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. Embo J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fawcett TW, Eastman HB, Martindale JL, Holbrook NJ. Physical and functional association between GADD153 and CCAAT/enhancer-binding protein beta during cellular stress. J Biol Chem. 1996;271:14285–14289. doi: 10.1074/jbc.271.24.14285. [DOI] [PubMed] [Google Scholar]
- 60.Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 61.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One. 2007;2:e835. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weiss C, Schneider S, Wagner EF, Zhang X, Seto E, Bohmann D. JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. Embo J. 2003;22:3686–3695. doi: 10.1093/emboj/cdg364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papa L, Germain D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. 2011;124:1396–1402. doi: 10.1242/jcs.078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem. 2008;105:1342–1351. doi: 10.1002/jcb.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 67.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 69.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2011 doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi KH, Licht S. Control of peptide product sizes by the energy-dependent protease ClpAP. Biochemistry. 2005;44:13921–13931. doi: 10.1021/bi0505060. [DOI] [PubMed] [Google Scholar]
- 72.Young L, Leonhard K, Tatsuta T, Trowsdale J, Langer T. Role of the ABC transporter Mdl1 in peptide export from mitochondria. Science. 2001;291:2135–2138. doi: 10.1126/science.1056957. [DOI] [PubMed] [Google Scholar]
- 73.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 76.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Copeland JM, Cho J, Lo T, Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19:1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 80.Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 81.Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 82.Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walter L, Baruah A, Chang HW, Pace HM, Lee SS. The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol. 2011;9:e1001084. doi: 10.1371/journal.pbio.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion. 2004;4:755–762. doi: 10.1016/j.mito.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 87.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 88.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, Marshall FF, Wallace DC. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu M, Zhou Y, Shi Y, Ning L, Yang Y, Wei X, Zhang N, Hao X, Niu R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life. 2007;59:450–457. doi: 10.1080/15216540701509955. [DOI] [PubMed] [Google Scholar]
- 90.Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, Baracca A, Tallini G, Martinuzzi A, Lenaz G, Rugolo M, Romeo G. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006;66:6087–6096. doi: 10.1158/0008-5472.CAN-06-0171. [DOI] [PubMed] [Google Scholar]
- 91.Putignani L, Raffa S, Pescosolido R, Aimati L, Signore F, Torrisi MR, Grammatico P. Alteration of expression levels of the oxidative phosphorylation system (OXPHOS) in breast cancer cell mitochondria. Breast Cancer Res Treat. 2008;110:439–452. doi: 10.1007/s10549-007-9738-x. [DOI] [PubMed] [Google Scholar]
- 92.Stankov K, Biondi A, D’Aurelio M, Gasparre G, Falasca A, Romeo G, Lenaz G. Mitochondrial activities of a cell line derived from thyroid Hurthle cell tumors. Thyroid. 2006;16:325–331. doi: 10.1089/thy.2006.16.325. [DOI] [PubMed] [Google Scholar]
- 93.Cappello F, Bellafiore M, Palma A, Marciano V, Martorana G, Belfiore P, Martorana A, Farina F, Zummo G, Bucchieri F. Expression of 60-kD heat shock protein increases during carcinogenesis in the uterine exocervix. Pathobiology. 2002;70:83–88. doi: 10.1159/000067304. [DOI] [PubMed] [Google Scholar]
- 94.Castle PE, Ashfaq R, Ansari F, Muller CY. Immunohistochemical evaluation of heat shock proteins in normal and preinvasive lesions of the cervix. Cancer Lett. 2005;229:245–252. doi: 10.1016/j.canlet.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 95.Ghosh JC, Dohi T, Kang BH, Altieri DC. Hsp60 regulation of tumor cell apoptosis. J Biol Chem. 2008;283:5188–5194. doi: 10.1074/jbc.M705904200. [DOI] [PubMed] [Google Scholar]
- 96.Tsai YP, Teng SC, Wu KJ. Direct regulation of HSP60 expression by c-MYC induces transformation. FEBS Lett. 2008;582:4083–4088. doi: 10.1016/j.febslet.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 97.Kirchhoff SR, Gupta S, Knowlton AA. Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation. 2002;105:2899–2904. doi: 10.1161/01.cir.0000019403.35847.23. [DOI] [PubMed] [Google Scholar]
- 98.Leav I, Plescia J, Goel HL, Li J, Jiang Z, Cohen RJ, Languino LR, Altieri DC. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am J Pathol. 2010;176:393–401. doi: 10.2353/ajpath.2010.090521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 100.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009;119:454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Farinas I, Karsenty G, Grosschedl R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]