

Abstract
AIMS
Among the main disadvantages of currently available Δ9-tetrahydrocannabinol (THC) formulations are dosing difficulties due to poor pharmacokinetic characteristics. Namisol® is a novel THC formulation, designed to improve THC absorption. The study objectives were to investigate the optimal administration route, pharmacokinetics (PK), pharmacodynamics (PD) and tolerability of Namisol®.
METHODS
This first in human study consisted of two parts. Panel I included healthy males and females (n = 6/6) in a double-blind, double-dummy, randomized, crossover study with sublingual (crushed tablet) and oral administration of Namisol® (5 mg THC). Based on these results, male and female (n = 4/5) participants from panel I received oral THC 6.5 and 8.0 mg or matching placebo in a randomized, crossover, rising dose study during panel II. PD measurements were body sway; visual analogue scales (VAS) mood, psychedelic and heart rate. THC and 11-OH-THC population PK analysis was performed.
RESULTS
Sublingual administration showed a flat concentration profile compared with oral administration. Oral THC apparent t1/2 was 72–80 min, tmax was 39–56 min and Cmax 2.92–4.69 ng ml−1. THC affected body sway (60.8%, 95% CI 29.5, 99.8), external perception (0.078 log mm, 95% CI 0.019, 0.137), alertness (−2.7 mm, 95% CI −4.5, −0.9) feeling high (0.256 log mm, 95% CI 0.093, 0.418) and heart rate (5.6 beats min–1, 95% CI 2.7, 6.5). Namisol® was well tolerated.
CONCLUSIONS
Oral Namisol® showed promising PK and PD characteristics. Variability and tmax of THC plasma concentrations were smaller for Namisol® than reported for studies using oral dronabinol and nabilone. This study was performed in a limited number of healthy volunteers. Therefore, future research on Namisol® should study clinical effects in patient populations.
Keywords: Δ9-tetrahydrocannabinol, cannabinoid, Namisol, pharmacodynamics, pharmacokinetic model, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Cannabis based medicines are registered as a treatment for various indications, such as pain and spasms in multiple sclerosis (MS) patients, and anorexia and nausea in patients with HIV or receiving cancer treatment.
the pharmacokinetics of the various administration routes of cannabis and cannabis based medicines are variable and dosing is hard to regulate.
WHAT THIS STUDY ADDS
Namisol is a new tablet containing pure THC (>98%) that has a beneficial pharmacokinetic profile after oral administration.
Namisol gives a quick onset of pharmacodynamic effects in healthy volunteers, which implies a rapid initiation of therapeutic effects in patients.
Introduction
Components of the Cannabis sativa L. plant, or cannabis, have been used for medical purposes for thousands of years. Nowadays, cannabis derived compounds, or cannabinoids, are registered in several countries for a variety of indications, including antinociception and muscle relaxation in patients suffering from multiple sclerosis [1]–[3], and anti-nausea and anti-emetic effects in cancer patients [4]–[6]. Cannabis consists of several cannabinoid compounds, some of which are still the subject of clinical research. For the registered products, Δ9-tetrahydrocannabinol (THC) is generally considered to be the active compound responsible for the clinical effects [7], [8].
THC induces its effects via activation of cannabinoid receptor types 1 and 2 (CB1 and CB2) [9]. CB1 are mainly located in the central nervous system, as well as in peripheral tissues such as the heart, adipose tissue and sympathetic ganglions, while CB2 are mainly present in immune cells [10]–[12]. The major metabolite of THC is 11-OH-THC [13]. This metabolite induces effects via CB1 receptors and has been described to be equally or up to seven times as potent as THC [14], [15]. This could mean that the clinical effects of THC are related to the combined activities of THC and 11-OH-THC.
The common medicinal cannabis administration routes are via smoking, after vaporizing and orally as tea or in baked goods. After smoking, THC plasma concentrations increase quickly [16]. However, smoking is not a very practical route and it can lead to stigmatization, which may be a limiting factor particularly for non-smokers. Also, cannabis, especially when co-administered with tobacco, contains a mixture of other compounds, some of which interact with the effects of THC, and some of which are noxious. Moreover, part of the active substances is not inhaled and will be lost. Also, depth and frequency of inhalations vary considerably between individuals. This lack of controlled dosing may reduce clinical efficacy or induce side effects and may also occur after vaporization of cannabis or THC. With regards to oral administration of THC using cannabis tea, a previous study found tea to have a different cannabinoid composition compared with non-decocted cannabis [17], affecting the clinical effects. To bypass these problems, methods have been developed to purify THC from cannabis and to formulate it in a stable dosage form.
Marinol® and Cesamet® are two oral THC formulations registered for anorexia in AIDS patients, and nausea and vomiting in cancer patients. Marinol® contains synthetic THC, or dronabinol, and is registered in Germany and the USA. Cesamet® contains nabilone, a THC analogue, and is registered in Canada and the USA. An oromucosal spray containing mainly THC and cannabidiol, a non-psychoactive cannabinoid, is registered in Canada and in some European countries as Sativex® against pain and spasms in MS. Disadvantages of the current administration forms are the long tmax-values for these formulations, ranging from 1 to 4 h for Marinol® and Cesamet®[18], [19], and 3.3 to 4.0 h for Sativex®[20]. Long times to reach a maximal concentration can be a disadvantage for on demand symptomatic treatment. Oral dronabinol formulations, such as Marinol®, have variable pharmacokinetics, as peak plasma concentration variations from 150% to 200% were observed in previous studies [21], [22]. This is unfavourable for accurate dose regulation.
In the current study, Namisol® was examined. It has a novel tablet formulation of pure THC that was produced using Alitra™ (Echo Pharmaceuticals b.v., Nijmegen, the Netherlands), an emulsifying drug delivery technology. This technology was designed to improve the uptake of poorly soluble lipophilic compounds, using less surfactant (less than 10% w/w). This is a first in human trial investigating the optimal administration route of Namisol®, the safety, pharmacokinetics, pharmacodynamics and tolerability. The first objective was to compare the sublingual and oral dosing routes of Namisol® tablets with respect to pharmacodynamic effects and pharmacokinetics of THC and its active metabolite 11-OH-THC and to choose the most favourable administration route. This was decided on factors such as a short time to maximal THC concentration and a high maximal concentration. The second objective was to use the most favourable administration route in a subsequent dose-ranging study, in order to evaluate the pharmacokinetics and pharmacodynamic effects of different doses. With these objectives, which intended to explore the pharmacokinetic and pharmacodynamic properties of Namisol®, no registered cannabis based medicines were taken as an additional treatment arm in at this early stage of development.
Methods
Design
The study consisted of two parts. In the first part of the study, the pharmacokinetic differences between oral and sublingual administration, and the most favourable administration route were determined, referred to as ‘panel I’. Panel I had a double blind, double dummy, two-way crossover design. Panel II refers to the dose-ranging part of the study, which was a randomized, double-blind, placebo-controlled, three-way dose-escalation trial. For both panels, the wash-out period between two treatments was at least 2 weeks. Subjects were medically screened within 3 weeks before dosing. Subjects had a follow-up visit after the 24 h PK sample of the last visit of panel II.
Sample size
This was an explorative study for which no sample size calculation was performed. For panel I, 12 healthy subjects (six male, six female) were included, and for panel II, nine subjects (mixed gender) were included. These numbers are usually sufficient to demonstrate significant dose-related pharmacodynamic effects of THC after inhalation [23], [24]. Participants from panel I were allowed to continue in panel II.
Inclusion and exclusion criteria
After signing the informed consent form, subjects were medically screened. Subjects were between 18 and 55 years old and had a body mass index between 18.0 and 28.5 kg m−2 (extremes included). They had to be cannabis users for at least 1 year, to minimize the risk of oversensitivity to THC in naive subjects. To prevent pharmacokinetic and pharmacodynamic tolerance, the maximal use was limited to once per week, and subjects were not allowed to have used cannabis from at least 2 weeks prior to the first treatment period to the end of the last study day. Subjects were not allowed to smoke more than 10 cigarettes per day and had to refrain from smoking during study days. Subjects using more than six units of (methyl)xanthine products (e.g. coffee, tea, cola, chocolate) were not included, and subjects had to stop using xanthine containing products from 12 h prior to dosing until discharge. An irregular diurnal rhythm and consumption of grapefruit (juice) were not allowed from 2 weeks prior to the first dose until the last study day. Quinine and alcohol use were not allowed from 2 days prior to dosing until discharge. Use of medication was not allowed from 1 week prior to dosing until the last study day. Use of illicit drugs was not allowed during the study, and each study day prior to dosing, illicit drug (including cannabis) use was tested using drug screening urine tests. In order to keep a consistent level of sex hormones, female subjects were only included if they used the Nuvaring® or one of the monophasic oral contraceptives, and were able and willing to skip the pill or ring-free week from screening until the end of the study. Pregnant and/or breastfeeding women were excluded, and urinary pregnancy tests were performed prior to study drug administration. The study was approved by the Ethics Review Board of Leiden University Medical Centre.
Treatments
Namisol® and matching placebo (Echo Pharmaceuticals b.v., Nijmegen, the Netherlands) were administered as 1.5 mg and 5 mg tablets. In panel I, one tablet (5.0 mg THC), and in panel II, three tablets (one 5.0 mg and two 1.5 mg tablets active or matching placebos) were used for the administration of 6.5 mg or 8.0 mg THC or placebo respectively. Oral administrations were done with 200 ml mineral water. Namisol® tablets were not designed for sublingual use. Due to a relatively long in vitro disintegration time of up to 15 min of this experimental formulation, tablets were crushed before sublingual administration using Pillmaster (Sell-Plan, Weesp, the Netherlands) to increase the surface area of the tablet and, as a result, improve sublingual absorption. The crushed tablet was then placed under the tongue using cigarette rolling paper.
In panel I, the following treatments were administered within 1 min of t = 0: (1) oral Namisol® 5 mg + sublingual matching placebo (2) sublingual Namisol® 5 mg + oral matching placebo. After panel I, an interim analysis of safety, pharmacokinetic and pharmacodynamic data was performed. Based on this analysis, the most favourable administration route of Namisol® was selected for panel II. The dose levels for panel II were also based on the interim results of panel I, leading to an oral dose selection of 6.5 mg, 8.0 mg or matching placebo.
Pharmacokinetics
For determination of the plasma concentration of THC and its active metabolite 11-OH-THC, venous blood was collected in EDTA tubes of 4 ml at the following time points: pre dose, 11 min, 30 min, 45 min, 1 h, 1.5 h and at 2, 3, 4, 6, 8, 12 and 24 h. The 24 h blood sample was only drawn in panel I. After blood collection the tubes were put in ice water in light-shielded containers and were centrifuged within 1 h (10 min, 2000 g, 4°C). The handling of THC samples was done at low ambient lighting. Plasma samples were stored at a temperature of at least −70°C and analyzed by Analytisch Biochemisch Laboratorium b.v. (Assen, the Netherlands) using liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) according to good laboratory practice procedures. The lower limit of quantification for both THC and 11-OH-THC was 0.100 ng ml−1.
Pharmacodynamics
Pharmacodynamic measurements were performed in ‘test-blocks’, in a quiet room with subdued lighting, with only one subject in the same room per session. Test-blocks were performed at the following time points: twice pre dose, 15 min, 32 min, 47 min, 1h 2 min, 1 h 32 min, and at 2 min past 2, 3, 4, 6 and 8 h. Within 3 weeks before the first occasion, subjects had a training session in order to get acquainted with the pharmacodynamic tests and to minimize learning effects during the study.
Body sway methodology
Measurements of postural stability for 2 min were performed using a body sway meter as described previously [23].
Visual analogue scales
The Bond & Lader visual analogue scales (VAS) were used to measure subjective alertness, mood, and calmness [25]. The Bowdle VAS of psychedelic effects were performed in order to measure subjective ‘feeling high’, and clustered scales that quantify effects on internal and external perception [23], [26]. Internal perception reflects inner feelings that do not correspond with reality, including mistrustful feelings, whereas external perception reflects a misperception of external stimuli or changes in the awareness of the subject's surroundings. The data were clustered and log transformed, and are expressed as units as described previously [23].
Heart rate
Electrocardiogram (ECG) measurements (Cardiofax V equipped with ECAPS12 analysis program, Nihon Kohden) were taken in triplicate after having been in a supine position for at least 5 min at the following time points: pre dose, 1 h 15 min and 24 h 08 min (panel II only). The QT-intervals were corrected for heart rate according to Bazett and Fridericia's QT correction. Blood pressure and heart rate measurements were performed using Nihon-Kohden (BSM-1101 K) or Colin (Pressmate BP 8800) automated device after sitting for at least 5 min. Safety heart rate and blood pressure measurements were performed at the following time points: pre dose, 1 h 03 min and 23 h 58 min (panel II only). Heart rate measurements were also recorded as pharmacodynamic endpoints, at time points described in that pertaining section.
Data analysis
As the first part of the study was not placebo-controlled, statistical analysis of safety and pharmacodynamics was performed for both study panels separately. For the pharmacokinetic parameters, all treatments were analyzed together. After panel I, an interim analysis was performed for adverse events, pharmacokinetics and pharmacodynamics, to adapt the design of panel II.
Non-compartmental pharmacokinetic analysis
Descriptive statistics were calculated for the plasma concentrations of THC, 11-OH-THC, and unbound active moiety (THC + 11-OH-THC) at each time point and for peak plasma concentration (Cmax), time to peak plasma concentration (tmax), apparent terminal half-life (t1/2), and area under the curve from t = 0 to infinity (AUC(0,∞)). Dose-proportionality was assessed for Cmax and AUC(0,∞). Pharmacokinetic parameters were compared with a mixed model analysis of variance and reported with 95% confidence intervals (CI) around the estimated differences. All effects were considered significant at the 5% level.
Compartmental pharmacokinetic analysis
A population pharmacokinetic model was developed for the most favourable Namisol® formulation, in order to make predictions of pharmacokinetic profiles for further clinical development. Pharmacokinetic modelling was conducted using NONMEM (version 7.1.2). Pharmacokinetics of THC and 11-OH-THC were described using a sequential compartmental modelling approach, which has been used previously [27], [28]. The model part of 11-OH-THC was linked to the individual empirical Bayes estimates determined for the THC pharmacokinetic parameters. Different absorption models were tested, including first order absorption and transit models, as well as different elimination models, including linear elimination and Michaelis-Menten elimination, which was used in a previous model [27]. Model discrimination was performed using the likelihood ratio test, using a difference in objective function values of 6.64 as significance criterion (chi-square test, α= 0.01, d.f. = 1). All models were also graphically evaluated using goodness of fit plots, depicting individual and population predicted vs. observed. Potential model misspecification was assessed using plots of residuals vs. time and the dependent variable. Predictive performance of the final models for internal validation was evaluated using a visual predictive check depicting the model simulated distribution together with the observed values vs. time.
Pharmacodynamic analyses
Average baseline values per subject and visit for each variable were obtained by calculation of the mean of two baseline assessments. Body sway was log transformed to correct for the log normal distribution. All pharmacodynamic parameters were analyzed by mixed model analyses of variance (using SAS PROC MIXED) with subject, subject by treatment and subject by time as random effects, with gender, treatment, occasion, time, treatment by gender and treatment by time as fixed effects, and the average baseline value was included as covariate. For panel I the contrast oral THC 5 mg vs.sublingual THC 5 mg was calculated. For panel II the calculated contrasts were: placebo vs. oral 6.5 mg, placebo vs. oral 8.0 mg and oral 6.5 mg vs. oral 8.0 mg. All effects were considered significant at the 5% level.
Results
Subjects
For panel I, 14 subjects (seven males and seven females) were included in order to get 12 complete data sets. Data sets from 13 subjects were used for pharmacodynamic and pharmacokinetic analysis. One subject dropped out after a vasovagal collapse and one subject for personal circumstances. Four males and five females from panel I continued the study in panel II. On average, the subjects were 21.4 years old, and had a body mass index of 21.7 kg m−2. Demographic details per panel can be found in Table 1.
Table 1.
Summary of subject demographics of panel I and panel II
| Variable | n | Mean | SD | Min | Max | |
|---|---|---|---|---|---|---|
| Panel I | Gender (M : F) | 7 : 7 | ||||
| Age (years) | 14 | 21.4 | 3.3 | 18 | 27 | |
| BMI (kg m−2) | 14 | 21.71 | 1.52 | 18.4 | 24.5 | |
| Height (m) | 14 | 1.783 | 0.103 | 1.62 | 1.96 | |
| Weight (kg) | 14 | 69.09 | 10.13 | 55.3 | 90.1 | |
| Panel II | Gender (M : F) | 4 : 5 | ||||
| Age (years) | 9 | 21.9 | 3.8 | 18 | 27 | |
| BMI (kg m−2) | 9 | 22.31 | 0.97 | 21.1 | 24.5 | |
| Height (m) | 9 | 1.766 | 0.099 | 1.62 | 1.91 | |
| Weight (kg) | 9 | 69.70 | 8.91 | 55.3 | 80.6 |
SD, standard deviation.
Adverse effects
All adverse events were of mild to moderate intensity and transitory in nature. A vasovagal syncope occurred during the first occasion, 32 min after administration of Namisol® oral 5 mg + placebo Namisol® sublingual, which was considered to be possibly related to treatment and led to the subject's withdrawal. In panel I, the frequencies and types of adverse events were similarly distributed over sublingual and oral administration. In panel II, compared with placebo, more subjects in the THC treatment groups had adverse events that were classified as nervous system disorders, especially in the 8.0 mg THC treatment group (9/9 subjects), 6.5 mg THC (7/9 subjects) and placebo (4/9 subjects), with dizziness as the most frequent adverse event. The same trend was found for the psychiatric disorder class (8.0 mg THC, 5/9; 6.5 mg THC, 3/9; placebo, 0/9), which mainly concerned self reported euphoric mood (‘feeling high’).
No clinically relevant changes in blood pressure, body temperature, haematology, biochemistry, urinalysis or any of the ECG intervals were found. Heart rate increase after treatment was analyzed as a pharmacodynamic parameter.
Noncompartmental pharmacokinetic analysis
Noncompartmental pharmacokinetic parameters of sublingual and oral THC are summarized in Table 2 and the concentration profiles of THC and 11-OH-THC are given in Figure 1. Based on the interim PK analysis, the oral administration route was chosen above the sublingual route. A shorter tmax and a higher Cmax of oral THC indicated a possibly larger effect with a faster onset compared with sublingual administration. These differences in tmax and Cmax between oral and sublingual administration were not statistically significant. Sublingual administration showed a significantly longer apparent t1/2 compared with oral administration (+122 min, 95% CI 64, 181, P = 0.0002). AUC(0,∞) and Cmax of oral THC were dose proportional and tmax and t1/2 were similar for all doses.
Table 2.
Pharmacokinetic parameters of THC and 11-OH-THC after sublingual and oral administration of Namisol®. All data are presented as means with coefficient of variation (%)
| Panel | I (n=13) | I (n=13) | II (n=9) | II (n=9) |
|---|---|---|---|---|
| Parameter | 5.0 mg sublingual | 5.0 mg oral | 6.5 mg oral | 8.0 mg oral |
| THC | ||||
| Cmax (ng ml−1)a | 2.30 (44) | 2.92 (51) | 4.43 (42) | 4.69 (62) |
| tmax (min) | 74.5 (52) | 56.0 (73) | 39.3 (20) | 43.6 (26) |
| AUC(0,∞) (ng ml−1 min)a | 235.8 (47) | 188.7 (40) | 286.6 (36) | 377.2 (46) |
| t1/2 (min) | 252.9 (98) | 71.9 (24) | 80.0 (22) | 78.8 (21) |
| 11-OH-THC | ||||
| Cmax (ng ml−1)a | 3.08 (42) | 4.68 (42) | 5.94 (44) | 6.10 (53) |
| tmax (min) | 83.6 (63) | 74.1 (68) | 46.1 (28) | 78.4 (63) |
| AUC(0,∞) (ng ml−1 min)a | 522.9 (50) | 648.1 (49) | 848.7 (42) | 1087.3 (50) |
| t1/2 (min) | 279.0 (51) | 196.0 (33) | 318.7 (54) | 314.1 (58) |
Cmax and AUC(0,∞) were dose-corrected for treatment P value calculation. Cmax, peak plasma concentration, tmax, time to peak plasma concentration, AUC(0,∞), area under the curve from t = 0 to infinity, t1/2, apparent terminal half-life.
Figure 1.
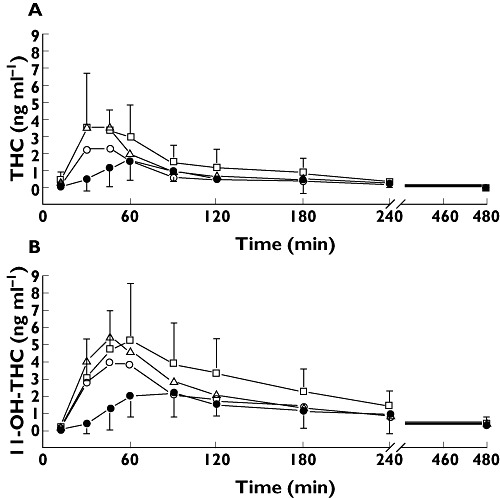
THC (A) and 11-OH-THC (B) concentrations after sublingual 5.0 mg and oral 5.0, 6.5 and 8.0 mg Namisol® administration as estimated with a mixed model. Closed circles are sublingual THC 5.0 mg, open circles are oral THC 5.0 mg, triangles are oral THC 6.5 mg and squares are oral THC 8.0 mg. Error bars represent SD
The difference between pharmacokinetic parameters for oral and sublingual THC 5 mg administration were not significantly different for 11-OH-THC, except for the dose corrected peak concentration (0.30 ng ml−1 mg−1, 95% CI 0.10, 0.49, P = 0.0047). Pharmacokinetic profiles for oral 5.0, 6.5 and 8.0 mg THC were also not different, except for t1/2, where 5 mg was shorter than both 6.5 and 8.0 mg (115 min, 95% CI 8, 222, P = 0.0366; and 110 min, 95% CI 3, 217, P = 0.0441 respectively).
Compartmental pharmacokinetic analysis
The two-compartment model for THC pharmacokinetics had first order absorption, linear elimination and a lag time. A proportional model was used for the residual error. The estimates for clearance and volumes are apparent values, i.e. CL/F and V/F, since this study had no intravenous administration and therefore absolute bioavailability (F) could not be determined. Peripheral volume of distribution of THC was approximately two times larger than central volume (1780 l vs. 889 l), while the peripheral volume of 11-OH-THC was approximately 19 times larger than the central volume of distribution (1010 l vs. 52.6 l). Inter-individual variability was estimated for clearance and central volume. THC clearance had a variability of 28.4%. 11-OH-THC had a large variability of clearance of 70.4%. Inter-individual variability of the central volume of distribution was large for THC with 56.3%, and was especially large for 11-OH-THC with 413%. Almost all parameters showed a relative standard error (RSE) that was smaller than 30%. An overview of the pharmacokinetic parameters after oral administration of Namisol® is given in Table 3. Visual predictive checks demonstrated that the predictive performance of the THC and 11-OH-THC models slightly overestimated the variability during wash-out. The visual predictive checks are shown in Figure 2.
Table 3.
THC population pharmacokinetic parameters after oral Namisol®
| THC | 11-OH-THC | |||
|---|---|---|---|---|
| Parameter | Estimate (RSE) | IIV | Estimate (RSE) | IIV |
| Clearance/F (l min−1)a | 26.5 (10.6) | 28.4 | 9.53 (25) | 70.4 |
| Central volume of distribution/F (l)a | 889 (22.5) | 56.3 | 52.6 (47.9) | 413 |
| Peripheral volume of distribution/F (l)a | 1790 (21.9) | – | 1010 (15.3) | 21.1 |
| Intercompartmental clearance/F (l min−1)a | 13.3 (17) | – | 4.46 (34.5) | 50.7 |
| Absorption rate constant (min−1) | 0.0401 (22) | – | – | – |
| Proportional residual error (sd mean−1) | 0.509 (8) | – | 0.461 (6.2) | – |
| Absorption lag time (min) | 11.5 (0.9) | – | – | – |
This parameter is an apparent parameter as bioavailability could not be calculated. RSE, relative standard error (%), IIV, inter-individual variability (coefficient of variation, %).
Figure 2.
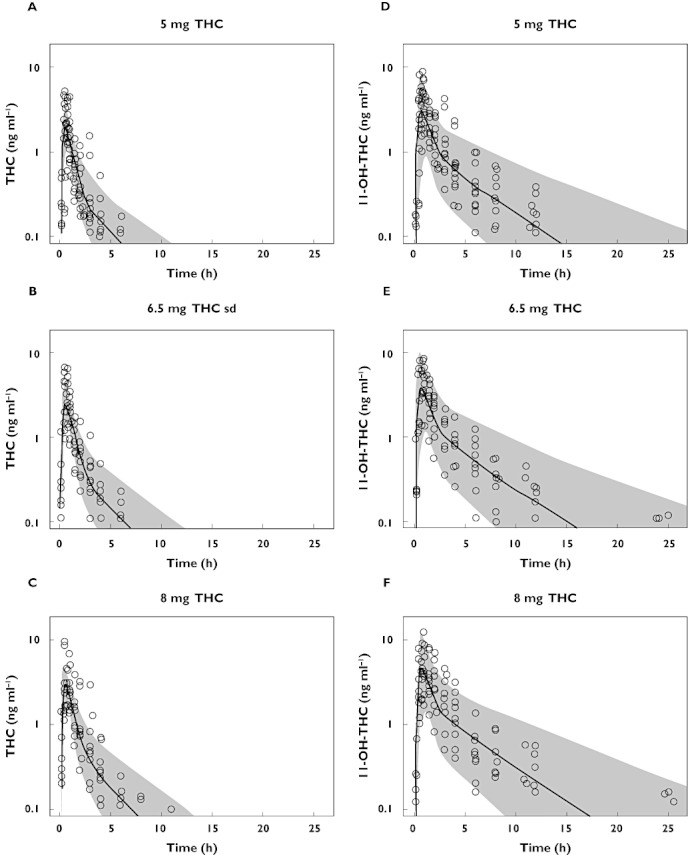
Visual predictive checks of THC concentrations after a single dose of THC 5.0, 6.5, and 8.0 mg (A, B and C) and of 11-OH-THC concentrations (D, E, and F). Lower limit of quantification for THC and 11-OH-THC is 0.1 ng ml−1
The pharmacokinetic model of THC was used for a stochastic simulation of THC and 11-OH-THC concentrations during a multiple dose design of two daily 5 mg THC doses. The graphical representation of this simulation can be found in Figure 3. In this simulation the plasma concentration of THC and 11-OH-THC will not drop below the lower limit of quantification (0.100 ng ml−1 for both THC and 11-OH-THC) in steady-state before the next dose is administered. The accumulation factor of the plasma concentration is 1.02 for THC, and 1.11 for the active metabolite as based on this single dose study.
Figure 3.
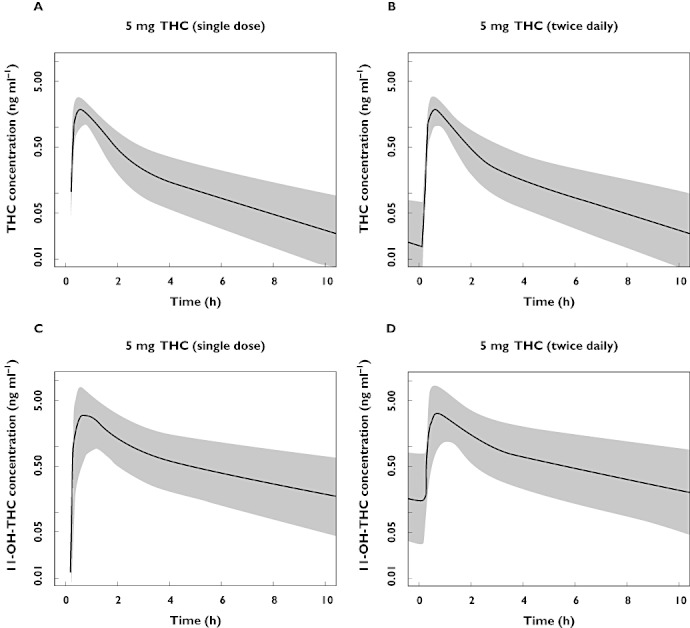
Stochastic simulations (n = 2000) of concentrations of THC after a single 5 mg dose (A), and after 21 dosages, 5 mg two times per day (B)and simulations of 11-OH-THC concentrations after a single 5 mg dose (C), and after 21 dosages, 5 mg two times per day (D)
Pharmacodynamics
Contrasts of pharmacodynamic parameters are summarized in Table 4. As an example of the graphical representation of the pharmacodynamic parameters, the effect of Namisol® on body sway is given in Figure 4. In panel I, oral THC administration gave a statistically significant increase in VAS calmness, compared with sublingual administration. This difference was not considered clinically relevant, as the absolute peak difference was 3 mm on a 100 mm scale. Between oral and sublingual administration, no clinically relevant differences in PD parameters were observed. In panel II, significant increases were found between THC 6.5 mg and placebo on VAS external perception, VAS feeling high and heart rate. THC 8.0 mg produced a decrease on VAS alertness and increases in body sway, VAS external perception, VAS feeling high, and heart rate compared with placebo. The THC effects changed in a dose-dependent way, which was significant for body sway when comparing THC 6.5 mg and 8.0 mg.
Table 4.
Pharmacodynamic effects after Namisol® dosing. Treatment differences are given in estimated differences of least square means with 95% confidence intervals and P values (significant P values are shown in bold). Log transformed VAS (scores in mm + 2) are given in units (U)
| Panel | I (n=13) | II (n=9) | II (n=9) | II (n=9) |
|---|---|---|---|---|
| Parameter | 5.0 mg oral vs. 5.0 mg sublingual | 6.5 mg oral vs. placebo | 8.0 mg oral vs. placebo | 8.0 mg vs. 6.5 mg oral |
| Body sway (%) | 7.66 (−4.62, 21.53) | 22.06 (−1.05, 50.57) | 60.82 (29.46, 99.79) | 31.76 (6.53, 62.96) |
| P = 0.2037 | P = 0.0610 | P = 0.0003* | P = 0.0145* | |
| VAS Alertness (mm) | −0.3 (−2.0, 1.5) | −1.4 (−3.2, 0.4) | −2.7 (−4.5, −0.9) | −1.3 (−3.1, 0.5) |
| P = 0.7124 | P = 0.1161 | P = 0.0057* | P = 0.1390 | |
| VAS Mood (mm) | 0.8 (−0.1, 1.6) | 0.1 (−0.3, 0.5) | 0.2 (−0.2, 0.6) | 0.1 (−0.4, 0.5) |
| P = 0.0653 | P = 0.5357 | P = 0.3686 | P = 0.7815 | |
| VAS Calmness (mm) | 1.8 (0.1, 3.5) | 0.7 (−0.1, 1.4) | 0.5 (−0.2, 1.2) | −0.1 (−0.9, 0.6) |
| P = 0.0443* | P = 0.0665 | P = 0.1246 | P = 0.7080 | |
| VAS Feeling high (U) | 0.111 (−0.042, 0.265) | 0.229 (0.073, 0.384) | 0.256 (0.093, 0.418) | 0.027 (−0.129, 0.183) |
| P = 0.1347 | P = 0.0071* | P = 0.0044* | P = 0.7145 | |
| VAS External perception (U) | 0.037 (−0.017, 0.090) | 0.061 (0.002, 0.121) | 0.078 (0.019, 0.137) | 0.017 (−0.042, 0.076) |
| P = 0.1482 | P = 0.0446* | P = 0.0141* | P = 0.5507 | |
| VAS Internal perception (U) | 0.006 (−0.014, 0.026) | 0.013 (−0.003, 0.029) | 0.002 (−0.015, 0.019) | −0.011 (−0.028, 0.005) |
| P = 0.5247 | P = 0.1057 | P = 0.8312 | P = 0.1632 | |
| Heart rate (beats min–1) | 0.2 (−3.6, 4.0) | 5.3 (2.4–8.2) | 5.6 (2.7–8.5) | 0.3 (−2.7, 3.2) |
| P = 0.9261 | P = 0.0019* | P = 0.0014* | P = 0.8524 |
P-values indicated with an
are statistically significant values (α= 0.05).
Figure 4.
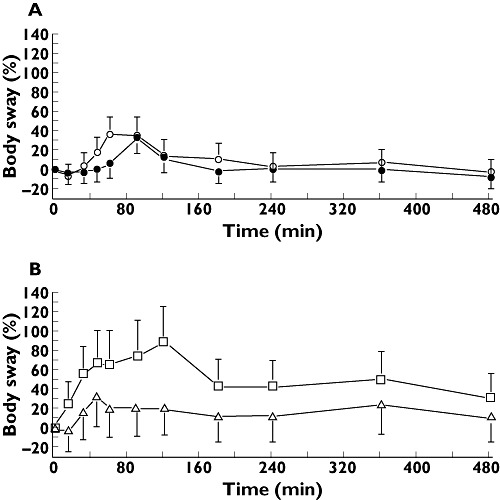
Effect−time profiles of baseline corrected body sway least square means in %, with 95% confidence interval error bars. A shows the results from panel I of the study, including sublingual THC 5.0 mg as closed circles and oral THC 5.0 mg as open circles. B has the results of panel II, with oral THC 6.5 mg as triangles and oral THC 8.0 mg as squares
Discussion
Available oral THC formulations and cannabis based medicines generally show disadvantageous pharmacokinetics that cause difficulties in dose regulation. Namisol® is a new THC formulation that was developed to achieve a more favourable pharmacokinetic profile. Since pharmacokinetic characteristics of THC ultimately determine its pharmacodynamic features, a fast onset of action and less variable response, as found in this study, are expected to lead to a more rapid and consistent clinical response. This study was designed to investigate two administration routes of Namisol® and three different oral doses of Namisol® in healthy volunteers.
Route of administration
The pharmacokinetic differences after oral and sublingual administration were small. Sublingual administration showed more flat concentration profiles of THC and 11-OH-THC, compared with oral administration, a late and small maximal concentration and a long apparent terminal half-life. This could be explained by a relatively small absorption constant of THC from the oral mucosa into the blood, with an absorption that could be slower than the elimination or distribution. The slow absorption from the oral mucosa after sublingual administration could be caused by the lipophilic character of THC. Furthermore, no in vitro data are available that support a slow absorption. The more favourable pharmacokinetic profile of the oral tablet compared with the sublingual route implies beneficial pharmacodynamic properties of oral Namisol®, such as an improvement of speed and accuracy of the onset and of the extent of the effects. Therefore, combined with the practical convenience of the administration procedure, the oral administration route was found to be more optimal.
Pharmacokinetics
Oral Namisol® showed a short time to reach maximal THC concentration (39–56 min) compared with reported values in previous studies using oral THC (60–240 min), nabilone (120–240 min), or oral-mucosal THC+CBD (Sativex®, 198–240 min) [18]–[21], [29]. Namisol® also had a shorter time to maximal concentration of the active metabolite 11-OH-THC (46–84 min) compared with what has been published for dronabinol (120–204 min) and Sativex® (216–234 min) [20], [21]. Although direct comparative studies are needed to corroborate these findings, the differences seem large enough to be realistic, and to be clinically relevant if the therapeutic effects follow the plasma concentrations reasonably directly. If so, Namisol® could give faster clinical effects compared with other oral formulations with THC or cannabis based medicines that are currently in clinical use. The short time to reach maximal THC and 11-OH-THC concentrations could be explained by a fast absorption of Namisol®. Inter-individual variability of Namisol® parameters was relatively large when compared with THC inhalation, as shown by compartmental analysis on THC pharmacokinetic parameters [27]. However, variability of THC maximal concentration was two to five times smaller than reported previously for dronabinol, which was based on non-compartmental analysis [21], [30]. This first in human study was primarily intended to explore the pharmacokinetic and pharmacodynamic properties of Namisol®. At this early stage of development, therefore, no registered cannabis based medicines were taken as an additional treatment arm. Although there are clear limitations to comparisons with literature data, in summary, the pharmacokinetic properties suggest that THC from Namisol® might have a faster absorption and a less variable maximal concentration. Therefore, the pharmacokinetics of Namisol® could be more favourable than currently registered oral dronabinol formulations and cannabis based medicines.
The pharmacokinetic model that was developed for THC and 11-OH-THC can be used to predict concentration−time profiles of alternative dosing scenarios. Hence, ‘what-if’ questions that are related to pharmacokinetics can be answered in further clinical development of this compound. Compartmental pharmacokinetic analysis assessed that the apparent terminal half-life of 11-OH-THC was shorter for oral 5.0 mg compared with 6.5 and 8.0 mg. This could be explained by the fact that the concentration after 5.0 mg drops below the lower limit of quantification more rapidly than for higher doses, and this does not necessarily imply that the actual half-life is different for oral than for sublingual administrations. A previous study administering 5 mg of labelled THC intravenously found that THC was still detectable in plasma 72 h after administration [31], while in the current study no THC or 11-OH-THC was detected in plasma at 24 h after administration. This confirms our implication that the limitations of the limit of quantification and the time frame of sampling in the current study thwarted an accurate estimation of the half-life of oral and sublingual Namisol®.
Compared with intravenous administration and inhalation, the concentration of the 11-OH metabolite after oral THC administration from Namisol® was relatively high [14], [29], [32]. The ratio of 11-OH-THC : THC (based on peak plasma concentrations) was 1 : 30 for intravenous administration and 1 : 7 for inhalation, while this ratio was 1 : 0.6–0.8 for Namisol®[13], [27], [33]. Previous studies with oral dronabinol and Sativex® also gave a lower metabolite concentration compared with Namisol® (11-OH-THC:THC was 1 : 1.2–2.0) [19], [20]. The relatively high concentrations of 11-OH-THC compared with the parent compound THC could be explained by several concomitant or alternative factors that could not be identified in this study. High concentrations of the metabolite suggest that considerable first-pass metabolism is taking place. Considering the absorption rate constant of 0.04 min–1 suggested by the PK model, it is possible that THC stays in the gastro-intestinal tract for a relatively long time where much of it is locally metabolized to 11-OH-THC. The metabolite is then absorbed from the gastrointestinal tract to the blood, where it is not as rapidly distributed to fatty tissues as THC, due to the less lipophilic character of the metabolite. At the same time, THC could rapidly disappear from blood into more fatty tissues, leading to low plasma concentrations. Long blood sampling schedules and very low detection thresholds for THC and its metabolites in plasma or mass balance studies would be needed to resolve the complex pharmacokinetics of THC in more detail.
Pharmacodynamics
Although the THC plasma concentrations after oral Namisol® administration were relatively low after completion of panel I, the pharmacodynamic effects were larger than we had expected, and comparable with those observed in a THC inhalation study in which high peak THC plasma concentrations were found [23]. This could reflect a large pharmacological effect of the 11-OH-metabolite. Preclinical studies have found 11-OH-THC to be a highly potent CB1-agonist [14], [15], and clinical studies also reported more rapid and larger effects after 11-OH-THC administration compared with THC [34]–[36]. In itself, this would have allowed us to predict the pharmacodynamic effects of higher doses in panel II, by reference to the results of other oral THC formulations in the literature which also produce high concentrations of 11-OH-THC. However, quantitative comparisons were quite difficult to make because of differences in methodology and study designs [37], [38]. Moreover, it was impossible to exclude the alternative (or additional) explanation that the large pharmacodynamic effects are due to a more efficient absorption of THC from the Namisol® formulation, with rapid redistribution to the CNS during the absorption phase. Since after panel I we could not be certain about the dose proportionality of Namisol® at higher doses, we decided to continue the study in panel II with two conservatively small dose increases (to 6.5 and 8.0 mg) for reasons of safety and tolerability, and to increase the dose further if necessary and possible.
The first pharmacodynamic effects of Namisol® 6.5 and 8.0 mg were already observed during the first assessments, 15 min after dosing. Namisol® had a faster onset of action than reported in a previous study with oral dronabinol (Marinol®), which had an onset of action between 0.5 and 1 h, and peak effects that were reached between 2 and 4 h [39]. The time profile of the pharmacodynamic effects was more similar to the concentration curve of 11-OH-THC than that of THC. A previous study reported that 11-OH-THC induced a quicker onset of the pharmacodynamic effects compared with THC [34]–[36]. These results in this study are quite promising for a fast onset of the clinical effects in a patient population, although future studies should carefully investigate the relation between pharmacodynamic effects in healthy volunteers and clinical effects in patients. Also, a more detailed analysis of the CNS effects of THC and 11-OH-THC should be done in humans to separate the contributions of both compounds to the effects. A future study where the effects of THC are compared with those of 11-OH-THC alone could provide meaningful information about the relative contributions of 11-OH-THC to the CNS effects of THC and cannabis.
In conclusion, Namisol® is a novel formulation of THC that is well-tolerated and absorbed quickly after ingestion, and reaches peak plasma concentrations within 1 h and maximal effects between 1 to 2 h after administration. Compared with the literature on registered dronabinol formulations and cannabis based medicines, these results imply that Namisol® may also have favourable pharmacokinetic and pharmacodynamic characteristics in patients. Further clinical studies are needed to show that these apparent advantages are also therapeutically relevant.
Acknowledgments
This study was made possible through a grant from the European Union, the European Fund for Regional Development (EFRO, ‘Here is an investment in your future’), awarded to Echo Pharmaceuticals.
Competing Interests
TB is an employee of Echo Pharmaceuticals, the Netherlands.
REFERENCES
- 1.Ungerleider JT, Andyrsiak T, Fairbanks L, Ellison GW, Myers LW. Delta-9-THC in the treatment of spasticity associated with multiple sclerosis. Adv Alcohol Subst Abuse. 1987;7:39–50. doi: 10.1300/j251v07n01_04. [DOI] [PubMed] [Google Scholar]
- 2.Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, Thompson A. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet. 2003;362:1517–26. doi: 10.1016/S0140-6736(03)14738-1. [DOI] [PubMed] [Google Scholar]
- 3.Zajicek JP, Sanders HP, Wright DE, Vickery PJ, Ingram WM, Reilly SM, Nunn AJ, Teare LJ, Fox PJ, Thompson AJ. Cannabinoids in multiple sclerosis (CAMS) study: safety and efficacy data for 12 months follow up. J Neurol Neurosurg Psychiatry. 2005;76:1664–9. doi: 10.1136/jnnp.2005.070136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang AE, Shiling DJ, Stillman RC, Goldberg NH, Seipp CA, Barofsky I, Simon RM, Rosenberg SA. Delata-9-tetrahydrocannabinol as an antiemetic in cancer patients receiving high-dose methotrexate. A prospective, randomized evaluation. Ann Intern Med. 1979;91:819–24. doi: 10.7326/0003-4819-91-6-819. [DOI] [PubMed] [Google Scholar]
- 5.Orr LE, McKernan JF, Bloome B. Antiemetic effect of tetrahydrocannabinol compared with placebo and prochlorperazine in chemotherapy-associated nausea and emesis. Arch Intern Med. 1980;140:1431–3. doi: 10.1001/archinte.140.11.1431. [DOI] [PubMed] [Google Scholar]
- 6.Sallan SE, Zinberg NE, Frei E., III Antiemetic effect of delta-9-tetrahydrocannabinol in patients receiving cancer chemotherapy. N Engl J Med. 1975;293:795–7. doi: 10.1056/NEJM197510162931603. [DOI] [PubMed] [Google Scholar]
- 7.Buccellato E, Carretta D, Utan A, Cavina C, Speroni E, Grassi G, Candeletti S, Romualdi P. Acute and chronic cannabinoid extracts administration affects motor function in a CREAE model of multiple sclerosis. J Ethnopharmacol. 2011;133:1033–8. doi: 10.1016/j.jep.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 8.Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, Layward L. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404:84–7. doi: 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- 9.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Janke J, Batkai S, Pacher P, Harvey-White J, Luft FC, Sharma AM, Jordan J. Activation of the peripheral endocannabinoid system in human obesity. Diabetes. 2005;54:2838–43. doi: 10.2337/diabetes.54.10.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herkenham M. Cannabinoid receptor localization in brain: relationship to motor and reward systems. Ann N Y Acad Sci. 1992;654:19–32. doi: 10.1111/j.1749-6632.1992.tb25953.x. [DOI] [PubMed] [Google Scholar]
- 12.Ishac EJ, Jiang L, Lake KD, Varga K, Abood ME, Kunos G. Inhibition of exocytotic noradrenaline release by presynaptic cannabinoid CB1 receptors on peripheral sympathetic nerves. Br J Pharmacol. 1996;118:2023–8. doi: 10.1111/j.1476-5381.1996.tb15639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42:327–60. doi: 10.2165/00003088-200342040-00003. [DOI] [PubMed] [Google Scholar]
- 14.Wilson RS, May EL. Analgesic properties of the tetrahydrocannabinols, their metabolites, and analogs. J Med Chem. 1975;18:700–3. doi: 10.1021/jm00241a012. [DOI] [PubMed] [Google Scholar]
- 15.Karler R, Turkanis SA. Different cannabinoids exhibit different pharmacological and toxicological properties. NIDA Res Monogr. 1987;79:96–107. [PubMed] [Google Scholar]
- 16.Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol. 1992;16:276–82. doi: 10.1093/jat/16.5.276. [DOI] [PubMed] [Google Scholar]
- 17.Hazekamp A, Bastola K, Rashidi H, Bender J, Verpoorte R. Cannabis tea revisited: a systematic evaluation of the cannabinoid composition of cannabis tea. J Ethnopharmacol. 2007;113:85–90. doi: 10.1016/j.jep.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Davis MP. Oral nabilone capsules in the treatment of chemotherapy-induced nausea and vomiting and pain. Expert Opin Investig Drugs. 2008;17:85–95. doi: 10.1517/13543784.17.1.85. [DOI] [PubMed] [Google Scholar]
- 19.Schwilke EW, Schwope DM, Karschner EL, Lowe RH, Darwin WD, Kelly DL, Goodwin RS, Gorelick DA, Huestis MA. Delta9-tetrahydrocannbinol (THC), 11-hydroxy-THC, and 11-nor-9-carboxy-THC plasma pharmacokinetics during and after continuous high-dose oral THC. Clin Chem. 2009;55:2180–9. doi: 10.1373/clinchem.2008.122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karschner EL, Darwin WD, Goodwin RS, Wright S, Huestis MA. Plasma cannabinoid pharmacokinetics following controlled oral {Delta}9-Tetrahydrocannabinol and oromucosal cannabis extract administration. Clin Chem. 2011;57:66–75. doi: 10.1373/clinchem.2010.152439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naef M, Curatolo M, Petersen-Felix S, Arendt-Nielsen L, Zbinden A, Brenneisen R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC-morphine combination in healthy subjects under experimental pain conditions. Pain. 2003;105:79–88. doi: 10.1016/s0304-3959(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 22.Wall ME, Perez-Reyes M. The metabolism of delta 9-tetrahydrocannabinol and related cannabinoids in man. J Clin Pharmacol. 1981;21:178S–89S. doi: 10.1002/j.1552-4604.1981.tb02594.x. [DOI] [PubMed] [Google Scholar]
- 23.Zuurman L, Roy C, Schoemaker R, Hazekamp A, den Hartigh J, Bender J, Verpoorte R, Pinquier J, Cohen A, van Gerven J. Effect of intrapulmonary tetrahydrocannabinol administration in humans. J Psychopharmacol. 2008;22:707–16. doi: 10.1177/0269881108089581. [DOI] [PubMed] [Google Scholar]
- 24.Bossong MG, van Berckel BN, Boellaard R, Zuurman L, Schuit RC, Windhorst AD, van Gerven JM, Ramsey NF, Lammertsma AA, Kahn RS. Delta 9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology. 2009;34:759–66. doi: 10.1038/npp.2008.138. [DOI] [PubMed] [Google Scholar]
- 25.Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974;47:211–8. [Google Scholar]
- 26.Bowdle TA, Radant AD, Cowley DS, Kharasch ED, Strassman RJ, Roy-Byrne RP. Psychedelic effects of ketamine in healthy volunteers. Relationship to steady-state plasma concentrations. Anesthesiology. 1998;88:82–8. doi: 10.1097/00000542-199801000-00015. [DOI] [PubMed] [Google Scholar]
- 27.Strougo A, Zuurman L, Roy C, Pinquier JL, van Gerven JM, Cohen AF, Schoemaker RC. Modelling of the concentration–effect relationship of THC on central nervous system parameters and heart rate – insight into its mechanisms of action and a tool for clinical research and development of cannabinoids. J Psychopharmacol. 2008;22:717–26. doi: 10.1177/0269881108089870. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30:387–404. doi: 10.1023/b:jopa.0000012998.04442.1f. [DOI] [PubMed] [Google Scholar]
- 29.Valeant Pharmaceuticals International. Cesamet Package Insert. 2006. Available at http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?id=13020 (last accessed May 2012)
- 30.Wall ME, Sadler BM, Brine D, Taylor H, Perez-Reyes M. Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clin Pharmacol Ther. 1983;34:352–63. doi: 10.1038/clpt.1983.179. [DOI] [PubMed] [Google Scholar]
- 31.Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK. Single dose kinetics of deuterium labelled delta 1-tetrahydrocannabinol in heavy and light cannabis users. Biomed Mass Spectrom. 1982;9:6–10. doi: 10.1002/bms.1200090103. [DOI] [PubMed] [Google Scholar]
- 32.Committee for medicinal products for human use. Guideline on the investigation of bioequivalence. 1-20-2010.
- 33.Naef M, Russmann S, Petersen-Felix S, Brenneisen R. Development and pharmacokinetic characterization of pulmonal and intravenous delta-9-tetrahydrocannabinol (THC) in humans. J Pharm Sci. 2004;93:1176–84. doi: 10.1002/jps.20037. [DOI] [PubMed] [Google Scholar]
- 34.Lemberger L, Crabtree RE, Rowe HM. 11-hydroxy-9-tetrahydrocannabinol: pharmacology, disposition, and metabolism of a major metabolite of marihuana in man. Science. 1972;177:62–4. doi: 10.1126/science.177.4043.62. [DOI] [PubMed] [Google Scholar]
- 35.Lemberger L. Tetrahydrocannabinol metabolism in man. Drug Metab Dispos. 1973;1:461–8. [PubMed] [Google Scholar]
- 36.Lemberger L, Martz R, Rodda B, Forney R, Rowe H. Comparative pharmacology of delta9-tetrahydrocannabinol and its metabolite, 11-OH-Delta9-tetrahydrocannabinol. J Clin Invest. 1973;52:2411–7. doi: 10.1172/JCI107431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curran HV, Brignell C, Fletcher S, Middleton P, Henry J. Cognitive and subjective dose-response effects of acute oral delta 9-tetrahydrocannabinol (THC) in infrequent cannabis users. Psychopharmacology (Berl) 2002;164:61–70. doi: 10.1007/s00213-002-1169-0. [DOI] [PubMed] [Google Scholar]
- 38.Zuurman L, Ippel AE, Moin E, van Gerven JM. Biomarkers for the effects of cannabis and THC in healthy volunteers. Br J Clin Pharmacol. 2009;67:5–21. doi: 10.1111/j.1365-2125.2008.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solvay Pharmaceuticals. Summary of Product Characteristics Marinol. 2004. Available at http://www.fda.gov/ohrms/dockets/dockets/05n0479/05N-0479-emc0004-04.pdf (last accessed May 2012)