

Abstract
AIMS
The aim of this analysis was to use a population approach to facilitate the understanding of the pharmacokinetics and pharmacodynamics of rivaroxaban in patients with acute coronary syndrome (ACS) and to evaluate the influence of patient covariates on the exposure of rivaroxaban in patients with ACS.
METHODS
A population pharmacokinetic model was developed using pharmacokinetic samples from 2290 patients in Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Standard Therapy in Subjects with Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 46. The relationship between pharmacokinetics and the primary pharmacodynamic end point, prothrombin time, was evaluated.
RESULTS
The pharmacokinetics of rivaroxaban in patients with ACS was adequately described by an oral one-compartment model. The estimated absorption rate, apparent clearance and volume of distribution were 1.24 h−1 (interindividual variability, 139%), 6.48 l h−1 (31%) and 57.9 l (10%), respectively. Simulations indicate that the influences of renal function, age and bodyweight on exposure in ACS patients are consistent with the findings in previous Phase 1 studies. Rivaroxaban plasma concentrations exhibit a close-to-linear relationship with prothrombin time in the ACS population, with little interindividual variability. The estimated pharmacokinetic and pharmacodynamic parameters for the ACS patients were comparable to those for venous thromboembolism prevention, deep vein thrombosis and atrial fibrillation patients.
CONCLUSIONS
The similarity in pharmacokinetics/pharmacodynamics of rivaroxaban among different patient populations and the low interindividual variability in the exposure–prothrombin time relationship indicate that the anticoagulant effect of rivaroxaban is highly predictable and consistent across all the patient populations studied.
Keywords: acute coronary syndrome, pharmacodynamics, population pharmacokinetics, prothrombin time, rivaroxaban
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Population pharmacokinetics and pharmacodynamics of rivaroxaban have been characterized in healthy subjects and in patients with total venous thromboembolism, deep vein thrombosis or atrial fibrillation.
WHAT THIS STUDY ADDS
This article is the first description of the population pharmacokinetics (PK) and pharmacodynamics (PD) of rivaroxaban in patients with acute coronary syndrome (ACS). It is the largest population pharmacokinetic and pharmacodynamic study on rivaroxaban conducted to date (n = 2290). The PK and PK–PD relationship of rivaroxaban in patients with ACS were similar to those in other patient populations. In addition, model-based simulations showed that the influence of renal function and age on the exposure to rivaroxaban in the ACS population were similar to the findings from Phase 1 special population studies. These findings suggest that rivaroxaban has highly predictable PK–PD and may provide a consistent anticoagulant effect across the studied patient populations, which allows an accurate prediction of the dose to control anticoagulation optimally.
Introduction
Rivaroxaban is an oral, direct inhibitor of Factor Xa. Rivaroxaban has previously demonstrated clinically meaningful, robust and statistically significant efficacy compared with enoxaparin for the prophylaxis of total venous thromboembolism (VTE) and major VTE in patients after elective hip replacement or elective knee replacement surgery with modestly increased rates of bleeding [1]–[4]. Rivaroxaban is now approved for the prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE) in patients undergoing knee or hip replacement surgery, in the USA, the European Union and many other countries and regions. In addition, rivaroxaban is under clinical development for the treatment of stroke prevention in nonvalvular atrial fibrillation (AF) [5], prevention of VTE in hospitalized medically ill patients [6], and treatment and secondary prevention of VTE [7]. The pharmacokinetic (PK) characteristics of rivaroxaban are characterized and summarized in the approved product label [8].
Previously, structural population PK and pharmacokinetic–pharmacodynamic (PK–PD) models were developed based on data-rich information from a multiple dose-escalation study in young healthy male subjects [9]. The models were further developed and applied in three VTE prevention trials using sparse sampling and prospectively cross-validated vs. an independent cohort of patients with intensively sampled data [10], [11]. Supportive evidence for the validity and robustness of the population PK and PK–PD models was provided by further analyses performed using sparse sampling data collected in patients with DVT [12] and in patients with AF [13].
Rivaroxaban is currently also under clinical development for the secondary prevention of major cardiovascular events in patients with acute coronary syndrome (ACS), a heterogeneous condition that includes ST-segment elevation myocardial infarction (STEMI), non-ST segment elevation myocardial infarction (NSTEMI) and unstable angina (UA). Inhibition of Factor Xa, because of its pivotal position in the coagulation cascade, has the potential to block the thrombotic process and, in so doing, to reduce the incidence of subsequent ACS events. In a recent study in patients with ACS Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Standard Therapy in Subjects with Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 46 (ATLAS ACS-TIMI 46; ClinicalTrials.gov number, NCT00402597), rivaroxaban was shown to reduce the main secondary efficacy end point of death, myocardial infarction or stroke compared with placebo [14]. The knowledge obtained from population PK and PD modelling and simulation can facilitate drug development, and may be used to optimize patient response within the therapeutic window. Although the PK and PD of rivaroxaban have been investigated in other patient populations, such knowledge and the influence of the disease and other subject demographics and pathophysiological factors on PK and PD remain unknown for patients with ACS. In addition, comparison of PK of rivaroxaban and its pharmacological effects for patients with ACS with those for other patient populations may provide information to facilitate the accurate prediction of dose and thereby provide optimal control of anticoagulation across a wide range of patient populations. The objectives of the present population analysis were as follows: (i) to describe the PK and PK–PD relationship of rivaroxaban using nonlinear mixed-effects modelling based on sparse and rich sampling data originating from the study in patients with ACS; (ii) to characterize the inter- and intra-individual variability in the PK and PK–PD model parameters of rivaroxaban in patients with ACS; (iii) to evaluate the influence of patient covariates on the PK and PK–PD of rivaroxaban in patients with ACS; and (iv) to compare the population PK and PK–PD models for rivaroxaban in the ACS patient population with those previously developed in other patient populations. Simulations were conducted to evaluate the magnitude of the covariate effects on drug exposure.
Patients and methods
Study design and treatment
The ATLAS ACS-TIMI 46 trial was a randomized, multicentre, double-blind, placebo-controlled study designed to evaluate the safety and efficacy of rivaroxaban in patients with recent ACS who received standard of care background acetyl salicylic acid (ASA) therapy without the intention to use thienopyridine therapy (Stratum 1, ASA only) or with the intention to use thienopyridine therapy (Stratum 2, ASA plus a thienopyridine). In Stratum 1, patients were randomized to the following seven treatment groups: oral rivaroxaban at 2.5, 5 or 10 mg twice daily [5, 10 or 20 mg total daily dose (TDD)], oral rivaroxaban at 5, 10 or 20 mg once daily in the evening (with a placebo dose in the morning), or placebo. In Stratum 2, patients were randomized to the following nine treatment groups: oral rivaroxaban at 2.5, 5, 7.5 or 10 mg twice daily (5, 10, 15 or 20 mg TDD), oral rivaroxaban at 5, 10, 15 or 20 mg once daily in the evening (with a placebo dose in the morning), or placebo. The planned duration of the double-blind treatment period was 180 days. Details regarding the design of the clinical study have previously been reported [14].
Blood sampling
Sparse PK and PD samples were taken from all randomized patients predose and 1–3, 3–6 and 8–24 h after dosing on day 1, predose on day 30 and predose and 3 ± 1 h after dosing on day 180. In addition, intensive PK samples were obtained from between 4 and 19 patients enrolled into each of the rivaroxaban dose groups predose and 1, 2, 3, 4, 6, 9 and 12 h after dosing on day 30.
Bioanalysis
Plasma concentrations of rivaroxaban were measured by using a validated and selective liquid chromatographic assay coupled to tandem mass spectrometric detection with a lower limit of quantification of 0.500 ng ml−1. Concentration data below the lower limit of quantification were flagged in the data set and not included in the analysis. The prothrombin time (PT) was measured using freeze-dried rabbit brain thromboplastin (STA Neoplastin C1 Plus®; Diagnostica Stago, Parsippany, NJ, USA). The absolute results are reported in seconds.
Population pharmacokinetic modelling
The structural population PK model was developed based upon a previous population PK analysis performed in patients with DVT, which was an oral one-compartment model, parameterized in terms of apparent oral clearance (CL/F), apparent volume of distribution (V/F), and a first-order absorption rate constant (KA) [12]. Nonlinear mixed-effects modelling of the sparse and rich data was conducted using NONMEM® VI level 1.1 (ICON, Ellicott City, MD, USA) [15], [16]. Log-transformed rivaroxaban plasma concentrations were fitted using the first-order conditional estimation method without interaction. Interindividual (IIV) and interoccasion (IOV) variability for pharmacokinetic parameters were evaluated using an exponential model [17]. For estimating IOV, occasions were defined by visits: day 1, day 30 and day 180. The magnitude of residual variability in the plasma concentrations was modelled using an additive error model in the log domain. The IIV and IOV were explored for each of the PK parameters in the model during model refinement.
Predefined covariates that are biologically meaningful and have been identified from previous population analyses were included to evaluate their influence on the PK of rivaroxaban in the ACS patients. As approximately one-third of rivaroxaban is excreted unchanged by the kidneys [18], [19], renal function, which tends to decrease in elderly patients compared with young adults, is expected to influence the PK of rivaroxaban [20]. Both age and renal function were found to be significant factors of rivaroxaban PK in healthy subjects, in patients with DVT and AF, and in patients receiving rivaroxaban for VTE prevention following major orthopaedic surgeries [9]–[13]. Significant effects of age and bodyweight on volume of distribution have also been reported in healthy subjects and the above-mentioned patient populations [9]–[13]. In addition, bioavailability of rivaroxaban was demonstrated to be dose dependent in both the DVT and VTE prevention patient populations [10]–[12]. Therefore, the covariates included in this PK model were as follows: age and renal function [assessed via serum creatinine (SCR)] on CL/F, and bodyweight [expressed via lean body mass (LBM)] and age on V/F. Dose dependence was estimated on relative bioavailability (F) using a 2.5 mg rivaroxaban dose as reference dose, where F equals 1.
Population PK/PD modelling
The PK–PD analysis was conducted in a subgroup of patients, from whom time-matched PK and PD samples were collected. Prothrombin time was the primary PD end point in the study. Matched samples imply that the PD blood samples to determine coagulation characteristics were taken at the same times as the PK blood samples were obtained. The resulting PK–PD data set for this analysis contained 1347 patients with 6644 observations for PT matched with corresponding PK samples. A linear intercept model with a declining exponent on plasma concentration (Cp), 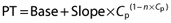 , was used to describe the relationship between PT and the drug exposure based on a previously developed model for DVT patients [12]. The effect of creatinine clearance was previously identified in the PK–PT model for patients with DVT [12], and was therefore included in the intercept (Base) and the exponent (n) terms.
, was used to describe the relationship between PT and the drug exposure based on a previously developed model for DVT patients [12]. The effect of creatinine clearance was previously identified in the PK–PT model for patients with DVT [12], and was therefore included in the intercept (Base) and the exponent (n) terms.
Model evaluation
The model improvement was evaluated based on goodness-of-fit criteria such as reduction in the objective function value, the agreement between the observed and predicted concentration values, and the reduction in patterns of conditional weighted residuals. To evaluate the predictive performance of the final model, population prediction corrected visual predictive checks were performed on the concentration–time data [21]. This method evaluates whether the majority (i.e. approximately 90%) of the observed concentrations fall within the 90% simulation interval of the individual PK profiles simulated using the final PK model. Plasma concentrations of rivaroxaban in the study population were simulated 500 times using the dose and covariate data from the patients that were used in the model development data set.
Monte Carlo simulation
To evaluate the clinical relevance of the covariates on model parameters and hence on exposure, simulations of steady-state rivaroxaban exposures [area under the curve (AUC), maximal plasma concentration (Cmax) and trough plasma concentration (Cmin)] were performed after multiple oral doses of 2.5 mg twice daily (i.e. a dose regimen used in the present ACS Phase 3 study) based on the individual parameter estimates. Steady-state AUC was calculated based on the dose and estimated individual CL/F. Steady-state Cmax was predicted as the maximal concentration during a dosing interval at steady state. Steady-state Cmin was predicted as the concentration at the end of the dose interval (C12). The simulated exposures were compared at different covariate levels.
Results
Patient characteristics and treatments
A summary of the patient characteristics and treatments is listed in Table 1. A total of 1784 (78%) male and 506 (22%) female patients were included in the PK data set. Of the 2290 patients in the PK analysis data set, 303 patients received a TDD of 5 mg rivaroxaban, 1037 patients received a TDD of 10 mg rivaroxaban, 353 patients received a TDD of 15 mg rivaroxaban, and 597 patients received a TDD of 20 mg rivaroxaban.
Table 1.
Demographic characteristics
| Characteristic | Value |
|---|---|
| Sex | Male (78%) |
| Female (22%) | |
| Race | White (95.8%) |
| Black (0.8%) | |
| Asian (1.7%) | |
| Others (1.7%) | |
| Age (years) | 57 (24–87)* |
| Bodyweight (kg) | 84 (36–181)* |
| Lean body mass (kg) | 60.7 (30.4–90.4)* |
| Serum creatinine (mg dl−1) | 0.95 (0.49–3.17)* |
| Creatinine clearance (ml min−1)† | 96.9 (22.4–298)* |
Values are expressed as medians (range).
Estimated with the [22] formula.
Pharmacokinetic model
The PK of rivaroxaban in patients with ACS was adequately described by an oral one-compartment model with first-order absorption and first-order elimination. Inclusion of IIV on KA and IOV on CL/F improved the model fit (P < 0.01). The parameter estimates for the final pharmacokinetic model are presented in Table 2. For a typical subject, the estimated values of CL/F, V/F and KA were 6.48 l h−1, 57.9 l and 1.24 h−1, respectively. The IIV of CL/F, V/F and KA were estimated at 31, 10 and 139% coefficient of variation (expressed as %CV), respectively. The estimated IOV on CL/F was 32%. An additive error model was used to describe the residual variability for the log-transformed data, and the variance was 0.35 (59%CV). The patient covariates included in the model were age and renal function (assessed via SCR) effects on CL/F, and bodyweight (expressed as LBM) and age effects on V/F. Similar covariate effects were also observed in the DVT and AF patient populations. As age increased by 1 year from the median age of 57 years, CL/F and V/F decreased 1.1 and 0.71%, respectively, whereas CL/F decreased 1.5% with 0.1 mg dl−1 increase in SCR. There was 0.83% increase in V/F per one kilogram increase in LBM. Since creatinine clearance (CRCL) is usually used to assess renal function, and consists of information regarding both SCR and age [22], CRCL was also tested as a covariate for CL/F, and was found to be statistically significant. However, the model that included age and SCR separately on CL/F reduced the objective function value by 48 points compared with the model with CRCL as the covariate. Bioavailability was found to be dependent on dose (P < 0.0001 based on the Wald test), and decreased 15% for doses ≤10 mg and 29% for doses above 10 mg, relative to a 2.5 mg dose. This significant dose dependence was confirmed by a likelihood ratio test, because the removal of dose as the covariate of bioavailability resulted in an 85 points increase in the objective function value compared with the final PK model. The estimated bioavailability relative to a 2.5 mg dose in the patients with ACS was comparable to that estimated in the VTE prevention patients following total knee replacement.
Table 2.
Population estimated pharmacokinetic parameters of rivaroxaban from the final pharmacokinetic model in acute coronary syndrome (ACS) treatment patients
| Parameter | Population mean (%SE) | Interindividual variability, %CV (%SE) |
|---|---|---|
| CL/F (l h−1) | 6.48 (2.21) | 31.3 (4.72) |
| V2/F (l) | 57.9 (1.16) | 10.0 (3.66) |
| KA (h−1) | 1.24 (3.28) | 139 (0.30) |
| F (dose ≤10 mg relative to 2.5 mg)* | 0.851 (8.91) | – |
| F (dose > 10 mg relative to 2.5 mg)* | 0.705 (11.2) | – |
| Covariates on CL/F† | ||
| Age (year−1) | −0.0112 (8.82) | – |
| Serum creatinine [(mg dl−1)−1] | −0.151 (20.3) | – |
| Covariates on V/F‡ | ||
| Lean body mass (kg−1) | 0.00833 (13.1) | – |
| Age (year−1) | −0.00707 (16.3) | – |
| Interoccasion variability on CL | – | 32.4 (5.39) |
| Additive error | 0.352 (1.09) | NA |
Abbreviations: %CV, coefficient of variation (%); KA, first-order absorption rate constant; LBM, lean body mass; NA, not applicable; SCR, serum creatinine; %SE, relative standard error (%).
Bioavailability (F) at 2.5 mg equals 1.
CL/F×[1 – 0.00112 × (Age – 57) – 0.151 × (SCR – 0.95)].
V/F×[1 + 0.00833 × (LBM – 60.7) – 0.00707 × (Age – 57)].
Model evaluation
The goodness-of-fit of the final model was tested by a graphic approach. Figure 1 indicates that population and individual predicted concentrations agreed well with observed concentrations. The conditional weighted residuals are randomly scattered across the range of population predictions and time, suggesting no bias or trends in the residual error model. The prediction corrected visual predictive checks stratified for dosing regimen are displayed in Figure 2. The visual predictive check was considered adequate because the majority of the observations lie within the 90% prediction interval and the extremes of the 90% prediction interval reflect the fifth and 95th percentiles of the observed data well. The fifth, 50th and 95th percentiles of the observations generally fall within the 90% prediction intervals of the simulated fifth, 50th and 95th percentiles. The visual predictive check plots show that the model predicted the overall concentration data in ACS treatment patients well.
Figure 1.
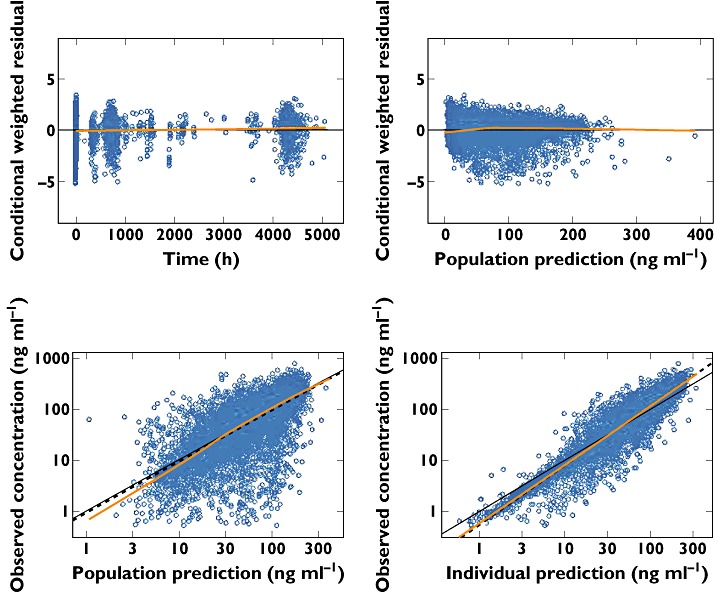
Diagnostic plots for the final phrmacokinetic (PK) model. The dashed line represents a locally weighted least squares. In the residual plots, the ordinate value of zero is presented (continuous horizontal line). In the plots of observed vs. population and individual predictions, the continuous line represents the line of identity
Figure 2.
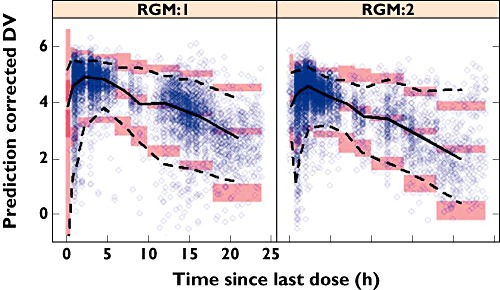
Visual predictive check plots for the final PK model, stratified for dosing regimen (RGM). Continuous black line is the median observed time course for rivaroxaban concentrations. The upper and lower dashed black lines represent the fifth and 95th percentiles of observed data, respectively. The semi-transparent red-shaded areas represent the simulation-based 90% confidence intervals for the fifth, 50th and 95th percentiles of the predicted data. The semi-transparent blue circles represent the observed concentrations. Abbreviations: DV, dependent variable (log-transformed concentration); RGM 1, once daily; RGM 2, twice daily
PK–prothrombin time (PT) model
Rivaroxaban concentrations correlated with PT in an almost linear fashion (Figure 3). Table 3 summarizes the parameter estimates obtained from the PK–PT structural model. The baseline PT in the study population was 14 s, and the slope of the correlation between PT and rivaroxaban plasma concentrations was 3.2 s (100 ng ml−1)−1. There was low overall IIV for the correlation between rivaroxaban plasma concentrations and PT, with IIV in the baseline of 9.32% and in the exponent on Cp of 6.61%. The effect of renal function (expressed as CRCL) on the PT model was statistically significant (P < 0.003), but the magnitude of its influence was small (<9% for the CRCL range in this study population). The visual predictive check plot of the PK–PT model (Figure 3) shows that the model predicted the PT–concentration data well in the ACS patients, because the majority of the observations lie within the 90% prediction interval.
Figure 3.
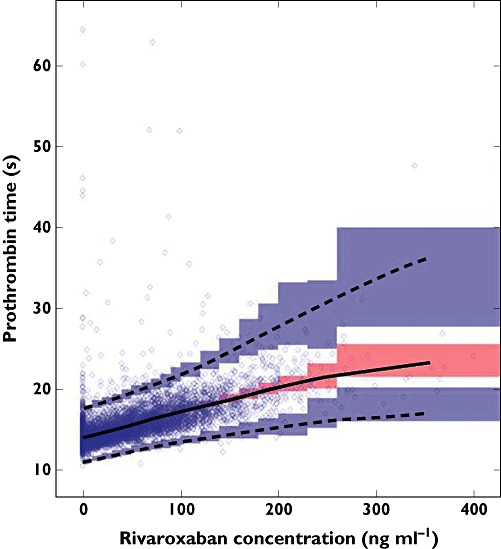
Visual predictive check plot for the pharmacokinetic–pharmacodynamic (PK–PD) model. Concinuous black line is the median prediction for rivaroxaban concentrations. The upper and lower dashed black lines represent the fifth and 95th percentiles of predicted data, respectively. The semi-transparent red-shaded area represents the simulation-based 90% confidence intervals for the median predicted data, while the semi-transparent blue-shaded areas represent the simulation-based 90% confidence intervals for the fifth and 95th percentiles of the predicted data. The semi-transparent blue circles represent the observed concentrations
Table 3.
Final parameter estimates from population PK/PT model
| Parameter | Typical value(%SE) | Interindividual variability (%SE) |
|---|---|---|
| Baseline (s) | 13.9 (0.26) | 9.32 (19.4) |
| Slope (s (ng ml−1)−1) | 0.032 (1.44) | NA |
| Slope, describing decline of exponent on Cp (n) [(ng ml−1)−1] | 0.0000593 (23.3) | 6.61 (17.9) |
| Creatinine clearance on baseline (% (ml min−1)−1) | −0.030 (23.1) | NA |
| Creatinine clearance on decline of exponent (% ml min−1)−1) | 2.33 (35.8) | NA |
| CV residual error (%SE) | 7.6 (12.1) |
Abbreviations: Cp, plasma concentration; CV, coefficient of variation (%); NA, not assessed; %SE, relative standard error (%).
Discussion
Clinical relevance of PK covariates
A full covariate modelling approach was implemented in this analysis, and all the covariates were included into the base PK model at once [23], [24]. Therefore, statistical significance of the covariates was not tested based on the likelihood ratio test approach. However, statistically significant parameters/covariates can be readily identified through the Wald test, which is asymptotically equivalent to the likelihood ratio test [25]. Based on the Wald test, all the included covariates are statistically significant (P < 0.0001), consistent with the findings from the previous population analyses in other patient populations [9]–[13].
The relevance of the model covariates on drug exposures was assessed by simulations of individual steady-state drug exposures (e.g. Cmax, Cmin and AUC) following oral administration of a 2.5 mg twice daily dosing regimen based on the empirical Bayes estimates of the model parameters for the ACS treatment patients in the study (n = 2290). Table 4 compares the median, 5 and 95% percentiles of the simulated drug exposures for subpopulations stratified for renal function (i.e. CRCL), age and body mass (i.e. LBM) for the simulated 2.5 mg twice daily dosing regimen.
Table 4.
Evaluation of influence of covariates on simulated steady-state exposure for rivaroxaban following administration of a 2.5 mg dose (twice daily) in ACS treatment patients
| Renal function | ||||
|---|---|---|---|---|
| Parameter | CRCL < 50 ml min−1 | 50 ≤ CRCL < 80 ml min−1 | CRCL ≥ 80 ml min−1 | Ratio (CRCL < 50 ml min−1/CRCL ≥ 80 ml min−1) |
| AUC (ng h ml−1) | 542 (295–865) | 426 (221–653) | 361 (209–589) | 1.50 |
| Cmax (ng ml−1) | 63.3 (38.9–90.3) | 51 (31.1–72.1) | 44 (27.6–66.2) | 1.44 |
| Cmin (ng ml−1) | 27.9 (8.54–53.3) | 19.7 (6.15–38.2) | 16.4 (5.95–33.5) | 1.70 |
| Age | ||||
|---|---|---|---|---|
| Age < 50 years | 50 ≤ age ≤ 75 years | Age > 75 years | Ratio (age > 75 years/age < 50 years) | |
| AUC (ng h ml−1) | 328 (183–527) | 397 (226–654) | 469 (277–901) | 1.43 |
| Cmax (ng ml−1) | 39.9 (23.9–56.9) | 48 (30.3–72) | 51.3 (38.9–92.9) | 1.29 |
| Cmin (ng ml−1) | 14 (5.18–30.1) | 18.3 (6.52–38) | 22.6 (7.7–49.9) | 1.61 |
| Lean body mass | ||||
|---|---|---|---|---|
| LBM < 53 kg | 53 ≤ LBM < 66 kg | LBM ≥ 66 kg | Ratio (LBM < 53 kg/LBM ≥ 66 kg) | |
| AUC (ng h ml−1) | 394 (215–671) | 373 (210–630) | 365 (216–588) | 1.08 |
| Cmax (ng ml−1) | 50.4 (31.2–77.4) | 45.1 (27.7–68) | 43.4 (28.1–63.4) | 1.16 |
| Cmin (ng ml−1) | 16.8 (5.3–36.4) | 17 (6.17–37.4) | 17.5 (6.74–34.9) | 0.96 |
Values are given as medians (5th–95th percentiles).
Renal function was identified as a significant covariate on the PK of rivaroxaban in VTE prevention, DVT and AF populations [10]–[12]. Likewise, renal function was also identified as an influential covariate on the PK of rivaroxaban in the ACS population. As CRCL is commonly used to classify impairment of renal function, the comparison of exposures to rivaroxaban was made based on CRCL, although SCR was used as the covariate for renal function in the model. The simulated median AUC in ACS treatment patients with moderate renal impairment (i.e. 30 ml min−1≤ CRCL < 50 ml min−1) was 1.5 times that in patients with normal renal function (Table 4). This result is consistent with the findings based on the Phase 1 renal impairment study where, on average, moderately renally impaired subjects had a 1.5-fold higher AUC than subjects with normal renal function [26]. As the influence of renal function on PT was minimal (<9% for the CRCL range in this study population), the effect of CRCL on the PD of rivaroxaban is mainly through its influence on the PK of rivaroxaban.
The simulated exposure to rivaroxaban increased with age. The simulated AUC in the elderly (i.e. >75 years old) was about 43% higher than that in young patients (i.e. <50 years old), which resembles the Phase 1 age comparison study, where the average AUC in subjects older than 75 years (up to 83 years) was 41% higher than that in young subjects (18–43 years old) [8], [27]. The elderly–young difference can be attributed mainly to reduced renal clearance in the elderly subjects.
According to the current US Prescribing Information for patients with AF and patients for VTE prevention following major orthopaedic surgeries, rivaroxaban should be avoided if their CRCL is <15 and <30 ml min−1, respectively [8]. Additionally, for AF patients with a CRCL level between 15 and 50 ml min−1, the recommended dose is reduced to 15 mg once daily, compared with 20 mg once daily for patients with CRCL greater than 50 ml min−1[8]. In the current Phase 2 study, the probability of treatment-emergent bleeding events was not statistically different between renally impaired ACS patients and the patients with normal renal function. This was confirmed by a post hoc analysis of a recent Phase 3 study (ATLAS ACS 2-TIMI 51), where no significant increase in the hazard ratio of Thrombolysis in Myocardial Infarction major bleeding events (not associated with coronary artery bypass grafting) was observed in ACS patients with renal impairment, and similar rates of the primary efficacy end point were observed among the subgroups of patients with different levels of renal function [28]. Therefore, no dose adjustment according to renal function or age was warranted. However, signs or symptoms of blood loss should be observed closely and evaluated promptly in patients with moderate renal impairment (CRCL 30 to <50 ml min−1).
The magnitude of the effect of lean body mass on the steady-state rivaroxaban exposure parameters (AUC, Cmax and Cmin) was generally small (Table 4). The difference in AUC and Cmin for patients with LBM < 53 kg (the 25th percentile of the ACS patients) and patients with LBM ≥ 66 kg (the 75th percentile) was no more than 10%, while the Cmax increased by 16% in patients with LBM < 53 kg relative to patients with LBM ≥ 66 kg (Table 4).
Comparison of parameter estimates across study populations
Model-estimated population mean values of the PK parameters for ACS patients were generally similar to those of VTE prevention patients [10], [11], DVT treatment patients [12] and patients with AF [13] (Table 5). Population mean values for KA in patients with ACS (1.24 h−1) were in close agreement with those obtained in patients with AF, DVT and the VTE prevention patients following knee and hip surgery (1.16, 1.23, 1.20 and 1.81 h−1, respectively). It should be noted that the definition of the typical subject (i.e. median of covariate values) in each study population was slightly different (Table 5). Therefore, the comparison of CL/F and V/F across studies was done after correcting the difference in median covariate values in different patient populations (Figures 4 and 5). Figure 4 shows that the typical values of CL/F and V/F in patients with ACS at a dose of 2.5 mg were comparable to those in VTE prevention patients following knee and hip surgery with a 2.5 mg dosing regimen. The estimated CL/F at 2.5 mg in patients following total knee and hip replacement (VTE prevention) were about 7 and 20%, respectively, lower than those estimated in the patients with ACS, whereas the estimated V/F at 2.5 mg was virtually identical between the ACS patients and the patients after knee and hip replacement (VTE prevention; Figure 4).
Table 5.
Comparison of model-based PK parameters of rivaroxaban across study populations [population mean (% SE)]*
| Parameter | ACS | VTE (knee) | VTE (hip) | AF | DVT |
|---|---|---|---|---|---|
| KA (h−1) | 1.24 (3.28) | 1.20 (8.9)§ | 1.81 (8.3)§ | 1.16 (14.1) | 1.23 (5.0) |
| CL/F (l h−1) | 6.48 (2.21) | 6.13 (4.5)¶ | 7.3 (4.0)¶ | 6.1 (3.9) | 5.67 (3.7) |
| V/F (l) | 57.9 (1.16) | 55.7 (5.8) | 49.1 (4.3) | 79.7 (6.1) | 54.4 (3.8) |
| Age effect on CL/F (year−1) | −0.0112 (8.82) | NA* | −0.015 (12.7) | −0.01050 (26.3) | −0.00692 (14.6) |
| SCR effect on CL/F[(mg dl−1)−1] | −0.151 (20.3) | NA* | −0.21 (21.6) | −0.19400 (34.0) | −0.269 (18.2) |
| LBM effect on V/F (kg−1) | 0.00833 (13.1) | NA* | 0.018 (13.5) | 0.01180 (32.4) | 0.0082 (17.8) |
| Age effect on V/F (year−1) | −0.00707 (16.3) | NA* | NA | −0.00133 (187.2) | −0.00486 (20.8) |
| F† | 0.85 (8.91) | 0.847 (4.4) | 0.740 (4.5) | NA†† | 0.79 (4.20) |
| F‡ | 0.71 (11.2) | 0.609 (4.6) | 0.533 (5.0) | NA†† | 0.63 (4.00) |
Definitions of typical subject: ACS (age = 57 years; SCR = 0.95 mg dl−1; LBM = 61 kg); VTE knee (CRCL = 104 ml min−1; BSA = 1.95 m2); VTE hip (age = 65 years; SCR = 0.78 mg dl−1; LBM = 51 kg); AF (age = 65 years; SCR = 1.05 mg dl−1; LBM = 57 kg); DVT (age = 61 years; SCR = 0.94 mg dl−1; LBM = 56 kg).
For ACS, relative bioavailability of 5, 7.5 and 10 compared with 2.5 mg; for VTE (knee) and VTE (kip), relative bioavailability (F) of 5 and 10 compared with 2.5 mg; for DVT, relative bioavailability of 20 compared with 10 mg.
For ACS, relative bioavailability of 15 and 20 compared with 2.5 mg; for VTE (knee) and VTE (hip), relative bioavailability of 20 and 30 compared with 2.5 mg; and for DVT, relative bioavailability of 30 and 40 compared with 10 mg.
Estimate for the fast absorption population based on a mixture model of KA for the VTE (hip and knee) populations only.
CL/F estimate for study day > 3.
Different covariates were used in this model (creatinine clearance on CL/F and body surface area on V/F).
Only 20 mg studied. Abbreviations: LBM, lean body mass; NA, not applicable; SCR, serum creatinine; %SE, relative standard error (%).
Figure 4.
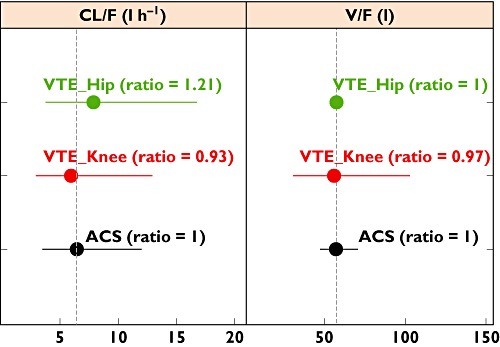
Comparison of estimated PK parameters for the acute coronary syndrome (ACS) population with those for a venous thromboembolism (VTE) prevention population at a 2.5 mg dose of rivaroxaban. The confidence intervals (horizontal bars) were calculated based on interindividual variability (IIV) with a log-normal distribution assumption. Ratio of population mean values of the PK parameters were calculated using the parameter estimates for the ACS population as the reference. The IIV of apparent volume of distribution (V/F) was not included in the model in VTE prevention patients following hip surgery
As the 2.5 mg dose was not used in the DVT and AF populations, and a 20 mg dose was used in the AF study, the comparison of the estimated CL/F and V/F across the ACS, AF, DVT and VTE prevention study populations was made at a 20 mg dose of rivaroxaban (Figure 5). The F-values in the ACS, VTE prevention and DVT populations at the 20 mg dose refer to those in Table 5. The differences in V/F at a 20 mg dose between patients with ACS and the other study populations were generally smaller than 15%. The estimated CL/F at a 20 mg dose for the ACS population was generally similar to that for DVT treatment and VTE prevention patients following knee surgery, because the difference was in the order of or less than 20%. The estimated CL/F at 20 mg in the AF population was approximately 30% lower than that in the ACS population. Greater discrepancy was observed for the CL/F and V/F at 20 mg between the ACS patients and the VTE prevention patients after hip surgery, where the differences were about 60 and 30%, respectively. As the differences in the PK parameters were much smaller at 2.5 mg (i.e. ∼20% difference in CL/F and virtually identical V/F; Figure 4), the larger discrepancy at 20 mg may be a result of overcorrection of relative bioavailability, which was estimated for the combined dose strengths of 20 and 30 mg for the VTE prevention patients undergoing hip replacement, because the relative bioavailability is expected to be lower at higher doses. Overall, the differences in population mean values across study populations are well within the interindividual variability, because the confidence intervals based on the estimated IIV for different populations largely overlapped (Figure 5).
Figure 5.
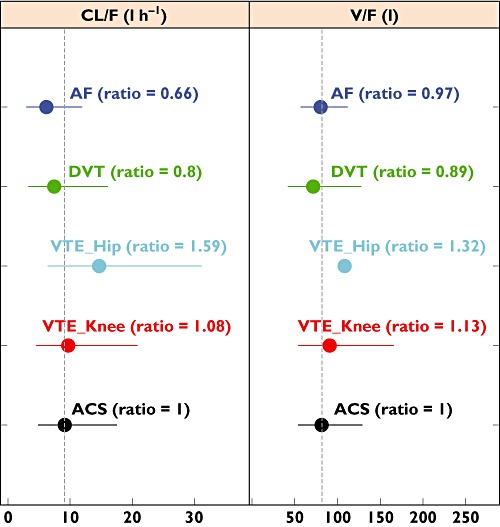
Comparison of estimated PK parameters across ACS, VTE prevention, deep vein thrombosis (DVT) and atrial fibrillation (AF) populations at a 20 mg dose. The confidence intervals (horizontal bars) were calculated based on interindividual variability with a log-normal distribution assumption. Ratio of population mean values of the PK parameters were calculated using the parameter estimates for the ACS population as the reference. The IIV of V/F was not included in the model in VTE prevention patients following hip surgery
The parameter estimates of the PK–PT model for the present ACS population are consistent with those reported for the DVT [12] and AF populations [13] (Table 6). Although the CRCL value for the typical subject in the ACS patient population was somewhat higher than those in the AF and DVT populations (97 vs. 76 and 87 ml min−1, respectively), its influence on the parameters of the typical subjects was small (<4%). Therefore, no corrections for the difference in CRCL values for the typical subjects in these three patient populations were made. The estimated mean slope, reflecting the sensitivity of this coagulation marker towards increases in rivaroxaban drug exposure, is similar to those observed in DVT patients ([3.6 s (100 ng ml−1)−1 of rivaroxaban plasma concentration] and AF patients [4.3 s (100 ng ml−1)−1 of rivaroxaban plasma concentration]. The estimated mean slopes are also similar to what was observed in the VTE prevention patients [3.2 s (100 ng ml−1)−1 in the hip study and 4.2 s (100 ng ml−1)−1 in the knee study][10], [11]. The close-to-linear relationship between rivaroxaban drug exposure and PT response also holds true for this ACS patient population, because the slope (n) on the exponent was very small [0.00006 (ng ml−1)−1]. Similar influences of CRCL on baseline and the slope of the exponent were observed for the ACS and the DVT populations and the ACS and the AF populations, respectively.
Table 6.
Comparison of PT model parameter estimates across patient populations*
| Parameter | AF population† typical values(%SE) | DVT population† typical values(%SE) | ACS population typical values(%SE) |
|---|---|---|---|
| Baseline (s) | 11.4 (2.0) | 12.5 (0.7) | 13.9 (0.26) |
| Slope (s (ng ml−1)−1) | 0.0426 (6.6) | 0.036 (2.8) | 0.032 (1.44) |
| Slope (n) [(ng ml−1)−1] | 0.0000551 (55.0) | 0.000096 (7.0) | 0.0000593 (23.3) |
| Creatinine clearance on baseline (% (ml min−1)−1) | 0.0192 (169.3) | −0.04 (23.2) | −0.030 (23.1) |
| Creatinine clearance on (n) (% (ml min−1)−1) | 1.74 (100.6) | 0.46 (24.4) | 2.33 (35.8) |
| CV residual error [%] | 12.85 (18.4) | 10.3 (8.6) | 7.6 (12.1) |
Clinical interpretation of the findings
Patients with a history of ACS are at higher risk for recurrent infarction, stroke and death [29]. A study found that subjects treated with the anticoagulant warfarin and ASA had a decreased rate of myocardial infarction, ischaemic stroke and revascularization compared with those treated with ASA alone [30]. However, warfarin dosing is complicated by dramatic interindividual variability in responsiveness of different patients to warfarin. While the polymorphism of CYP2C9 is associated with the highly variable metabolism of warfarin [31], [32], genetic variations of the vitamin K epoxide reductase complex, the VKORC1 gene, have been found to be related to individual sensitivity to the anticoagulant response to warfarin therapy [33], [34]. In addition, other confounding factors, such as individual dietary habits, may further increase the inter- and/or intra-individual variability in pharmacological effects of warfarin, because genetic variants accounted for about only a third of the interindividual variability [33]. Moreover, based on the data from 48 healthy subjects, the variability in the warfarin concentrations contributed at most only 40% of the observed variability in the pharmacological anticoagulation response [35]. Bleeding events were found to be associated with the intensity of anticoagulation, and prothrombin time ratio was demonstrated to be the most predictive risk factor [36]. As a result of its large unexplained variability, the PK and PD (anticoagulation effect and its associated bleeding risk) of warfarin are not predictable. Therefore, the use of the same fixed dose of warfarin for all patients is not feasible. The anticoagulant effect of warfarin is usually measured by a standardized prothrombin time, and the dose is adjusted accordingly [37]. The effective daily dose of warfarin typically ranges from 0·5 to 60 mg [38], [39]. Nevertheless, due to the widespread variation in the intensity of anticoagulation effect associated with warfarin, although patients taking warfarin routinely monitored their international normalized ratio readings to guide dose adjustments, they spend about a third of the treatment time outside the standard target therapeutic international normalized ratio range (2.0–3.0) [40].
Rivaroxaban exhibits only low-to-moderate interindividual variability in V/F (∼10%) and CL/F (∼30%) in patients with ACS. Furthermore, the prolongation in PT correlated in an almost linear fashion with plasma rivaroxaban concentration in the patient population, with little interindividual variability (<10%). This is consistent with previous Phase 1 studies, where the observed rivaroxaban concentrations explained approximately 90% of the variability in PT prolongation [9], [41]. Consequently, compared with warfarin, rivaroxaban is a more predictable and convenient oral anticoagulant, and can be prescribed in fixed doses without the need for routine coagulation monitoring. The similarity in the PK and PK–PD among different patient populations (i.e. VTE, DVT, AF and ACS) suggests that rivaroxaban can provide a predictable and consistent anticoagulant effect across all the patient populations studied. Therefore, the high predictability of rivaroxaban PK–PD, along with the low interindividual variation may facilitate the accurate prediction of its dose and thereby provide optimal control of anticoagulation to improve clinical outcomes and to reduce unwanted adverse events without the need to adjust the dose.
Conclusion
The PK parameter estimates for ACS patients were comparable to those for VTE prevention patients, DVT treatment patients and AF patients. The demographic covariates identified to affect rivaroxaban pharmacokinetics (CL/F and V/F) were age, renal function (assessed via serum creatinine) and bodyweight (expressed as lean body mass). The covariate estimates were consistent with those observed in VTE, DVT and AF patients. Model-based simulations showed that the influence of renal function and age on the exposure to rivaroxaban in the ACS population were similar to the findings from Phase 1 special population studies. The effect of body size (i.e. lean body mass) on the drug exposure was minimal. Prothrombin time correlated in an almost linear fashion with rivaroxaban concentrations observed in the study, with little interindividual variability. These findings suggest that rivaroxaban has highly predictable PK–PD and may provide a consistent anticoagulant effect across the studied patient populations, which allows an accurate prediction of the dose to control anticoagulation optimally. This may improve clinical outcomes and reduce unwanted bleeding events without a need for dose adjustments.
Acknowledgments
The authors appreciate the helpful review and constructive comments from Dr Partha Nandy.
Competing Interests
M. Gibson reports receiving research grant support from Janssen R&D and Bayer Pharma AG, and lecture fees from Janssen R&D. The other authors are employees of Janssen R&D or Bayer Pharma AG. The analyses and studies described in this report were funded by Janssen R&D and Bayer Pharma AG. Steven Xu is an adjunct assistant professor in the School of Public Health at the University of Medicine and Dentistry of New Jersey.
REFERENCES
- 1.Lassen MR, Ageno W, Borris LC, Lieberman JR, Rosencher N, Bandel TJ, Misselwitz F, Turpie AG. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med. 2008;358:2776–86. doi: 10.1056/NEJMoa076016. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson BI, Borris LC, Friedman RJ, Haas S, Huisman MV, Kakkar AK, Bandel TJ, Beckmann H, Muehlhofer E, Misselwitz F, Geerts W. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med. 2008;358:2765–75. doi: 10.1056/NEJMoa0800374. [DOI] [PubMed] [Google Scholar]
- 3.Kakkar AK, Brenner B, Dahl OE, Eriksson BI, Mouret P, Muntz J, Soglian AG, Pap AF, Misselwitz F, Haas S. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet. 2008;372:31–9. doi: 10.1016/S0140-6736(08)60880-6. [DOI] [PubMed] [Google Scholar]
- 4.Turpie AG, Lassen MR, Davidson BL, Bauer KA, Gent M, Kwong LM, Cushner FD, Lotke PA, Berkowitz SD, Bandel TJ, Benson A, Misselwitz F, Fisher WD. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial. Lancet. 2009;373:1673–80. doi: 10.1016/S0140-6736(09)60734-0. [DOI] [PubMed] [Google Scholar]
- 5.Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, Breithardt G, Halperin JL, Hankey GJ, Piccini JP, Becker RC, Nessel CC, Paolini JF, Berkowitz SD, Fox KA, Califf RM. Rivaroxaban versus Warfarin in Nonvalvular Atrial Fibrillation. N Engl J Med. 2011;365:883–91. doi: 10.1056/NEJMoa1009638. [DOI] [PubMed] [Google Scholar]
- 6.Cohen A, Spiro T, Buller H, Haskell L, Hu D, Hull R, Mebazaa A, Merli G, Schellong S, Spyropoulos A, Tapson V. Rivaroxaban Compared with Enoxaparin for the Prevention of Venous Thromboembolism in Acutely Ill Medical Patients. New Orleans, LA: American College of Cardiology; 2011. [Google Scholar]
- 7.Bauersachs R, Berkowitz SD, Brenner B, Buller HR, Decousus H, Gallus AS, Lensing AW, Misselwitz F, Prins MH, Raskob GE, Segers A, Verhamme P, Wells P, Agnelli G, Bounameaux H, Cohen A, Davidson BL, Piovella F, Schellong S. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499–510. doi: 10.1056/NEJMoa1007903. [DOI] [PubMed] [Google Scholar]
- 8.Xarelto (R) Product Label. 2011. Available at: http://www.xareltohcp.com/sites/default/files/pdf/xarelto_0.pdf#zoom=100 (last accessed 9 May 2011)
- 9.Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct factor xa inhibitor – in healthy subjects. Int J Clin Pharmacol Ther. 2007;45:335–44. doi: 10.5414/cpp45335. [DOI] [PubMed] [Google Scholar]
- 10.Mueck W, Borris LC, Dahl OE, Haas S, Huisman MV, Kakkar AK, Kalebo P, Muelhofer E, Misselwitz F, Eriksson BI. Population pharmacokinetics and pharmacodynamics of once- and twice-daily rivaroxaban for the prevention of venous thromboembolism in patients undergoing total hip replacement. Thromb Haemost. 2008;100:453–61. [PubMed] [Google Scholar]
- 11.Mueck W, Eriksson BI, Bauer KA, Borris L, Dahl OE, Fisher WD, Gent M, Haas S, Huisman MV, Kakkar AK, Kalebo P, Kwong LM, Misselwitz F, Turpie AG. Population pharmacokinetics and pharmacodynamics of rivaroxaban – an oral, direct factor Xa inhibitor – in patients undergoing major orthopaedic surgery. Clin Pharmacokinet. 2008;47:203–16. doi: 10.2165/00003088-200847030-00006. [DOI] [PubMed] [Google Scholar]
- 12.Mueck W, Lensing AW, Agnelli G, Decousus H, Prandoni P, Misselwitz F. Rivaroxaban: population pharmacokinetic analyses in patients treated for acute deep vein thrombosis and exposure simulations in patients with atrial fibrillation treated for stroke prevention. Clin Pharmacokinet. 2011;50:675–86. doi: 10.2165/11595320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13.Girgis IG, Peters G, Moore K, Patel MR, Mahaffey KW, Nessel CC, Paolini JF, Halperin JL, Califf RM, Fox KAA, Becker RC. Population pharmacokinetics and pharmacodynamics of rivaroxaban in patients with non-valvular atrial fibrillation. 2011. American Heart Association Annual Meeting; 2011 Nov 12–16, 2011; Orlando, Fla. [DOI] [PubMed]
- 14.Mega JL, Braunwald E, Mohanavelu S, Burton P, Poulter R, Misselwitz F, Hricak V, Barnathan ES, Bordes P, Witkowski A, Markov V, Oppenheimer L, Gibson CM. Rivaroxaban versus placebo in patients with acute coronary syndromes (ATLAS ACS-TIMI 46): a randomised, double-blind, phase II trial. Lancet. 2009;374:29–38. doi: 10.1016/S0140-6736(09)60738-8. [DOI] [PubMed] [Google Scholar]
- 15.Boeckman A, Sheiner LB, Beal SL. NONMEM VI. Ellicott City, MD: GloboMax, Icon Development Solutions; 2007. [Google Scholar]
- 16.Beal SL, Sheiner LB. NONMEM Users Guides. Ellicott City, MD: GloboMax, Icon Development Solutions; 1989. p. 1998. [Google Scholar]
- 17.Karlsson MO, Sheiner LB. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J Pharmacokinet Biopharm. 1993;21:735–50. doi: 10.1007/BF01113502. [DOI] [PubMed] [Google Scholar]
- 18.Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos. 2009;37:1056–64. doi: 10.1124/dmd.108.025569. [DOI] [PubMed] [Google Scholar]
- 19.Lang D, Freudenberger C, Weinz C. In vitro metabolism of rivaroxaban, an oral, direct factor Xa inhibitor, in liver microsomes and hepatocytes of rats, dogs, and humans. Drug Metab Dispos. 2009;37:1046–55. doi: 10.1124/dmd.108.025551. [DOI] [PubMed] [Google Scholar]
- 20.Clark B. Biology of renal aging in humans. Adv Ren Replace Ther. 2000;7:11–21. doi: 10.1016/s1073-4449(00)70002-1. [DOI] [PubMed] [Google Scholar]
- 21.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–51. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 23.Agoram B, Heatherington AC, Gastonguay MR. Development and evaluation of a population pharmacokinetic-pharmacodynamic model of darbepoetin alfa in patients with nonmyeloid malignancies undergoing multicycle chemotherapy. AAPS J. 2006;8:E552–63. doi: 10.1208/aapsj080364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu C, Zhou H. An improved approach for confirmatory phase III population pharmacokinetic analysis. J Clin Pharmacol. 2008;48:812–22. doi: 10.1177/0091270008318670. [DOI] [PubMed] [Google Scholar]
- 25.Engle RF. Wald, likelihood ratio, and lagrange multiplier tests in econometrics. In: Intriligator MD, Griliches Z, editors. Handbook of Econometrics: Elsiever. Amsterdam, The Netherlands: North-Holland Publishing Company; 1983. pp. 796–801. [Google Scholar]
- 26.Kubitza D, Becka M, Mueck W, Halabi A, Maatouk H, Klause N, Lufft V, Wand DD, Philipp T, Bruck H. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct Factor Xa inhibitor. Br J Clin Pharmacol. 2010;70:703–12. doi: 10.1111/j.1365-2125.2010.03753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubitza D, Becka M, Roth A, Mueck W. Dose-escalation study of the pharmacokinetics and pharmacodynamics of rivaroxaban in healthy elderly subjects. Curr Med Res Opin. 2008;24:2757–65. doi: 10.1185/03007990802361499. [DOI] [PubMed] [Google Scholar]
- 28.Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook-Bruns N, Fox KA, Goto S, Murphy SA, Plotnikov AN, Schneider D, Sun X, Verheugt FW, Gibson CM. Rivaroxaban in Patients with a Recent Acute Coronary Syndrome. N Engl J Med. 2012;366:9–19. doi: 10.1056/NEJMoa1112277. [DOI] [PubMed] [Google Scholar]
- 29.Moss AJ, Benhorin J. Prognosis and management after a first myocardial infarction. N Engl J Med. 1990;322:743–53. doi: 10.1056/NEJM199003153221107. [DOI] [PubMed] [Google Scholar]
- 30.Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med. 2005;143:241–50. doi: 10.7326/0003-4819-143-4-200508160-00005. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi H, Kashima T, Nomizo Y, Muramoto N, Shimizu T, Nasu K, Kubota T, Kimura S, Echizen H. Metabolism of warfarin enantiomers in Japanese patients with heart disease having different CYP2C9 and CYP2C19 genotypes. Clin Pharmacol Ther. 1998;63:519–28. doi: 10.1016/S0009-9236(98)90103-5. [DOI] [PubMed] [Google Scholar]
- 32.Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353:717–9. doi: 10.1016/S0140-6736(98)04474-2. [DOI] [PubMed] [Google Scholar]
- 33.D'Andrea G, D'Ambrosio RL, Di Perna P, Chetta M, Santacroce R, Brancaccio V, Grandone E, Margaglione M. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood. 2005;105:645–9. doi: 10.1182/blood-2004-06-2111. [DOI] [PubMed] [Google Scholar]
- 34.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, Muller CR, Strom TM, Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–41. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 35.Pitsiua M, Parkera EM, Aarons L, Rowland M. Population pharmacokinetics and pharmacodynamics of warfarin in healthy young adults. Eur J Pharm Sci. 1993;1:151–7. [Google Scholar]
- 36.Fihn SD, Callahan CM, Martin DC, McDonell MB, Henikoff JG, White RH. The risk for and severity of bleeding complications in elderly patients treated with warfarin. The National Consortium of Anticoagulation Clinics. Ann Intern Med. 1996;124:970–9. doi: 10.7326/0003-4819-124-11-199606010-00004. [DOI] [PubMed] [Google Scholar]
- 37.Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 1995;108(4) Suppl.:231S–46S. doi: 10.1378/chest.108.4_supplement.231s. [DOI] [PubMed] [Google Scholar]
- 38.James AH, Britt RP, Raskino CL, Thompson SG. Factors affecting the maintenance dose of warfarin. J Clin Pathol. 1992;45:704–6. doi: 10.1136/jcp.45.8.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hallak HO, Wedlund PJ, Modi MW, Patel IH, Lewis GL, Woodruff B, Trowbridge AA. High clearance of (S)-warfarin in a warfarin-resistant subject. Br J Clin Pharmacol. 1993;35:327–30. doi: 10.1111/j.1365-2125.1993.tb05703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones M, McEwan P, Morgan CL, Peters JR, Goodfellow J, Currie CJ. Evaluation of the pattern of treatment, level of anticoagulation control, and outcome of treatment with warfarin in patients with non-valvar atrial fibrillation: a record linkage study in a large British population. Heart. 2005;91:472–7. doi: 10.1136/hrt.2004.042465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin Pharmacol Ther. 2005;78:412–21. doi: 10.1016/j.clpt.2005.06.011. [DOI] [PubMed] [Google Scholar]