

Abstract
AIM
To investigate whether hepatic impairment affects linagliptin pharmacokinetics, pharmacodynamics and tolerability.
METHOD
This open label, parallel group, single centre study enrolled patients with mild (n = 8), moderate (n = 9) or severe (n = 8) hepatic impairment and healthy subjects (n = 8). Groups were matched with regard to age, weight and gender. Primary endpoints were linagliptin exposure following 5 mg linagliptin once daily for 7 days in patients with mild and moderate hepatic impairment vs. healthy subjects or after a single 5 mg dose for patients with severe hepatic impairment vs. healthy subjects.
RESULTS
In mild hepatic impairment, steady-state linagliptin exposure was slightly lower than in healthy subjects [AUCτ,ss geometric mean ratio (GMR) 75.5%, 90% confidence interval (CI) 61.6%, 92.5%, and Cmax,ss GMR 64.4%, 90% CI 43.2%, 96.0%]. Exposure also tended to be lower in moderate hepatic impairment (AUCτ,ss GMR 85.5%, 90% CI 70.2%, 104.2% and Cmax,ss GMR 92.3%, 90% CI 62.8%, 135.6%). After a single dose, AUC(0,24 h) in patients with severe hepatic impairment was similar to that in healthy subjects (GMR 100.4%, 90% CI 75.0%, 134.3%) and Cmax was lower (GMR 77.0%, 90% CI 44.9%, 132.3%). Accumulation based on AUC or Cmax and renal excretion of unchanged linagliptin (≤7%) were comparable across groups. Median plasma DPP-4 inhibition was similar in healthy subjects (91%), and patients with mild (90%) and moderate (89%) hepatic impairment at steady-state trough concentrations, and in patients with severe hepatic impairment 24 h after a single dose (84%). Linagliptin was well tolerated.
CONCLUSION
Mild, moderate or severe hepatic impairment did not result in an increase in linagliptin exposure after single and multiple dosing compared with normal hepatic function. Dose adjustment with linagliptin is not required in patients with hepatic impairment.
Keywords: DPP-4 inhibitor, hepatic impairment, linagliptin, type 2 diabetes mellitus
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Linagliptin is an oral, highly selective dipeptidyl peptidase-4 (DPP-4) inhibitor that was approved in the United States, Europe and elsewhere in 2011 for the treatment of type 2 diabetes mellitus.
The elimination of linagliptin is primarily non-renal. Therefore, a potential effect of hepatic impairment on the elimination of linagliptin may have important implications for dosing recommendations.
WHAT THIS STUDY ADDS
This study shows that mild, moderate or severe hepatic impairment did not result in an increase in linagliptin exposure after single and multiple dosing as compared with normal hepatic function.
No linagliptin dose adjustment is required in patients with any degree of hepatic impairment.
Introduction
The global prevalence of diabetes, and in particular type 2 diabetes mellitus (T2DM), is rising rapidly, from an estimated 366 million in 2011 (8.3% of the world's adult population) to a predicted 552 million by 2030 [1]. Therefore, the incidence and prevalence of diabetes complications will also increase over the coming decades.
Renal impairment is common among subjects with T2DM. Thus, there is a need for novel drugs that address the specific issues of this large patient group, including the development of new drugs with non-renal elimination. Drugs that are mainly excreted through routes other than the kidneys are predominantly eliminated through the entero-hepatic pathway. Therefore, their metabolism in subjects with impaired liver function must be thoroughly clarified to provide solid, safe and reliable dosing recommendations.
Subjects with T2DM frequently suffer not only from impaired renal function, but also from various forms of liver disease. In fact, approximately 70% of patients with T2DM have non-alcoholic fatty liver disease, which is characterized by insulin resistance and hepatic fat accumulation in the absence of other causes, such as alcohol abuse, or viral or autoimmune hepatitis [2]. Furthermore, cirrhosis is potentially both a cause and consequence of diabetes [3] and approximately 30% of patients with cirrhosis have diabetes [4].
Linagliptin is a dipeptidyl peptidase-4 (DPP-4) inhibitor with a xanthine-based structure that differs from those of other agents in its therapeutic class [5]. Similar to other DPP-4 inhibitors, linagliptin is an effective oral antidiabetic drug with a low risk of hypoglycaemia and no association with weight gain [6]. Linagliptin exhibits non-linear pharmacokinetic characteristics [7]. It is assumed that this is due to target-mediated drug disposition resulting from high affinity, concentration dependent and saturable binding of linagliptin to DPP-4. Plasma protein binding of linagliptin decreased from about 99% at 1 nmol l−1 to 75%–89% at ≥30 nmol l−1 and 70%–80% at >100 nmol l−1[8]. Thus, at low plasma concentrations, linagliptin is predominantly bound to its target DPP-4 and there is a low free fraction that can be eliminated. With increasing plasma concentrations, linagliptin readily saturates DPP-4. Its free fraction then increases, and consequently its volume of distribution and clearance increase. Therefore, the pharmacokinetic parameters, which remain constant regardless of dose for drugs with linear pharmacokinetics (e.g. clearance, volume of distribution and fraction excreted renally), increase with higher doses in the case of linagliptin. As a result, the terminal half-life of linagliptin is long (>100 h) since it mainly reflects the slow release of linagliptin from the DPP-4 enzyme. Despite this long terminal half-life, time to steady-state is usually reached within 2–4 days and the accumulation ratio is usually around 1.3 for a 5 mg dose [9].
Linagliptin is predominantly non-renally excreted. In people with normal hepatic function, <10% of the linagliptin dose undergoes renal clearance [9]. In contrast, 80–85% of sitagliptin and vildagliptin are excreted via the renal route, whereas saxagliptin is excreted by both renal and hepatic routes [10]–[13]. Because linagliptin is only renally excreted to a minor extent, the major route of elimination is via the entero-hepatic system. Hepatic metabolism of linagliptin is minimal and its metabolites, including its main metabolite CD1790, are pharmacologically inactive [14], [15]. Linagliptin is not a clinically relevant inhibitor or inducer of cytochrome P450 isoenzymes (although it is a weak inhibitor of CYP3A4) [15]. It is also a substrate and a weak inhibitor (that is not of clinical significance at therapeutic doses) of P-glycoprotein [14], [15].
Given the predominantly non-renal route of elimination, it is particularly important to characterize the pharmacokinetics of linagliptin in patients with hepatic impairment, in order to clarify potential risks and dosing implications. Thus, the primary objective of this study was to investigate the influence of hepatic impairment on the pharmacokinetics and pharmacodynamics of linagliptin after single or multiple oral administration of 5 mg linagliptin.
Methods
Subjects
Subjects, aged 18–70 years, with normal liver function or mild, moderate or severe hepatic impairment were eligible for this study. The participants, enrolled from the pool of subjects at IFE Human Pharmacology SRL, Timisoara, Romania, were matched with regard to gender, age ±10 years and weight ±20%. The body mass index of the participants was 18.5–29.9 kg m−2. As recommended in the guidance for clinical trials in patients with impaired hepatic function from the United States Food and Drug Administration (FDA), this study included patients from all three Child-Pugh categories (mild, moderate and severe) as well as control subjects with normal hepatic function [16]. Hepatic impairment was classified as mild (Child-Pugh class A score 5 or 6 points), moderate (Child-Pugh class B score 7–9 points) or severe (Child-Pugh class C score 10–15 points) [17]. Using the Cockcroft−Gault formula to calculate creatinine clearance, the minimum creatinine clearance for inclusion was ≥80 ml min−1 for healthy controls and patients with mild or moderate hepatic impairment and ≥40 ml min−1 for patients with severe hepatic impairment.
Other exclusion criteria included abdominal surgery (except appendectomy and oesophageal varices), diseases of the central nervous system, psychiatric and neurological disorders (except hepatoportal encephalopathy), history of relevant orthostatic hypotension, fainting spells or blackouts, chronic or relevant acute infections (except nonprogressive chronic hepatitis), relevant allergy or hypersensitivity, use of drugs that might influence the results, smoking >10 cigarettes, three cigars or three pipes per day and consuming >60 g alcohol per day.
Alcohol consumption throughout the study period and smoking on study days was prohibited. Caffeine, theobromine and methylxanthine-containing food or drink (coffee, tea, chocolate, cola, energy drinks), certain fruit juices (apple, orange, grapefruit), vegetables in the mustard green family (kale, broccoli, watercress, kohlrabi, Brussels sprouts) and charbroiled meat were not permitted on the main study days and for 4 h after drug intake on other days. Citrus fruits, in particular grapefruit and Seville oranges and their juices, were not permitted from 5 days before the first intake of study drug until after the last sample from each period had been collected. Fruit juices were not permitted since these have been known to interfere with transporters potentially involved in drug disposition [18].
Study design
This open label, phase I, parallel group comparison, single centre study evaluated 5 mg linagliptin once daily (the licensed therapeutic dose in T2DM) in healthy subjects and patients with mild, moderate and severe hepatic impairment. After a 28 day screening period, eligible subjects underwent a baseline evaluation (day −1) and received a 5 mg oral dose of linagliptin on day 1 (all groups), followed by a further six doses at 24 h intervals (healthy subjects and patients with mild and moderate hepatic impairment only). Participants were admitted to the study centre at least 14 h prior to the first linagliptin administration and were discharged no earlier than 25 h after administration of the final dose. Following an overnight fast of at least 10 h, linagliptin tablets were administered with 240 ml of water while subjects were in a standing position. All doses of study medication were witnessed. Since there was a theoretical risk of a prolonged plasma half-life of linagliptin in patients with severe hepatic impairment, these subjects received a single dose of 5 mg linagliptin on day 1 only.
All subjects were kept under close medical surveillance until 25 h after drug administration and were not allowed to lie down during the 2 h following drug administration except for medical examination or electrocardiogram (ECG) recording. The observation time after the last administration of drug was at least 4 days.
The primary pharmacokinetic objective was to compare linagliptin exposure in patients with mild and moderate hepatic impairment with healthy subjects. The exposure was based on the area under the concentration–time curve (AUC) at steady-state over a uniform dosing interval (AUCτ,ss) and the maximum plasma concentration at steady-state (Cmax,ss). Additionally, the study compared linagliptin exposure in patients with severe hepatic impairment with healthy subjects based on AUC over the 24 h period after dosing (AUC(0,24 h)) and the maximum plasma concentration (Cmax) after a single dose. The AUC over a 24 h period was selected over longer time periods such as 72 h, as linagliptin has a long terminal half-life due to its slow dissociation rate from DPP-4. Linagliptin's tight target binding results in effects on clearance that are more visible in the first hours, due to initial rapid clearance of non-DPP-4 bound drug, whereas beyond 24 h the pharmacokinetics of linagliptin mainly reflect the binding to DPP-4 and slow dissociation of the linagliptin/DPP-4 complex. Therefore AUC(0,24 h) was considered a more sensitive parameter to detect any effect of severe hepatic impairment on linagliptin exposure. Secondary endpoints comprised additional pharmacokinetic parameters. Steady-state pharmacokinetic parameters were measured only in healthy subjects and patients with mild and moderate hepatic impairment, and were predicted using a population pharmacokinetic model in patients with severe hepatic impairment.
The study was conducted according to the principles of the Declaration of Helsinki and in adherence to the principles of Good Clinical Practice. Signed, written, informed consent was obtained from all participants. The trial protocol and other relevant study documents were reviewed and approved by the local independent ethics committee (the National Ethics Committee for Drug Clinical Trials, Bucharest, Romania) and by the national competent authority (National Medicine Agency, Bucharest, Romania). This trial was registered in the European Clinical Trials Database (EudraCT), registration number 2008-000922-38.
Pharmacokinetic and pharmacodynamic methods
Blood samples for the measurement of plasma concentrations of linagliptin and CD1790 (the pharmacologically inactive main metabolite of linagliptin [14]) were taken pre dose and at regular intervals post dose (0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8 and 12 h) on day 1 for all groups, and for the control, mild and moderate hepatic impairment groups on day 7. Samples were also taken at 24 h intervals on the intervening days 2 to 6 (pre dose for the control, mild and moderate hepatic impairment groups) and on days 8 to 12 after the last dose. For subjects with severe hepatic impairment, samples were taken at 24 h intervals on days 2 to 6. For pharmacokinetic assessments, approximately 5 ml of blood was collected in ethylenediaminetetraacetic acid (EDTA) tubes and centrifuged immediately at 2000–4000 g for 10 min at 4–8°C. The plasma was collected and frozen immediately at approximately −18°C.
Urine collections for determination of urinary linagliptin and CD1790 concentrations were obtained after the first dose on day 1 and at steady-state on day 7 at the following time points: 0.25 h pre dose and 0–4, 4–8, 8–12, 12–24 and 24–48 h post dose. Urine collected during each time period was pooled and 1 ml aliquots were frozen.
Plasma and urine concentrations of linagliptin and CD1790 were analyzed by fully validated methods using high performance liquid chromatography coupled to tandem mass spectrometry with [13C3]linagliptin and [13C3]CD1790 as internal standards as described previously [14]. No interference of endogenous compounds was observed in blank human plasma or urine. The calibration curves of undiluted plasma samples (150 µl) were linear over the range of concentrations from 0.100 to 20 nmol l−1 for linagliptin and from 0.050 to 10 nmol l−1 for CD1790. The calibration curves of undiluted acidified urine (40 µl) were linear over the range of concentrations from 1.00 to 1000 nmol l−1 for linagliptin and from 0.50 to 500 nmol l−1 for CD1790. In-study assay validation for linagliptin at three nominal concentrations yielded assay inaccuracy of 0.7% to 4.8% (plasma) and 0.3% to 5.6% (urine), and imprecision of 4.9% to 5.5% (plasma) and 1.9% to 5.1% (urine). For the CD1790 assay validation, three nominal concentrations resulted in assay inaccuracy of −1.5% to 4.0% (plasma) and −0.2% to 5.6% (urine), and imprecision of 2.4% to 3.5% (plasma) and 2.6% to 5.1% (urine).
For the assessment of plasma DPP-4 activity, blood samples (approximately 3.0 ml) were taken at the same time points and plasma was obtained in the same way as for the pharmacokinetic samples. Activity of DPP-4 in EDTA-plasma was analyzed by a validated semi-quantitative enzyme activity assay with fluorescence detection based on the reaction with H-alanine-proline-7-aminoamido-4-trifluoromethylcoumarin, as described previously [9]. The in-study assay precision using in-house controls was 4.4% to 9.4% (accuracy was not assessed since no international standards with known concentration exist for DPP-4 activity).
For the determination of protein binding of linagliptin and CD1790, on day 1 before drug administration, 27 ml of blood was collected in three vials coated with EDTA. Blood was centrifuged at ∼2500 g for 10 min at 4°C. Plasma samples obtained were frozen at −20°C. In vitro protein binding was determined by equilibrium dialysis using radiolabelled compounds [8].
Pharmacokinetic analysis – non-compartmental analysis
The non-compartmental pharmacokinetic analysis of the linagliptin and CD1790 plasma/urine concentration–time data was carried out using WinNonlin® software (Professional, version 4.1, Pharsight Corporation; Mountain View, CA, USA) and standard non-compartmental methods as described previously [9]. As the pre dose analyte concentration was generally below the limit of quantification, the concentration at time zero was assumed to be zero.
Pharmacokinetic analysis – model-based analysis
In addition, a model-based analysis was performed to predict the linagliptin exposure at steady-state in patients with severe hepatic impairment. The analysis was conducted using the non-linear mixed effect modelling software NONMEM V (GloboMax LLC, Hanover, Maryland). A population pharmacokinetic model, developed previously to describe the non-linear pharmacokinetics of linagliptin in patients with T2DM after a single dose and at steady-state, served as the basis for the current analysis [19]. The model parameters were adjusted for the current analysis using linagliptin plasma concentrations after a single dose (from healthy subjects and patients with mild, moderate and severe hepatic impairment) and at steady-state (from healthy subjects and patients with mild and moderate hepatic impairment only). The individual linagliptin AUCτ,ss and Cmax,ss values for patients with severe hepatic impairment were predicted using their individual model estimates as derived in the population analysis and the same sampling scheme as applied for healthy subjects and patients with mild and moderate hepatic impairment.
Safety
Safety and tolerability were evaluated on the basis of adverse events, blood pressure and pulse rate, 12-lead ECGs, clinical laboratory tests (haematology, clinical chemistry and urinalysis), medical examination and investigator assessment of global tolerability. All subjects who received at least one dose of study drug were included in the safety evaluation.
Statistical analysis
The pharmacokinetic parameters for linagliptin were compared between groups using an analysis of variance (anova) model. These parameters were natural log transformed before fitting this model. The difference between the expected means for log(test)− log(reference) was estimated by the difference in the corresponding least square means, and two-sided 90% confidence intervals (CI) based on the t-distribution were computed. The results were back-transformed to derive the geometric mean (gMean) and interval estimates for the median inter-subject ratio between responses under test and reference.
The study planned to recruit 32 subjects (eight per group). This sample size was not based on a formal power calculation, but was judged adequate to achieve the study objective and is in agreement with regulatory guidance from the FDA for evaluable subjects in the control and the moderate-impairment arms of such studies [16].
Results
Subject disposition
Thirty-three subjects were treated (eight each in the healthy, mild and severe hepatic impairment groups and nine with moderate hepatic impairment). The safety analysis included all 33 subjects who received at least one dose of linagliptin (Table 1). Two patients in the moderate hepatic impairment group had creatinine clearance values ≤80 ml min−1 (60 ml min−1 and 69 ml min−1) and, therefore, violated the renal function inclusion criterion. Pharmacokinetic analyses were performed with and without inclusion of these two patients. Since results were comparable, data are shown for the entire cohort including these patients. One patient in the mild hepatic impairment group had no detectable linagliptin plasma or urine concentrations on day 1, but concentrations were in the expected range on subsequent days. Therefore, there were seven evaluable plasma profiles in this group after single doses but eight after multiple doses of linagliptin. For another patient from this group, no urine samples were available for day 1, resulting in assessment of six patients after single dose and eight patients at steady-state for urine pharmacokinetic parameters. One patient in the moderate hepatic impairment group withdrew informed consent after the sixth dose. Therefore, in this group, single dose data are available from nine patients and multiple dose data from eight patients.
Table 1.
Demographics and baseline characteristics of healthy subjects and patients with different degrees of hepatic impairment
| Hepatic impairment group | ||||
|---|---|---|---|---|
| Healthy (n = 8) | Mild (n = 8) | Moderate (n = 9) | Severe (n = 8) | |
| Gender [n (%)] | ||||
| Male | 5 (62.5) | 5 (62.5) | 5 (55.6) | 5 (62.5) |
| Female | 3 (37.5) | 3 (37.5) | 4 (44.4) | 3 (37.5) |
| Age (years) | ||||
| Mean ± SD | 49.5 ± 6.7 | 51.8 ± 3.9 | 55.9 ± 7.5 | 52.9 ± 7.1 |
| Range | 41–59 | 44–57 | 43–66 | 44–66 |
| White race [n (%)] | 8 (100) | 8 (100) | 9 (100) | 8 (100) |
| Height (cm) | ||||
| Mean ± SD | 168.9 ± 7.3 | 168.5 ± 8.4 | 165.9 ± 10.3 | 168.4 ± 11.6 |
| Range | 160–179 | 152–180 | 153–186 | 152–188 |
| Weight (kg) | ||||
| Mean ± SD | 72.8 ± 9.9 | 71.4 ± 14.1 | 72.2 ± 10.5 | 74.5 ± 17.8 |
| Range | 56–88 | 56–92 | 56–94 | 55–105 |
Plasma concentration–time profiles
Linagliptin plasma concentration–time profiles were broadly similar across all groups (Figure 1). Linagliptin concentrations following a single dose increased rapidly. The median times to maximum plasma concentration (tmax) were 1.5, 1.5, 1.0 and 0.9 h in the healthy, mild, moderate and severe hepatic impairment groups, respectively, and the profiles showed at least biphasic disposition. Visual inspection suggested that steady-state trough concentrations were reached after the second dose. Linagliptin showed a long terminal half-life after the last dose. Plasma concentrations 24 h after dosing in patients with mild, moderate and severe hepatic impairment were almost congruent. Healthy subjects showed higher exposure during the first 6 h after dosing than patients with hepatic impairment, after which exposure was comparable.
Figure 1.
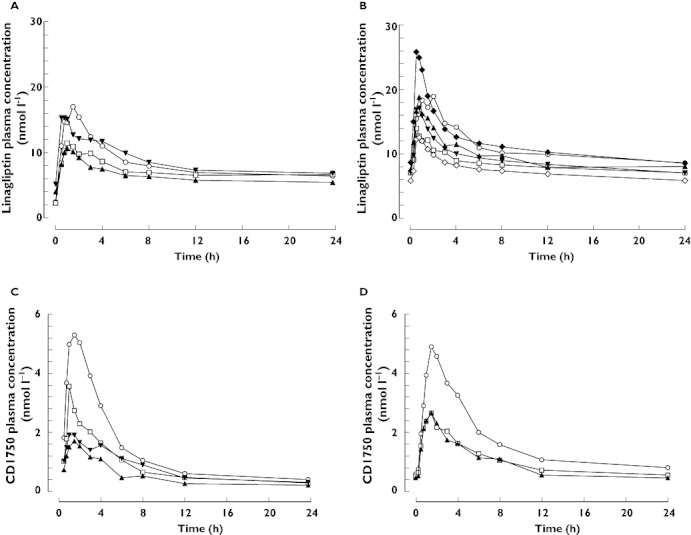
Arithmetic mean plasma concentration–time profiles of linagliptin 5 mg after single oral doses (A) and at steady-state (B), and of CD1790 after single oral doses of linagliptin (C) and at steady-state (D) in healthy subjects (normal hepatic function) and patients with mild, moderate or severe hepatic impairment. (A and C) Normal hepatic function (n = 8) ( ); Mild hepatic impairment (n = 8) (
); Mild hepatic impairment (n = 8) ( ); Moderate hepatic impairment (n = 9) (
); Moderate hepatic impairment (n = 9) ( ); Severe hepatic impairment (n = 8) (
); Severe hepatic impairment (n = 8) ( ); (B and D) Normal hepatic function (n = 8) (
); (B and D) Normal hepatic function (n = 8) ( ); Mild hepatic impairment (n = 8) (
); Mild hepatic impairment (n = 8) ( ); Moderate hepatic impairment (n = 8*) (
); Moderate hepatic impairment (n = 8*) ( ); Severe hepatic impairment (median prediction†) (
); Severe hepatic impairment (median prediction†) ( ); Severe hepatic impairment (5th percentile prediction†) (
); Severe hepatic impairment (5th percentile prediction†) ( ); Severe hepatic impairment (95th percentile prediction†) (
); Severe hepatic impairment (95th percentile prediction†) ( ). *Eight patients only completed the multiple dosing period in the moderate impairment group. †Distribution of predicted arithmetic mean steady-state profiles for severe hepatic impairment group (1000 simulated studies with n = 8)
). *Eight patients only completed the multiple dosing period in the moderate impairment group. †Distribution of predicted arithmetic mean steady-state profiles for severe hepatic impairment group (1000 simulated studies with n = 8)
Following single doses of 5 mg linagliptin, plasma concentrations of the inactive metabolite CD1790 were generally detected between 15 and 30 min after dosing, reaching Cmax after 0.75–4 h. Plasma concentrations of CD1790 were highest in healthy subjects, followed by patients with mild hepatic impairment. CD1790 exposure was comparable in patients with moderate and severe hepatic impairment (Figure 1). Plasma concentrations of CD1790 24 h post dosing were almost congruent in all cohorts. After multiple dosing, plasma concentrations of CD1790 were higher in healthy subjects than in patients with mild or moderate hepatic impairment, where concentrations were comparable.
Pharmacokinetic analysis
Following a single oral dose of 5 mg linagliptin, the rate and extent of absorption and exposure as indicated by AUC(0,24 h), Cmax and tmax in patients with different degrees of hepatic impairment and healthy subjects showed no trend to increase with more severe hepatic impairment (Table 2). The AUC(0,24 h) and Cmax tended to be lower in patients with mild and moderate hepatic impairment than in healthy subjects. Pharmacokinetic parameters in patients with moderate hepatic impairment were comparable with and without inclusion of the two patients with abnormal renal function (AUC(0,24 h) 148 nmol l−1 h vs. 155 nmol l−1 h and Cmax 12.1 nmol l−1vs. 12.0 nmol l−1). The severe hepatic impairment and healthy groups exhibited similar AUC(0,24 h) and Cmax. The geometric mean ratio (GMR) for AUC(0,24 h) was 100.4% (90% CI 75.0%, 134.3%) and for Cmax was 77.0% (90% CI 44.9%, 132.3%).
Table 2.
Non-compartmental pharmacokinetic parameters of linagliptin after single oral doses of 5 mg linagliptin in patients with different degrees of hepatic impairment compared with healthy subjects
| Hepatic impairment group | ||||||||
|---|---|---|---|---|---|---|---|---|
| Healthy (n = 8) | Mild (n = 7†) | Moderate (n = 9) | Severe (n = 8) | |||||
| gMean | gCV | gMean | gCV | gMean | gCV | gMean | gCV | |
| AUC(0,24 h) (nmol l−1 h) | 189 | 27.8 | 164 | 33.3 | 148 | 21.3 | 190 | 39.4 |
| Cmax (nmol l−1) | 17.3 | 56.9 | 11.9 | 45.2 | 12.1 | 31.2 | 13.3 | 77.8 |
| tmax (h)* | 1.50 | 0.50–3.00 | 1.50 | 0.25–3.00 | 1.00 | 0.25–2.00 | 0.875 | 0.50–6.00 |
| C24 (nmol l−1) | 6.26 | 24.5 | 6.45 | 26.9 | 5.28 | 27.0 | 6.67 | 23.7 |
| fe(0,24 h) (%) | 1.31 | 148 | 0.705‡ | 336 | 0.483 | 162 | 0.923§ | 275 |
| CLR,(0,24 h) (ml min−1) | 12.2 | 123 | 7.31‡ | 215 | 5.75 | 145 | 8.74§ | 161 |
| t1/2 (h) | – | – | – | – | – | – | 124 | 61.2 |
For tmax, the median and range (min–max) is given.
Plasma linagliptin concentrations only available throughout day 1 for seven patients in the mild impairment group.
Urine linagliptin concentrations only available for whole of day 1 for six patients in the mild impairment group.
Urine linagliptin concentrations only available for whole of day 1 for seven patients in the severe impairment group. gMean geometric mean, gCV geometric coefficient of variation, AUC(0,24 h) area under the plasma concentration–time curve over the time interval 0 to 24 h, Cmax maximum plasma concentration, tmax time from dosing to the maximum plasma concentration, C24 plasma concentration at 24 h, fe(0,24 h) fraction excreted unchanged in urine in the time interval 0 to 24 h, CLR(0,24 h) renal clearance in the time interval 0 to 24 h, t1/2 terminal half-life in plasma.
The inter-individual variability in single dose pharmacokinetic parameters was highest among patients with severe hepatic impairment. Concentrations 24 h after the first dose were comparable in all groups and showed no trend to greater exposure with increasing severity of hepatic impairment. Renal excretion was <1.4% within the first 24 h after dosing in all groups and was independent of the degree of hepatic impairment.
Steady-state pharmacokinetic parameters were generally comparable between patients with mild and moderate hepatic impairment and healthy subjects (Table 3). Individual steady-state AUCτ,ss and Cmax values showed a large overlap between patients with mild and moderate hepatic impairment and healthy subjects. In patients with mild hepatic impairment, the linagliptin exposure at steady-state tended to be lower than in healthy controls. The GMR for AUCτ,ss was 75.5% (90% CI 61.6%, 92.5%) and the GMR for Cmax,ss was 64.4% (90% CI 43.2%, 96.0%). In patients with moderate hepatic impairment, exposure at steady-state was comparable with that in the healthy controls. The GMR for AUCτ,ss was 85.5% (90% CI 70.2%, 104.2%) and for Cmax,ss was 92.3% (90% CI 62.8%, 135.6%). In patients with moderate hepatic impairment, steady-state pharmacokinetic parameters were generally comparable with and without the two patients with abnormal renal function (AUCτ,ss 217 nmol l−1 h vs. 207 nmol l−1 h and Cmax,ss 19.2 nmol l−1vs. 17.9 nmol l−1). However, accumulation half-life and accumulation factors based on AUCτ,ss and Cmax tended to be higher when these two patients were included. Steady-state renal excretion was ≤7% during the 24 h dosing interval and comparable in all cohorts.
Table 3.
Steady-state non-compartmental pharmacokinetic parameters of linagliptin after multiple oral doses of 5 mg linagliptin in patients with mild or moderate hepatic impairment compared with healthy subjects
| Hepatic impairment group | ||||||
|---|---|---|---|---|---|---|
| Healthy (n = 8) | Mild (n = 8) | Moderate (n = 8§) | ||||
| gMean | gCV | gMean | gCV | gMean | gCV | |
| AUCτ,ss (nmol l−1 h) | 254 | 18.9 | 191 | 27.2 | 217 | 26.0 |
| Cmax,ss (nmol l−1) | 20.8 | 38.6 | 13.4 | 55.8 | 19.2 | 52.5 |
| tmax,ss (h)* | 1.50 | 0.50–2.00 | 1.00 | 0.50–3.00 | 0.625 | 0.25–2.00 |
| C24,ss (nmol l−1) | 8.41 | 18.2 | 6.75 | 28.2 | 7.85 | 18.8 |
| t1/2,ss (h) | 77.7 | 32.6 | 95.0 | 18.0 | 96.1 | 54.7 |
| fe(0,24 h),ss (%) | 7.12 | 50.3 | 4.84† | 57.8 | 6.13 | 51.2 |
| CLR,(0,24 h),ss (ml min−1) | 49.5 | 40.8 | 44.7† | 40.1 | 49.8 | 50.8 |
| RA,AUC(0,24 h) | 1.34 | 22.2 | 1.25‡ | 23.9 | 1.46 | 28.4 |
 |
1.20 | 53.9 | 1.22‡ | 64.3 | 1.53 | 65.8 |
| Accumulation t1/2 (h) | 10.9 | 66.2 | 8.11‡ | 86.8 | 13.1 | 61.7 |
For tmax, the median and range (min–max) is given.
Urine linagliptin concentrations only available for whole of day 1 for seven patients in the mild impairment group.
Single dose and steady-state pharmacokinetic data only available for seven patients in the mild impairment group.
Eight patients only completed full 7 days of study in the moderate impairment group. AUCτ,ss area under the plasma concentration–time curve at steady-state over the dosing interval τ, Cmax,ss maximum plasma concentration at steady-state, tmax,ss time from last dosing to maximum plasma concentration at steady-state over the dosing interval τ, C24,ss plasma concentration at 24 h at steady-state, t1/2,ss terminal half-life in plasma at steady-state, fe(0,24 h),ss fraction excreted unchanged in urine in the time interval 0 to 24 h at steady-state; CLR(0,24 h),ss renal clearance in the time interval 0 to 24 h at steady-state, RA,AUC(0,24 h) accumulation ratio based on AUC(0,24 h), 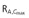 accumulation ratio based on Cmax, Accumulation t1/2 equals t× ln(2)/ln(RA,AUC(0,24 h)/[RA,AUC(0,24 h)– 1]).
accumulation ratio based on Cmax, Accumulation t1/2 equals t× ln(2)/ln(RA,AUC(0,24 h)/[RA,AUC(0,24 h)– 1]).
After single oral doses of 5 mg linagliptin, CD1790 exposure appeared to be lower in subjects with hepatic impairment compared with healthy subjects (Table S1). In the severe hepatic impairment vs. healthy group, the GMR for AUC(0,24 h) was 43.1% (90% CI 21.1%, 88.2%) and for Cmax was 32.3% (90% CI 14.2%, 73.5%).
At steady-state for linagliptin, there were some decreases in CD1790 exposure in patients with mild and moderate hepatic impairment compared with healthy subjects (Table S2). In the mild hepatic impairment vs. healthy group, the GMR for AUCτ,ss was 62.0% (90% CI 39.1%, 98.0%) and for Cmax was 50.7% (90% CI 25.3%, 101.7%). In the moderate hepatic impairment vs. healthy group, the GMR for AUCτ,ss was 49.9% (90% CI 27.8%, 89.4%) and for Cmax was 53.0% (90% CI 26.6%, 105.3%).
Model analysis
Based on the individual parameter estimates from the population pharmacokinetic analysis, steady-state predictions were performed for patients with severe hepatic impairment (Figure 1). The predicted gMean was 233 nmol l−1 h (gCV 33%) for AUCτ,ss and 19 nmol l−1 (gCV 67%) for Cmax,ss. Thus, the prediction of the linagliptin exposure for patients with severe hepatic impairment was only slightly lower than those observed in healthy controls.
Protein binding
A concentration-dependent effect of [3H]-linagliptin binding to plasma proteins in samples from healthy controls and patients with hepatic impairment was observed in vitro at three concentrations (0.5, 10 and 200 nmol l−1). Across the groups, mean plasma protein binding ranged from 73% to 81% at concentrations of 200 nmol l−1, 87% to 91% at 10 nmol l−1 and 98% to 99% at 0.5 nmol l−1.
Pharmacodynamic analysis
Median plasma DPP-4 inhibition 24 h after a single dose exceeded 80% (the therapeutic threshold) in patients with mild, moderate and severe hepatic impairment and in healthy subjects (77%, 80%, 84% and 85%, respectively) (Figure 2). At steady-state trough concentration, median DPP-4 inhibition was maintained at high levels in patients with mild and moderate hepatic impairment and in healthy subjects (90%, 89% and 91%, respectively). The pharmacokinetic–pharmacodynamic relationship was comparable and unaffected by hepatic function (Figure 3).
Figure 2.
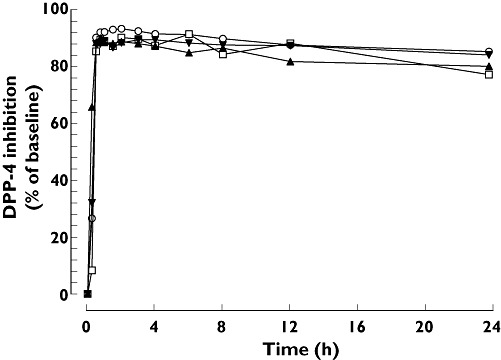
Median DPP-4 inhibition after single oral doses of 5 mg linagliptin in patients with different degrees of hepatic function. Normal hepatic function (n = 8) ( ); Mild hepatic impairment (n = 8) (
); Mild hepatic impairment (n = 8) ( ); Moderate hepatic impairment (n = 9) (
); Moderate hepatic impairment (n = 9) ( ); Severe hepatic impairment (n = 8) (
); Severe hepatic impairment (n = 8) ( )
)
Figure 3.
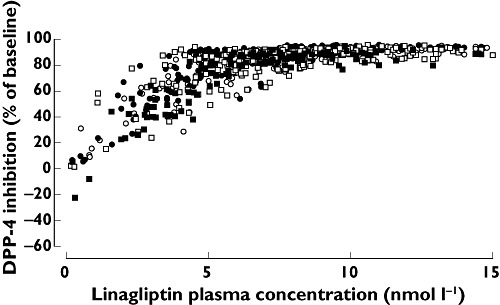
Correlation between plasma DPP-4 inhibition and plasma linagliptin concentrations in patients with different degrees of hepatic function. Normal hepatic function (○); Mild hepatic impairment (●); Moderate hepatic impairment (□); Severe hepatic impairment (n = 8) ( )
)
Safety and tolerability
Linagliptin was well tolerated in this study. Four patients (44.4%) with moderate hepatic impairment and one patient (12.5%) with mild hepatic impairment reported at least one adverse event, compared with three healthy subjects (37.5%). Myocardial ischaemia without clinical symptoms was reported in one healthy subject (12.5%). This adverse event was deemed unrelated to study medication. The diagnosis was based on T wave inversions observed on the ECG recorded 2 h after dosing on day 1. No further T wave inversions were detectable on subsequent ECGs and there was no further diagnostic or therapeutic follow-up. After a single dose of 5 mg linagliptin, five patients (62.5%) with severe hepatic impairment reported at least one adverse event, one of which the investigator considered possibly related to the study medication (prolonged QTc interval). The most frequently reported MedDRA system organ class was gastrointestinal disorders (in four subjects).
Discussion
In contrast to all other DPP-4 inhibitors currently approved or in clinical development for the treatment of diabetes, the elimination of linagliptin is primarily non-renal. This unique elimination pathway is likely to be an advantage in patients with T2DM and renal impairment. However, since linagliptin is predominantly eliminated via the entero-hepatic system, it was considered to be of crucial importance to elucidate its hepatic excretion, not only in subjects with normal liver function but also in subjects with hepatic impairment and clarify whether dose reductions would be necessary in this patient population.
In general, it is expected that the elimination of drugs primarily excreted via the liver would be influenced by hepatic impairment. However, as demonstrated in this study, linagliptin exposure was not increased following single or multiple doses of 5 mg linagliptin in subjects with mild, moderate or severe hepatic impairment when compared with controls with normal liver function. Linagliptin exposure was slightly higher (14–25%) in healthy subjects than in subjects with various degrees of hepatic impairment.
The relatively lower linagliptin exposure in patients with hepatic impairment was somewhat surprising, but there may be several explanations for this observation. First, it has been reported that P-glycoprotein activity in the intestine and the liver may be increased/up-regulated in hepatic impairment [20]. This could then limit the absorption of linagliptin from the intestine and enhance biliary excretion, contributing to the lower exposure in the patient groups with reduced liver function. Secondly, linagliptin exposure in the control group was at the higher end of the normal distribution when compared with previously performed linagliptin studies [9], so it cannot be excluded that the higher exposure in this group may be due to variability and small sample size. Thirdly, an increase in renal excretion may have compensated for any deficit in hepatic excretory capacity. However, no increase in renal excretion of linagliptin was evident in the hepatic impairment groups in the current study compared with controls with normal liver function. Fourthly, direct excretion of linagliptin into the gut may be an alternative excretion route in the presence of hepatic impairment (unpublished observations).
However, the most likely reason why linagliptin exposure is not increased in subjects with hepatic impairment may be related to the pharmacokinetic and pharmacodynamic properties of the drug. Linagliptin has a very high affinity for its target, DPP-4, which is present in plasma and tissues, and it is only released slowly from the enzyme [5], with unbound, free drug being eliminated quickly [19]. Thus, linagliptin exhibits high, concentration-dependent plasma protein binding (99% at concentrations below 1 nmol l−1 in in vitro plasma protein binding assays [8]), which results in very low plasma concentrations of unbound compound [i.e. the free average linagliptin concentration is only approximately 0.7 nmol l−1 (unpublished data)]. It is only this small unbound plasma fraction that is directly exposed to hepatic metabolism and excretion. In liver disease, a potential impairment in drug metabolism may occur through decreased capacity of the metabolizing enzymes, decreased liver blood flow or intra/extra-hepatic shunting. In addition, hepatic impairment may also affect drug excretion due to increased capacity of hepatic drug transporters to eliminate linagliptin via the bile. The metabolites of linagliptin have been shown to play only a minor role in the overall disposition and elimination of the drug [14]. Therefore, because linagliptin is predominantly eliminated without involvement of the hepatic metabolizing function, we suggest that the high enzyme binding, in conjunction with a low rate of hepatic metabolism results in hepato-biliary excretion of predominantly unchanged linagliptin. The low concentrations of unbound linagliptin in the circulation suggest that even patients with severe hepatic impairment may have sufficient residual liver capacity to meet the limited metabolic and hepatic excretory needs to eliminate this small fraction of unbound drug efficiently.
The most important observation from this study is that no linagliptin dose reduction seems to be necessary in subjects with hepatic impairment, despite the fact that 85% of this DPP-4 inhibitor is excreted non-renally. Plasma DPP-4 inhibition after single (severe hepatic impairment) and multiple (mild and moderate hepatic impairment) dosing was >80%, the level considered to be required for adequate glycaemic control in T2DM [21]. This may be because the in vivo efficacy of linagliptin is determined by the drug concentration bound to the therapeutic target tissues, and not by the unbound plasma fraction. From a treatment perspective, these results provide support that linagliptin has a reassuring safety profile and that the dosing recommendations should be the same, independent of the presence of any degree of hepatic impairment.
The protein binding of [3H]-linagliptin observed in this study was comparable with previous reports in rat and human plasma [9], [22]. Thus, the plasma protein binding of [3H]-linagliptin did not seem to be altered in patients with different degrees of hepatic impairment.
Linagliptin 5 mg was well tolerated and all adverse events were of mild to moderate intensity. This is consistent with recently completed pivotal trials in patients with T2DM receiving up to 24 weeks of linagliptin treatment [23], [24]. In one subject with severe hepatic impairment, a prolonged QTc interval was observed. This is a well known effect in hepatic impairment, which has previously been observed in patients in Child-Pugh categories from A to C [25]. Thus, it was not considered related to linagliptin treatment, particularly since linagliptin doses up to 20 times the therapeutic dose have previously been reported not to influence the QTc interval in a thorough QT study [26].
In conclusion, linagliptin exposure after single and multiple dosing in patients with mild, moderate or severe hepatic impairment was comparable with healthy subjects. These results indicate that no dose adjustment is required in patients with any degree of hepatic impairment and that linagliptin is a suitable treatment alternative in T2DM, not only in patients with renal impairment, but also in those with hepatic impairment.
Acknowledgments
We would like to thank the subjects and staff who participated in this study and Dr Vincent Duval (an employee of Boehringer Ingelheim at the time of planning and reporting the study) for his valuable contribution to the population pharmacokinetic analysis. Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany sponsored the study. Medical writing and editorial assistance was provided by Mark Greener and Gail Busza of PHASE II International during the preparation of this paper and was supported by Boehringer Ingelheim. A medical accuracy review of this manuscript was undertaken by Fredrik Lönnqvist, M.D., Ph.D., of Translational Medicine Bridges B.V., Amsterdam, the Netherlands, who received funding from Boehringer Ingelheim for this activity. This review did not alter either the content or the authors' interpretation of the data presented in the final draft of the manuscript. The authors were fully responsible for all content and editorial decisions, and were involved at all stages of manuscript development.
Competing Interests
UG-M, PR, SR and H-JW are employees of Boehringer Ingelheim. LW was a freelance medical writer under contract with Boehringer Ingelheim. AR was an employee of Boehringer Ingelheim at the time of planning and reporting the study. RC was the Principal Investigator at IFE Human Pharmacology SRL, Timisoara, Romania, where the study, sponsored by Boehringer Ingelheim, was conducted.
Supporting Information
Table S1
Non-compartmental pharmacokinetic parameters of CD1790 after single oral doses of 5 mg linagliptin in patients with different degrees of hepatic impairment compared with healthy subjects
Table S2
Steady-state non-compartmental pharmacokinetic parameters of CD1790 after multiple oral doses of 5 mg linagliptin in patients with mild or moderate hepatic impairment compared with healthy subjects
REFERENCES
- 1.Silink M. International Diabetes Federation Diabetes Atlas. Foreword [online] fifth edition. 2009. Available at http://www.idf.org/diabetesatlas/5e/foreword (last accessed 10 February 2012)
- 2.Cusi K. Nonalcoholic fatty liver disease in type 2 diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 2009;16:141–9. doi: 10.1097/MED.0b013e3283293015. [DOI] [PubMed] [Google Scholar]
- 3.Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl. 3):S206–14. doi: 10.1016/S1590-8658(10)60507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Compean D, Jaquez-Quintana JO, Gonzalez-Gonzalez JA, Maldonado-Garza H. Liver cirrhosis and diabetes: risk factors, pathophysiology, clinical implications and management. World J Gastroenterol. 2009;15:280–8. doi: 10.3748/wjg.15.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas L, Eckhardt M, Langkopf E, Tadayyon M, Himmelsbach F, Mark M. R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008;325:175–82. doi: 10.1124/jpet.107.135723. [DOI] [PubMed] [Google Scholar]
- 6.Scheen AJ. Pharmacokinetics of dipeptidylpeptidase-4 inhibitors. Diabetes Obes Metab. 2010;12:648–58. doi: 10.1111/j.1463-1326.2010.01212.x. [DOI] [PubMed] [Google Scholar]
- 7.Retlich S, Duval V, Ring A, Staab A, Hüttner S, Jungnik A, Jaehde U, Dugi KA, Graefe-Mody U. Pharmacokinetics and pharmacodynamics of single rising intravenous doses (0.5 mg-10 mg) and determination of absolute bioavailability of the dipeptidyl peptidase-4 inhibitor linagliptin (BI 1356) in healthy male subjects. Clin Pharmacokinet. 2010;49:829–40. doi: 10.2165/11536620-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs H, Tillement J-P, Urien S, Greischel A, Roth W. Concentration-dependent plasma protein binding of the novel dipeptidyl peptidase 4 inhibitor BI 1356 due to saturable binding to its target in plasma of mice, rats and humans. J Pharm Pharmacol. 2009;61:55–62. doi: 10.1211/jpp/61.01.0008. [DOI] [PubMed] [Google Scholar]
- 9.Heise T, Graefe-Mody EU, Hüttner S, Ring A, Trommeshauser D, Dugi KA. Pharmacokinetics, pharmacodynamics and tolerability of multiple oral doses of linagliptin, a dipeptidyl peptidase-4 inhibitor in male type 2 diabetes patients. Diabetes Obes Metab. 2009;11:786–94. doi: 10.1111/j.1463-1326.2009.01046.x. [DOI] [PubMed] [Google Scholar]
- 10.Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. Januvia® (sitagliptin) tablets. Prescribing information. January 2011. Available at http://www.merck.com/product/usa/pi_circulars/j/januvia/januvia_pi.pdf (last accessed 10 February 2012)
- 11.Neumiller JJ. Differential chemistry (structure), mechanism of action, and pharmacology of GLP-1 receptor agonists and DPP-4 inhibitors. J Am Pharm Assoc. 2009;49(Suppl. 1):S16–29. doi: 10.1331/JAPhA.2009.09078. [DOI] [PubMed] [Google Scholar]
- 12.Novartis Pharmaceuticals Corporation. Galvus (vildagliptin) 50 mg tablets: Summary of product characteristics. Electronic Medicines Compendium, 28 February 2011. Available at http://www.medicines.org.uk/EMC/medicine/20734/SPC/Galvus+50+mg+Tablets/ (last accessed 10 February 2012)
- 13.Bristol-Myers Squibb Company. ONGLYZA™ (saxagliptin) tablets. Prescribing information. February 2011. Available at http://packageinserts.bms.com/pi/pi_onglyza.pdf (last accessed 10 February 2012)
- 14.Blech S, Ludwig-Schwellinger E, Gräfe-Mody EU, Withopf B, Wagner K. The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans. Drug Metab Dispos. 2010;38:667–78. doi: 10.1124/dmd.109.031476. [DOI] [PubMed] [Google Scholar]
- 15.Boehringer Ingelheim Pharmaceuticals Inc. Tradjenta (linagliptin) tablets. Prescribing information. July 2011. Available at http://bidocs.boehringer-ingelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing+Information/PIs/Tradjenta/Tradjenta.pdf (last accessed 10 February 2012)
- 16.Food and Drug Administration. Guidance for Industry: Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) May 2003. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072123.pdf (last accessed 10 February 2012)
- 17.Pugh RNH, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 18.Farkas D, Greenblatt DJ. Influence of fruit juices on drug disposition: discrepancies between in vitro and clinical studies. Expert Opin Drug Metab Toxicol. 2008;4:381–93. doi: 10.1517/17425255.4.4.381. [DOI] [PubMed] [Google Scholar]
- 19.Retlich S, Duval V, Graefe-Mody U, Jaehde U, Staab A. Impact of target-mediated drug disposition on linagliptin pharmacokinetics and DPP-4 inhibition in type 2 diabetic patients. J Clin Pharmacol. 2010;50:873–85. doi: 10.1177/0091270009356444. [DOI] [PubMed] [Google Scholar]
- 20.Ikemura K, Iwamoto T, Okuda M. Altered functions and expressions of drug transporters in liver, kidney and intestine in disorders of local and remote organs: possible role of oxidative stress in the pathogenesis. Expert Opin Drug Metab Toxicol. 2009;5:907–20. doi: 10.1517/17425250903008525. [DOI] [PubMed] [Google Scholar]
- 21.Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, Snyder K, Hilliard D, Tanen M, Tanaka W, Wang AQ, Zeng W, Musson D, Winchell G, Davies MJ, Ramael S, Gottesdiener KM, Wagner JA. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther. 2005;78:675–88. doi: 10.1016/j.clpt.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs H, Binder R, Greischel A. Tissue distribution of the novel DPP-4 inhibitor BI 1356 is dominated by saturable binding to its target in rats. Biopharm Drug Dispos. 2009;30:229–40. doi: 10.1002/bdd.662. [DOI] [PubMed] [Google Scholar]
- 23.Del Prato S, Barnett AH, Huisman H, Neubacher D, Woerle H-J, Dugi KA. Effect of linagliptin monotherapy on glycaemic control and markers of β-cell function in patients with inadequately controlled type 2 diabetes: a randomized controlled trial. Diabetes Obes Metab. 2011;13:258–67. doi: 10.1111/j.1463-1326.2010.01350.x. [DOI] [PubMed] [Google Scholar]
- 24.Taskinen M-R, Rosenstock J, Tamminen I, Kubiak R, Patel S, Dugi KA, Woerle H-J. Safety and efficacy of linagliptin as add-on therapy to metformin in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled study. Diabetes Obes Metab. 2011;13:65–74. doi: 10.1111/j.1463-1326.2010.01326.x. [DOI] [PubMed] [Google Scholar]
- 25.Genovesi S, Prata Pizzala DM, Pozzi M, Ratti L, Milanese M, Pieruzzi F, Vincenti A, Stella A, Mancia G, Stramba-Badiale M. QT interval prolongation and decreased heart rate variability in cirrhotic patients: relevance of hepatic venous pressure gradient and serum calcium. Clin Sci. 2009;116:851–9. doi: 10.1042/CS20080325. [DOI] [PubMed] [Google Scholar]
- 26.Ring A, Port A, Graefe-Mody EU, Revollo I, Iovino M, Dugi KA. The DPP-4 inhibitor linagliptin does not prolong the QT interval at therapeutic and supratherapeutic doses. Br J Clin Pharmacol. 2011;72:39–50. doi: 10.1111/j.1365-2125.2011.03931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.