

Abstract
AIM
Anacetrapib is currently being developed for the treatment of dyslipidaemia. Since warfarin, an anticoagulant with a narrow therapeutic index, is expected to be commonly prescribed in this population, a drug interaction study was conducted.
METHODS
In a randomized, open-label, two-period fixed-sequence design, 12 healthy male subjects received two different treatments (treatment A followed by treatment B). In treatment A, a single oral dose of 30 mg warfarin (3 × 10 mg CoumadinTM) was administered on day 1. After a washout interval, subjects began treatment B, where they were given daily 100 mg doses of anacetrapib (1 × 100 mg) beginning on day −14 and continuing through day 7, with concomitant administration of 30 mg warfarin (3 × 10 mg) on day 1. All anacetrapib and warfarin doses were administered with a standard low fat breakfast. After warfarin concentrations and prothrombin time were measured, standard pharmacokinetic, pharmacodynamic and statistical (linear mixed effects model) analyses were applied.
RESULTS
Anacetrapib was generally well tolerated when co-administered with warfarin in the healthy males in this study. The geometric mean ratios (GMRs) for warfarin + anacetrapib : warfarin alone and 90% confidence interval (CIs) for warfarin AUC(0–∞) were 0.94 (0.90, 0.97) for the R(+) warfarin enantiomer and 0.93 (0.87, 0.98) for the S(−) warfarin enantiomer, both being contained in the interval (0.80, 1.25), supporting the primary hypothesis of the study. The GMRs warfarin + anacetrapib : warfarin alone and 90% CIs for the statistical comparison of warfarin Cmax were 1.01 (0.97, 1.05) for both the R(+) warfarin and the S(−) warfarin enantiomers, and were also contained in the interval (0.80, 1.25). The GMR (warfarin + anacetrapib : warfarin alone) and 90% CI for the statistical comparison of INR AUC(0–168 h) was 0.93 (0.89, 0.96).
CONCLUSION
The single dose pharmacokinetics and pharmacodynamics of orally administered warfarin were not meaningfully affected by multiple dose administration of anacetrapib, indicating that anacetrapib does not affect CYP 2C9 clinically. Thus, no dosage adjustment for warfarin is necessary when co-administered with anacetrapib.
Keywords: anacetrapib, CETP, dyslipidaemia, warfarin
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Inhibition of cholesteryl ester transfer protein (CETP) is a potential new mechanism for the treatment of dyslipidaemia. Anacetrapib is a novel CETP inhibitor in development. Warfarin is a commonly prescribed anticoagulant that has a narrow therapeutic index. A drug interaction study for warfarin with a novel CETP inhibitor is expected to be helpful in defining dosing regimens.
WHAT THIS STUDY ADDS
This is the first study to show that there is no clinically meaningful pharmacokinetic interaction between anacetrapib and warfarin. The single dose pharmacokinetics and pharmacodynamics of orally administered warfarin were not meaningfully affected by multiple dose administration of anacetrapib, indicating that anacetrapib does not affect CYP 2C9 clinically. Thus, no dosage adjustment for warfarin is necessary when co-administered with anacetrapib.
Introduction
The residual clinical event rate of cardiovascular risk, despite the use of HMG-CoA reducase inhibitors, still remains high and hence, the search continues for agents that can yield both additional LDL-C lowering and beneficial effects on other biomarkers or targets, such as high density lipoprotein cholesterol (HDL-C) that also may impact disease [1]. A pharmacologic lipid-altering agent that can safely and effectively lower LDL-C concentrations and raise HDL-C concentrations would answer a significant unmet medical need. One such novel mechanism is the cholesteryl ester transfer protein (CETP) target, which is a plasma protein that catalyzes the heteroexchange of cholesteryl esters from HDL and triglycerides to apo B-containing lipoproteins [2]. Anacetrapib is an investigational orally active and potent CETP inhibitor that is currently under development for the treatment of dyslipidaemias including primary hypercholesterolaemia and mixed dyslipidaemia, which in clinical trials to date has exhibited an acceptable safety, tolerability and lipid altering profile to warrant continued clinical investigation [3]–[10].
Warfarin is a coumarin-based anticoagulant that is commonly prescribed to patients at risk of thrombotic and embolic disorders [11]–[13]. It is a racemic mixture of R(+) and S(−) warfarin enantiomers, with the anticoagulant potency of the S(−) enantiomer being up to ∼6 times greater than the R(+) enantiomer [11]. The efficacy of warfarin therapy is commonly monitored by the measurement of prothrombin time (PT) converted to the standardized parameter of the International Normalized Ratio (INR) [14], [15]. Changes in PT/INR, a commonly used measure of pharmacodynamics, are correlated with the clinical consequences of excessive bleeding and thrombosis, and thus, this parameter is monitored closely [14], [15]. Additionally, warfarin has a narrow therapeutic index and alterations in its pharmacokinetics can often lead to clinically important changes in warfarin pharmacodynamics [11]. Warfarin is primarily metabolized by the cytochrome P-450 (CYP) 2C9 pathway which is responsible for the oxidative conversion of the (S)-enantiomer to (S)-7-hydroxywarfarin and, to a more limited extent, (S)-6-hydroxywarfarin [16]–[19]. The oxidative metabolism of (R)-warfarin is more balanced and known to be mediated by CYP 3A4, 2C19 and 1A2 [16]–[19].
The metabolic and drug interaction profile of anacetrapib has been characterized [8], [20]. Anacetrapib is a moderately sensitive CYP3A substrate [8], [9]. Therefore, CYP3A4 metabolism is a major pathway of elimination of anacetrapib in humans. A probe study using midazolam in healthy subjects indicated that anacetrapib does not inhibit or induce CYP3A4 activity [8]. There was also no clinically meaningful interaction with simvastatin or digoxin [10], [21]. Since the CYP2C9 pathway is not affected by anacetrapib based on information to date, no CYP2C9-mediated interaction was a priori expected for this combination. However, the purpose of this study was to exclude the potential for a drug–drug interaction by examining the potential of multiple dose anacetrapib to influence single dose warfarin pharmacodynamics (i.e. INR values) in addition to its pharmacokinetics. To ensure that plasma concentrations of anacetrapib reached apparent steady-state prior to the administration of single dose warfarin in this study, a single dose of warfarin was co-administered following multiple once daily dosing of anacetrapib. A 100 mg dose of anacetrapib was chosen in this study because it represented the highest dose being used in the phase III programme [3]. The main objective of this study was to evaluate the potential effects of anacetrapib 100 mg dosed once daily on the pharmacokinetics (primary endpoint: AUC(0–∞), secondary endpoint Cmax) and the pharmacodynamics (PT/INR) of single dose warfarin in healthy male and female subjects. The safety and tolerability profile of the combined administration of anacetrapib and warfarin was also examined in this study.
Methods
The study protocol and informed consent documents were approved by the Celerion Pharma Services Institutional Review Board, which is fully independent from the MDS Pharma Services clinical site (now Celerion) where the study was conducted. The study was conducted in accordance with the guidelines established by the Declaration of Helsinki and in compliance with Good Clinical Practice.
Subjects
Healthy, non-smoking, male subjects and non-pregnant female subjects of non-childbearing potential (i.e. hysterectomy, bilateral oophorectomy, tubal ligation postmenopausal) between the ages of 18 to 50 years with a body mass index between 18 and 33 kg m−2 who agreed to comply with all study restrictions were eligible to participate in this study. The ranges in age and BMI above were selected based on available information for the study drugs. For example, as there was no meaningful effect of age or BMI on the pharmacokinetics of anacetrapib, a wider BMI range was considered [6]. Similarly, the study was open to a healthy subject of any ethnicity. Although the protocol specified that both male and female subjects were eligible to participate in the study and accordingly, it did not specify that a specific number of a particular gender needed to be recruited. After screening per protocol, healthy male subjects participated in this study. As only male subjects participated, no serum pregnancy tests were performed.
Each subject provided written informed consent prior to the administration of study procedures. Additional entry criteria included normal pre-study laboratory test results for PT, activated partial thromboplastin time (aPTT), platelet count and negative stool occult blood test. Subjects could not be involved with any activities that would place them at high risk of haemorrhage (e.g. contact sports). Subjects also had to agree to restrict their intake of alcohol, caffeinated beverages, grapefruit and grapefruit juice and quinine containing beverages. Subjects were excluded if they had any relevant history of pulmonary, hepatic, gastrointestinal, psychiatric or neurological disease, diabetes, any condition predisposing them to immunodeficiency and any condition contraindicating use of warfarin (e.g. haemorrhagic tendencies, recent or pending surgery, ulceration or overt bleeding of the gastrointestinal system). Subjects with an estimated creatinine clearance ≤60 ml min−1 or serum creatinine >1.5 mg dl–1 were excluded. Additional exclusion criteria included a history of multiple and/or severe allergies to drugs or foods. The use of prescription and non-prescription medications was not allowed within 14 days of study start and throughout the entire study period.
Study design
A randomized, open-label, two-period fixed-sequence design was used. Twelve healthy male subjects received two different treatments (treatment A followed by treatment B). In treatment A, a single oral dose of 30 mg warfarin (3 × 10 mg CoumadinTM) was administered on day 1. After a washout interval comprising at least 10 days, subjects began treatment B, where they were given daily 100 mg doses of anacetrapib (1 × 100 mg) beginning on day −14 and continuing through day 7, with concomitant administration of 30 mg warfarin (3 × 10 mg) on day 1. All anacetrapib and warfarin doses were administered with 240 ml of water, following consumption of a standard low fat breakfast. Blood samples for characterizing the pharmacokinetics (warfarin R(+) and S(−) enantiomers) and pharmacodynamics (prothrombin time measured as PT and INR) were collected at pre-dose and at selected time points over the 168 h interval following warfarin administration in both treatment periods.
Bioanalytical and pharmacokinetic assessments
Blood (4 ml) for measurement of S(−) and R(+) enantiomers of warfarin was collected in sodium heparin containing tubes at pre dose on day 1 of each treatment and post dose at 0.5, 1, 2, 4, 12, 24, 48, 72, 96, 120, 144 and 168 h. The samples were immediately centrifuged (within 30 min) at 3000 rev min–1 for 10 min at 4°C. The plasma was then separated into polypropylene crytubes and stored at −20°C until assayed and shipped on dry ice for analysis. A sensitive, specific, accurate and reproducible analytical method was developed by Advion BioSciences (Advion), Inc., Ithaca, New York to quantitate total (R)-warfarin and (S)-warfarin in heparinized human plasma samples. Plasma samples were diluted with citric acid, centrifuged and injected onto a column switching system where the two enantiomers were separated chromatographically using a chiral column following clean up on an SPS-Ph trapping column. Samples were analyzed by turbo ion spray, column switching, liquid chromatography/tandem mass spectrometry (LC/LC/MS/MS) in the negative ion mode. The assay demonstrated a lower limit of quantitation (LLQ) of 10 ng ml−1 using 0.1 ml plasma sample aliquots. The calibration curves were linear from 10 ng ml−1 to 2500 ng ml−1 for (R)- and (S)-warfarin. Plasma concentrations above the lower limit of quantification (LLOQ) of 10 ng ml−1 were determined with a precision of ≤7.5% for (R)-warfarin, ≤6.7% for (S)-warfarin and accuracies of −3.3 to 2.0% for (R)-warfarin and −5.5 to –0.8% for (S)-warfarin.
The pharmacokinetic parameters were computed from the individual plasma concentrations using actual blood draw times for R(+) warfarin and S(−) warfarin enantiomers employing a noncompartmental approach using WinNonlin® Professional Version 5.0.1. (Pharsight Corporation, Mountain View, CA). The parameter values were then imported into SAS® and all descriptive statistics were calculated in SAS® Version 8.2. Plasma R(+) warfarin and S(−) warfarin concentrations below the lower limit of quantitation were set to 0 (BLLQ = 10 ng ml−1). The apparent terminal rate constant (λz) was calculated by linear regression of the terminal log-linear portion of the individual plasma concentration–time profiles. The apparent first order terminal elimination half-life (apparent terminal t1/2) was calculated as the quotient of ln(2) and λz. The area under the concentration–time curve extrapolated through infinity [AUC(0–∞)], calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations (linear-up/log-down), was estimated from the sum of AUC to last measured concentration and the extrapolated area given by the quotient of the last measured concentration and λz. Peak plasma concentration [Cmax (ng ml−1)] and its time of occurrence [tmax (h)] were generated by WinNonlin from the individual plasma concentration–time data for each analyte. Individual λz values and the subsequently derived pharmacokinetic parameters [apparent terminal t1/2 and AUC(0–∞)] were not reported in cases where the terminal phase of the log concentration vs. time profile did not exhibit an apparent linear decline with regression coefficient >0.8. At least three data points (excluding Cmax) in the terminal phase were used for λz calculations.
Pharmacodynamic assessments
The pharmacodynamic parameters of warfarin were evaluated at various time points throughout the study through measurement of PT and calculation of INR using a single lot of thromboplastin with international sensitivity index. All measurements of PT and calculations of INR were done at the site's local laboratory. The LLOQ for PT was 9.0 s, and there were control runs, with at least one set of controls run each day of use; a normal control and an abnormal high control. The same lot of control was used throughout this study. The CVs for the controls were as follows: level 1 (normal), the CV was 1.4 and level 2 (abnormal high), the CV was 2.1.
Blood samples (4.5 ml) for the determination of PT/INR were collected at pre dose and at 0.5, 1, 2, 4, 12, 24, 48, 72, 96, 120, 144 and 168 h post warfarin dose in each treatment. Plasma was prepared and used for PT determination in duplicate within 2 h of collection. PTs were reported both as raw data in absolute time (s) and as INRs. The LLOQ for PT was 9.0 s, and there were control runs, with at least one set of controls run each day of use; a normal control and an abnormal high control. The same lot of control was used throughout this study. The CVs for the controls were as follows: level 1 (normal), the CV was 1.4 and level 2 (abnormal high), the CV was 2.1.
Statistical analysis
Power
The sample size for the study was rationalized as follows. The probability that the overall primary hypothesis will be supported [that both confidence intervals (CIs) for R(+) warfarin and S(−) warfarin AUC(0–∞) will fall within the interval (0.80, 1.25)] was considered to be approximately 97%. This calculation assumed a sample size of 12 subjects in a two-period, fixed-sequence design, a within-subject standard deviation of 0.1018 and 0.1239 (ln ng ml−1 h), respectively, for R(+) warfarin and S(−) warfarin, a type I error rate of 0.05, the true geometric mean ratios (GMRs) of 1.00 (assumed to be 1.00 because a priori, an interaction was not expected based on available data) and that warfarin R(+) and S(−) AUC(0–∞) are uncorrelated.
Analysis of warfarin pharmacokinetics
The AUC(0–∞) and Cmax values were analyzed after transformation to the natural log scale. A linear mixed-effects model with treatment as a fixed effect and subject as a random effect was applied on the natural log (ln)-transformed pharmacokinetic parameters AUC(0–∞) and Cmax. Ninety percent (90%) CIs were constructed for the difference in least-squares (LS) means for ln-transformed AUC(0–∞) and Cmax. Exponentiating the log scale 90% CI provided the 90% CI for the GMR (warfarin with anacetrapib/warfarin alone) of AUC(0–∞) and Cmax.
tmax was summarized by providing medians. Harmonic mean was provided for apparent terminal t1/2. Terminal half-lives were presented in this study for information purposes only, and no statistical analysis was planned or reported for this parameter since boundary effects for bioequivalence are for rate and extent of absorption alone.
The primary hypothesis, that the co-administration of warfarin with anacetrapib did not influence the pharmacokinetics of warfarin was to be considered satisfied if the 90% CIs for the AUC(0–∞) GMRs for both enantiomers were contained within the 0.80, 1.25 interval. The secondary hypothesis was similar, but assessed comparability of warfarin pharmacokinetics based on 90% CIs for the Cmax GMR for both enantiomers being contained within the 0.80, 1.25 interval.
The statistical analysis was done with a mixed model in SAS® version 8.2 using PROC MIXED. This statistical method was considered to be the ‘best’ method to handle missing data as the analysis uses standard ANOVA procedures and hypothesis testing to estimate the LS means and the differences between the LS means in the log scale. This difference was back transformed into the normal scale, which was the mean ratio. It was ensured that the extrapolated part of the AUC(0–∞) was <20%. For S-warfarin the extrapolated component of the AUC(0–∞) was well below 20% in all subjects. For R-warfarin, the extrapolated component was also below 20% in all but one subject (one exception was in a subject who discontinued early from the study, at 72 h).
Analysis of warfarin pharmacodynamics
The influence of multiple dose anacetrapib on the pharmacodynamics of single dose warfarin was assessed through the measurement of INR AUC(0–168 h) and INRmax for prothrombin time and analyzed using the same linear mixed effects model as described above for the pharmacokinetic analyses. The natural log transformation was applied to both of these parameters prior to analysis. Summary statistics and 90% CIs for INR AUC(0–168 h) and INRmax GMRs warfarin + anacetrapib : warfarin were provided.
Safety measurements
The safety and tolerability of study drugs were assessed by clinical evaluation of adverse experiences and by physical examinations, vital signs, routine laboratory safety measurements (haematology, blood chemistry and urinalysis), serum β-human chorionic gonadotropin (β-hCG) and 12-lead electrocardiograms (ECG). Adverse experiences were monitored throughout the study and evaluated in terms of intensity (mild, moderate or severe), duration, severity, outcome and relationship to study drug. All subjects who took at least one dose of study medication were included in the safety and tolerability analyses.
Results
Study population
Twelve healthy male subjects were enrolled in the study and 10 subjects completed the study. All 12 subjects completed treatment A and 11 subjects completed a sufficient number of pharmacokinetic and pharmacodynamic assessments to be included in the pharmacokinetic, pharmacodynamic and statistical analyses. All 12 subjects were included in the evaluation of safety.
One subject withdrew from the study prior to period 2 (treatment B) and hence his data were excluded from the statistical analyses. Another subject withdrew from the study after 72 h of period 2 (treatment B), and hence his data were included in all the analyses, as appropriate. Of the remaining 10 subjects, four had one missed blood collection time point in treatment B (three of which were after 96 h). These missing data points had no meaningful impact on the study.
Twelve subjects completed all assessments as specified in the protocol for treatment A. While only 10 subjects completed all assessments for treatment B, one of the two subjects who discontinued did so after completing the 72 h assessments. Therefore, PK and PD data from this subject were sufficiently complete to evaluate warfarin PK and PD in treatment B for this subject. However, one parameter, INR AUC(0–168 h), was not evaluable in this subject since he did not have data through to 168 h.
The per protocol population on this study was specified in the protocol as the subset of subjects who complied with the protocol sufficiently to ensure that their data were likely to exhibit the effects of treatment, according to the underlying scientific model. All subjects who were compliant with the study procedure and had available data from at least one treatment were to have been included in the primary analysis dataset. This population was to have been used for the PK and PD analyses. For this reason in the statistical model data from all 12 subjects were included. Furthermore, PK and PD data were calculated for treatment B for one of the two subjects who discontinued the study early, as the data were deemed sufficiently complete to evaluate his PK and PD profiles in that treatment arm.
Pharmacokinetics
The arithmetic mean plasma R(+) warfarin and S(−) warfarin concentration vs. time profiles following the single-dose administration of 30 mg warfarin alone (treatment A) and co-administered with multiple, once daily 100 mg anacetrapib doses (treatment B) are presented in Figures 1 and 2, respectively. Mean R(+) warfarin and S(−) warfarin concentrations following single doses of warfarin were similar between administration of 30 mg warfarin alone (treatment A) and co-administered with multiple, once daily 100 mg anacetrapib doses (treatment B).
Figure 1.
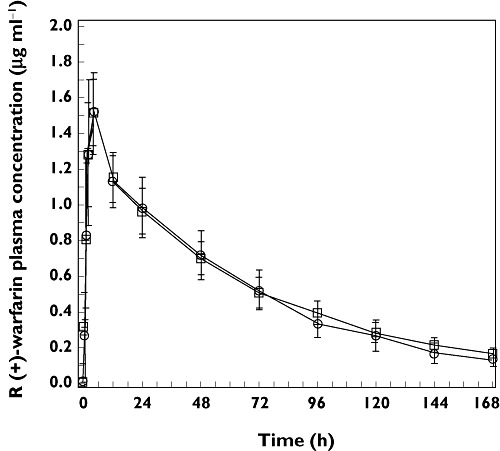
Arithmetic mean (SD) plasma concentration–time profiles of plasma R(+) warfarin following the administration of a single oral dose of 30 mg warfarin alone (day 1, treatment A, □) and co-administered with multiple once daily doses of 100 mg anacetrapib (day 1, treatment B, ○) in healthy adult subjects (n = 12 for treatment A and n = 11 for treatment B)
Figure 2.
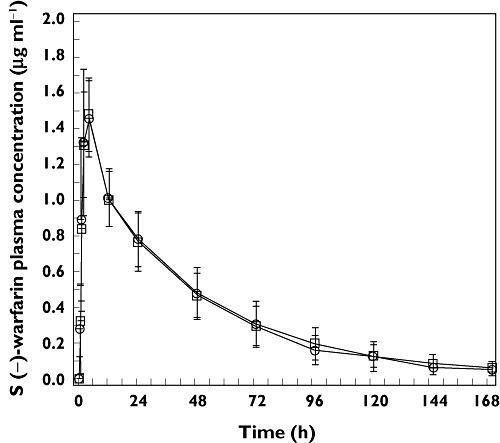
Arithmetic mean (SD) plasma concentration–time profiles of plasma S(−) warfarin following the administration of a single oral dose of 30 mg warfarin alone (day 1, treatment A, □) and co-administered with multiple once daily doses of 100 mg anacetrapib (day 1, treatment B, ○) in healthy adult subjects (n = 12 for treatment A and n = 11 for treatment B)
There were no apparent differences between the two treatments in peak mean R(+) warfarin and S(−) warfarin concentrations, the times to reach these peak mean concentrations or in the apparent post-peak rates of decline in these mean concentrations.
The GMRs warfarin + anacetrapib : warfarin alone and 90% CIs for the statistical comparison of warfarin AUC(0–∞) were 0.94 (0.90, 0.97) for the R(+) warfarin enantiomer and 0.93 (0.87, 0.98) for the S(−) warfarin enantiomer. Since the 90% CIs for the GMRs for the plasma AUC(0–∞) of warfarin [S(−) and R(+)] enantiomers were contained in the interval (0.80, 1.25), the primary hypothesis was supported (Table 1). The GMRs for warfarin + anacetrapib : warfarin alone and 90% CIs for the statistical comparison of warfarin Cmax were 1.01 (0.97, 1.05) for both the R(+) warfarin and the S(−) warfarin enantiomers. Since the 90% CIs for the GMRs for the plasma Cmax of warfarin [S(−) and R(+)] enantiomers were contained in the interval 0.80, 1.25, the secondary hypothesis was supported.
Table 1.
Statistical comparison of plasma pharmacokinetics of warfarin [R(+) and S(−)] enantiomers following the administration of a single oral dose of 30 mg warfarin alone (day 1, treatment A) and co-administered with multiple once daily doses of 100 mg anacetrapib (day 1, treatment B) in healthy adult subjects (n = 12 for treatment A and n = 11 for treatment B)
| Warfarin + anacetrapib | Warfarin alone | Warfarin + anacetrapib : warfarin alone | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pharmacokinetic parameter | n | GM | 95% CI | n | GM | 95% CI | GMR | 90% CI | rMSE† |
| R(+) enantiomer | |||||||||
| AUC(0–∞)‡ (µg ml−1 h) | 11 | 95.55 | (86.46, 105.61) | 12 | 101.77 | (92.16, 112.39) | 0.94 | (0.90, 0.97) | 0.0480 |
| Cmax‡ (µg ml−1) | 11 | 1.54 | (1.41, 1.68) | 12 | 1.52 | (1.39, 1.66) | 1.01 | (0.97, 1.05) | 0.0520 |
| tmax§ (h) | 11 | 4.0 | (2.0, 4.1) | 12 | 4.0 | (1.0, 4.0) | |||
| Apparent terminal t1/2¶ (h) | 11 | 46.7 | 8.5 | 12 | 53.3 | 6.8 | |||
| S(−) enantiomer | |||||||||
| AUC(0–∞)‡ (µg ml−1 h) | 11 | 58.14 | (49.09, 68.85) | 12 | 62.74 | (53.04, 74.20) | 0.93 | (0.87, 0.98) | 0.0781 |
| Cmax‡ (µg ml−1) | 11 | 1.51 | (1.38, 1.65) | 12 | 1.50 | (1.37, 1.64) | 1.01 | (0.97, 1.05) | 0.0513 |
| tmax§ (h) | 11 | 4.0 | (1.0, 4.1) | 12 | 4.0 | (1.0, 4.0) | |||
| Apparent terminal t1/2¶ (h) | 11 | 32.2 | 4.4 | 12 | 38.2 | 7.1 | |||
rMSE: Root mean square error on log scale. When multiplied by 100, provides estimate of the pooled within subject coefficient of variation.
Back-transformed least squares mean and confidence interval from mixed effects model performed on natural log transformed values.
Median (min, max) reported for tmax.
Harmonic mean (pseudo SD) reported for apparent terminal t1/2. GM geometric mean, GMR geometric mean ratio.
Pharmacodynamics
The arithmetic mean prothrombin time INR vs. time profiles following the single dose administration of 30 mg warfarin alone (treatment A) and co-administered with multiple, once daily 100 mg anacetrapib doses (treatment B) are presented in Figure 3.
Figure 3.
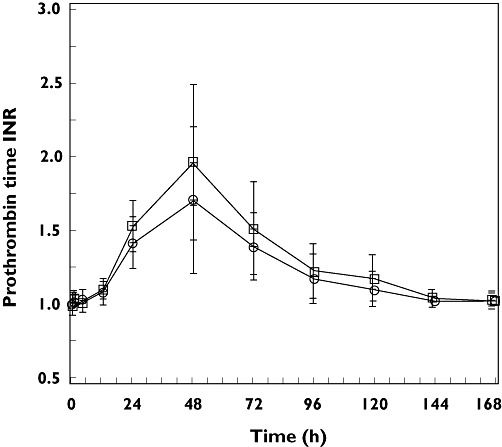
Arithmetic mean (SD) prothrombin time INR -time profiles following the administration of a single oral dose of 30 mg warfarin alone (day 1, treatment A, □) and co-administered with multiple once daily doses of 100 mg anacetrapib (day 1, treatment B, ○) in healthy adult subjects (n = 12 for treatment A and n = 11 for treatment B)
The overall shapes of the mean prothrombin time INR vs. time profiles were similar. Peak mean prothrombin time INR, which occurred at 48 h post dose in both treatments, was somewhat higher following single dose administration of 30 mg warfarin alone (treatment A) relative to when co-administered with multiple, once-daily 100 mg anacetrapib doses (treatment B). The GMRs for warfarin + anacetrapib : warfarin alone and 90% CIs for the statistical comparison of INR AUC(0–168 h) and INRmax were 0.93 (0.89, 0.96) and 0.85 (0.76, 0.96), respectively (Table 2).
Table 2.
Statistical comparison of prothrombin time INR endpoints following the administration of a single oral dose of 30 mg warfarin alone (day 1, treatment A) and co-administered with multiple once daily doses of 100 mg anacetrapib (day 1, treatment B) in healthy adult subjects (n = 12 for treatment A, n = 11 for INRmax in treatment B and n = 10 for INR AUC(0–168 h) in treatment B)
| Warfarin + anacetrapib | Warfarin alone | Warfarin + anacetrapib : warfarin alone | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | n | GM | 95% CI | n | GM | 95% CI | GMR | 90% CI | rMSE† |
| INR AUC(0–168 h)‡ | 10 | 206.77 | (190.44, 224.50) | 12 | 223.25 | (206.05, 241.89) | 0.93 | (0.89, 0.96) | 0.0464 |
| INRmax‡ | 11 | 1.63 | (1.38, 1.93) | 12 | 1.92 | (1.63, 2.26) | 0.85 | (0.76, 0.96) | 0.1497 |
rMSE: Root mean square error on log scale. When multiplied by 100, provides estimate of the pooled within-subject coefficient of variation.
Back-transformed least squares mean and confidence interval from mixed effects model performed on natural log transformed values. INR AUC(0–168 h) value was not calculable for subject AN 0002, Treatment B, as subject withdrew consent after 72 h. GM geometric mean, GMR geometric mean ratio.
Safety
Anacetrapib was generally well tolerated when co-administered with warfarin in the healthy subjects. There were no serious adverse experiences reported and no subjects discontinued due to an adverse experience. There were no meaningful changes in laboratory safety, vital signs or ECG parameters. Of the 12 subjects dosed in this study, four (33%) reported a total of nine clinical adverse experiences. The clinical adverse experiences were each reported by only one (8%) subject. The investigator considered all nine clinical adverse experiences to be mild to moderate in intensity, and probably not or definitely not related to study drug. One subject experienced presyncope which was associated with venipuncture.
Discussion
As anacetrapib may be dosed in patients with cardiovascular disease being treated with a narrow therapeutic index drug such as warfarin, understanding the effect of steady-state anacetrapib, a novel and potent CETP inhibitor, on the pharmacokinetics and pharmacodynamics of warfarin was considered essential in the programme. A 100 mg dose of anacetrapib was used in this study, which is the highest dose being used in phase III [3]. Since it is anticipated that anacetrapib will be administered with a meal, likely corresponding to the American Heart Association's therapeutic lifestyle diet, both warfarin and anacetrapib were administered with a standard low fat breakfast. Exposure to anacetrapib is increased in the presence of a meal [6], [7]. Thus, in this study, the concentrations of anacetrapib that were studied were higher than those expected in the fasted state.
Available in vitro and clinical data have indicated that anacetrapib is primarily metabolized by CYP3A and is negligibly eliminated in the urine [20]. Consistent with in vitro human hepatocyte data, anacetrapib did not meaningfully alter the pharmacokinetics of midazolam, a probe substrate of CYP3A4, indicating that anacetrapib does not induce or inhibit CYP3A activity in vivo[8]. The metabolic pathways thought to be responsible for the metabolism of the R(+) warfarin enantiomer include CYP3A4, CYP1A2 and CYP2C19, whereas the more potent S(−) warfarin enantiomer is oxidized primarily by CYP2C9 [11]. Anacetrapib was not a potent reversible inhibitor of human CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 or 3A4 in human liver microsomal incubations, given that the IC50 values in all cases were greater than 100 µm[8]. Thus, based on the known disposition of anacetrapib and warfarin, co-administration of anacetrapib dosed to apparent steady-state with a single 30 mg dose of warfarin was not expected to have a clinically meaningful effect on the pharmacokinetics or pharmacodynamics of warfarin.
Consistent with this hypothesis, the findings of this study are that the pharmacokinetics [AUC(0–∞), Cmax, tmax and apparent terminal t1/2 of S(−) and R(+) warfarin] and pharmacodynamics [INR AUC(0–168 h) and INRmax] of warfarin were not meaningfully affected by anacetrapib. Specifically, the GMRs for warfarin + anacetrapib : warfarin alone and 90% CIs for warfarin (both enantiomers) AUC(0–∞) (primary endpoint) and Cmax (secondary endpoint) were all contained within the interval 0.80, 1.25. These results indicate that anacetrapib does not inhibit CYP 2C9, the primary enzyme responsible for the disposition of S(−) warfarin.
In addition, there were no meaningful differences in tmax and apparent terminal t1/2 observed between the two treatments. These findings indicate that no clinically meaningful effects on blood coagulation are expected. This is consistent with the observation that the GMR for warfarin + anacetrapib : warfarin alone and 90% CI for the statistical comparison of INR AUC(0–168 h) were close to unity, specifically, 0.93 (0.89, 0.96). Therefore, the dose of warfarin does not need to be adjusted when co-administered with anacetrapib. Concomitant administration of anacetrapib and warfarin was generally well tolerated in this population of healthy subjects. There were no instances of clinically significant bleeding or unusual changes in INR with either treatment and no subjects discontinued from this study due to adverse experiences.
Multiple doses of anacetrapib were administered in this study. One potential consideration would be whether the given study design would distinguish between potential inducing and inhibiting effects of anacetrapib. While it is theoretically possible to have simultaneous inducing and inhibiting effects of anacetrapib on warfarin, there are three reasons that would argue against it: 1) available information with anacetrapib indicate that while it is a substrate for CYP3A4 it is not an inhibitor or inducer of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 2E1 and 3A4. Further, a midazolam probe clinical study revealed anacetrapib was not an inducer or inhibitor of CYP3A [8]. This was the reason why the study design did not include tracking surrogates for enzyme induction, 2) given the overall PK profiles of R- and S-warfarin between the two treatments as well as relative profiles of the two enantiomers to each other, it is unlikely to see induction perfectly matched by inhibition, and for both enantiomers and 3) in addition to the absence of an effect on warfarin exposure (i.e. PK), INR efficacy endpoint did not seem to be affected to any notable extent by multiple dose administration of anacetrapib.
One assumption in this study was that the single 30 mg dose of warfarin was sufficiently representative of the clinical situation, i.e. producing a stable anticoagulant effect in vivo. However, due to the healthy subjects being exposed to warfarin in this study, the approach was to use a representative dose of warfarin that would result in adequate drug concentrations for pharmacokinetic analysis but one at which there was minimal anticoagulation. Some authors have used a single dose of warfarin as low as 7.5 mg [22], [23]. Because warfarin pharmacodynamics are also of interest in the study and because it has been shown that a single 30 mg dose of warfarin sufficiently increases the INR value to be able to detect a clinically meaningful pharmacodynamic interaction [24], we chose to study a single 30 mg dose of warfarin under carefully monitored study conditions. In warfarin drug interaction studies performed at Merck, we have adequately established the sensitivity of a single 30 mg dose of warfarin [24]–[26]. The single dose study design is favoured because of the advantage of allowing for the investigation of possible drug interactions while reducing the safety risks associated with exposing healthy participants to multiple doses of warfarin.
In conclusion, administration of multiple 100 mg doses of anacetrapib concomitantly with a 30 mg dose of warfarin does not influence the pharmacokinetics and pharmacodynamics of warfarin to a clinically meaningful extent, indicating that anacetrapib does not affect CYP 2C9 clinically. Thus, no dosage adjustment for warfarin is necessary when co-administered with anacetrapib. Concomitant administration of multiple doses of anacetrapib with a single dose of warfarin 30 mg appeared to be generally well tolerated when administered to healthy male subjects in this study.
Competing Interests
This study was sponsored by Merck & Co., Inc. RK, AG, JC, AM, YL, SL, JW and SAS are employees of Merck and may hold stock or stock options in the company.
Acknowledgments
We would like to acknowledge the assistance of Walter Kline, Jin Zhang and Man-Wai Lo for their help in coordinating the outsourcing of warfarin assay at Advion.
REFERENCES
- 1.Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, Joyal SV, Hill KA, Pfeffer MA, Skene AM Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 2.Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res. 1993;34:1255–74. [PubMed] [Google Scholar]
- 3.Cannon CP, Shah S, Dansky HM, et al. for the DEFINE Investigators. Safety of Anacetrapib in Patients with or at High Risk for Coronary Heart Disease. N Engl J Med. 2010;363:2406–15. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 4.Bloomfield D, Carlson GL, Sapre A, Tribble D, McKenney JM, Littlejohn TW, III, Sisk CM, Mitchel Y, Pasternak RC. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157:352.e2–60.e2. doi: 10.1016/j.ahj.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Krishna R, Anderson MS, Bergman AJ, Jin B, Fallon M, Cote J, Rosko K, Chavez-Eng C, Lutz R, Bloomfield DM, Gutierrez M, Doherty J, Bieberdorf F, Chodakewitz J, Gottesdiener KM, Wagner JA. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–14. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 6.Krishna R, Garg A, Jin B, Cote J, Bergman A, Von Hoydonck P, Laethem T, Van Dyck K, Chavez-Eng C, Archer L, Lutz R, Hilliard D, Snyder K, Panebianco D, Bortel L, Lasseter K, Al-Huniti N, Dykstra K, Gottesdiener K, Wagner JA. Single-dose pharmacokinetics and pharmacodynamics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol. 2009;68:535–45. doi: 10.1111/j.1365-2125.2009.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishna R, Bergman A, Jin B, Fallon M, Cote J, Van Hoydonck P, Laethem T, Gendrano IN, III, Van Dyck K, Hilliard D, Laterza O, Snyder K, Chavez-Eng C, Lutz R, Chen J, Bloomfield DM, De Smet M, Van Bortel LM, Gutierrez M, Al-Huniti N, Dykstra K, Gottesdiener KM, Wagner JA. Multiple-dose pharmacodynamics and pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Clin Pharmacol Ther. 2008;84:679–83. doi: 10.1038/clpt.2008.109. [DOI] [PubMed] [Google Scholar]
- 8.Krishna R, Bergman AJ, Jin B, Garg A, Roadcap BA, Chiou RH, Dru JD, Cote J, Laethem T, Vets E, Gottesdiener KM, Wagner JA. Assessment of the CYP3A-mediated drug interaction potential of anacetrapib, a potent cholesteryl ester transfer protein (cetp) inhibitor, in healthy subjects. J Clin Pharmacol. 2009;49:80–7. doi: 10.1177/0091270008326718. [DOI] [PubMed] [Google Scholar]
- 9.Garg A, Maes A, Corr C, Jin B, Wadhwa T, Handa N, Van Dyck K, De Lepeleire I, Shah J, Wagner JA, Krishna R. Effect of diltiazem, a moderate CYP3A inhibitor, on the pharmacokinetics of anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. J Clin Pharmacol. 2011;51:436–9. doi: 10.1177/0091270010368676. [DOI] [PubMed] [Google Scholar]
- 10.Krishna R, Garg A, Jin B, Keshavarz SS, Bieberdorf F, Chodakewitz JA, Wagner JA. Assessment of a pharmacokinetic and pharmacodynamic interaction between simvastatin and anacetrapib, a potent cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br J Clin Pharmacol. 2009;67:520–6. doi: 10.1111/j.1365-2125.2009.03385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bristol-Myers Squibb. Coumadin tablets/Coumadin for injection [Prescribing information] Princeton, NJ: Bristol-Myers Squibb; 2007. [Google Scholar]
- 12.Hirsh J. Oral anticoagulant drugs. N Engl J Med. 1991;324:1865–75. doi: 10.1056/NEJM199106273242606. [DOI] [PubMed] [Google Scholar]
- 13.Hirsh J, Dalen J, Anderson DR, Poller L, Bussey H, Ansell J, Deykin D. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119:8S–21S. doi: 10.1378/chest.119.1_suppl.8s. [DOI] [PubMed] [Google Scholar]
- 14.Kirkwood TB. Calibration of reference thromboplastins and standardisation of the prothrombin time ratio. Thromb Haemost. 1983;49:238–44. [PubMed] [Google Scholar]
- 15.Johnston M, Harrison L, Moffat K, Willan A, Hirsh J. Reliability of the international normalized ratio for monitoring the induction phase of warfarin: comparison with the prothrombin time ratio. J Lab Clin Med. 1996;128:214–7. doi: 10.1016/s0022-2143(96)90014-1. [DOI] [PubMed] [Google Scholar]
- 16.Serlin MJ, Breckenridge AM. Drug interactions with warfarin. Drugs. 1983;25:610–20. doi: 10.2165/00003495-198325060-00004. [DOI] [PubMed] [Google Scholar]
- 17.Herman D, Locatelli I, Grabnar I, Peternel P, Stegnar M, Mrhar A, Breskvar K, Dolzan V. Influence of CYP2C9 polymorphisms, demographic factors and concomitant drug therapy on warfarin metabolism and maintenance dose. Pharmacogenomics J. 2005;5:193–202. doi: 10.1038/sj.tpj.6500308. [DOI] [PubMed] [Google Scholar]
- 18.Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73:67–74. doi: 10.1016/s0163-7258(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 19.Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol. 1992;5:54–9. doi: 10.1021/tx00025a009. [DOI] [PubMed] [Google Scholar]
- 20.Kumar S, Tan EY, Hartmann G, Biddle Z, Bergman AJ, Dru J, Ho JZ, Jones AN, Staskiewicz SJ, Braun MP, Karanam B, Dean DC, Gendrano IN, Graves MW, Wagner JA, Krishna R. Metabolism and disposition of 14C-anacetrapib, a potent inhibitor of cholesteryl ester transfer protein, in humans. Drug Metab Dispos. 2010;38:474–83. doi: 10.1124/dmd.109.028704. [DOI] [PubMed] [Google Scholar]
- 21.Krishna R, Stypinski D, Ali M, Garg A, Gendrano IN, Maes AL, Degroot B, Liu Y, Connolly S, Wagner JA, Stoch SA. Lack of a meaningful effect of anacetrapib on the pharmacokinetics of digoxin in healthy subjects. American Society for Clinical Pharmacology and Therapeutics 112th Annual Meeting, Dallas, TX, March 2011 (Abstract)
- 22.Washington C, Hou SY, Hughes NC, Campanella C, Berner B. Ciprofloxacin prolonged-release tablets do not affect warfarin pharmacokinetics and pharmacodynamics. J Clin Pharmacol. 2007;47:1320–6. doi: 10.1177/0091270007305504. [DOI] [PubMed] [Google Scholar]
- 23.Frymoyer A, Shugarts S, Browne M, Wu AB, Frassetto L, Benet LZ. Effect of single-dose rifampin on the pharmacokinetics of warfarin in healthy volunteers. Clin Pharmacol Ther. 2010;88:540–7. doi: 10.1038/clpt.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Hecken A, Depre M, Verbesselt R, Wynants K, De Lepeleire I, Arnout J, Wong PH, Freeman A, Holland S, Gertz B, De Schepper PJ. Effect of montelukast on the pharmacokinetics and pharmacodynamics of warfarin in healthy volunteers. J Clin Pharmacol. 1999;39:495–500. [PubMed] [Google Scholar]
- 25.Schwartz JI, Dunbar S, Yuan J, Li S, Gipson A, Rosko K, Johnson-Levonas AO, Lasseter KC, Addy C, Stoch AS, Wagner JA. Influence of taranabant, a cannabinoid-1 receptor inverse agonist, on pharmacokinetics and pharmacodynamics of warfarin. Adv Ther. 2008;25:1175–90. doi: 10.1007/s12325-008-0116-9. [DOI] [PubMed] [Google Scholar]
- 26.Wright DH, Herman GA, Maes A, Liu Q, Johnson-Levonas AO, Wagner JA. Multiple doses of sitagliptin, a selective DPP-4 inhibitor, do not meaningfully alter pharmacokinetics and pharmacodynamics of warfarin. J Clin Pharmacol. 2009;49:1157–67. doi: 10.1177/0091270009341653. [DOI] [PubMed] [Google Scholar]