

Abstract
AIM
Angiotensin II receptor blockers (ARBs) improve endothelial cell (EC)-dependent vasodilation in patients with hypertension through suppression of angiotensin II type 1 receptors but may have additional and differential effects on endothelial nitric oxide (NO) synthase (eNOS) function. To investigate this question, we tested the effects of various ARBs on NO release in ECs from multiple donors, including those with eNOS genetic variants linked to higher cardiovascular risk.
METHODS
The effects of ARBs (losartan, olmesartan, telmisartan, valsartan), at 1 µm, on NO release were measured with nanosensors in human umbilical vein ECs obtained from 18 donors. NO release was stimulated with calcium ionophore (1 µm) and its maximal concentration was correlated with eNOS variants. The eNOS variants were determined by a single nucleotide polymorphism in the promoter region (T-786C) and in the exon 7 (G894T), linked to changes in NO metabolism.
RESULTS
All of the ARBs caused an increase in NO release as compared with untreated samples (P < 0.01, n = 4–5 in all eNOS variants). However, maximal NO production was differentially influenced by eNOS genotype. Olmesartan increased maximal NO release by 30%, which was significantly greater (P < 0.01, n = 4–5 in all eNOS variants) than increases observed with other ARBs.
CONCLUSIONS
The ARBs differentially enhanced NO release in ECs in a manner influenced by eNOS single nucleotide polymorphisms. These findings provide new insights into the effects of ARBs on EC-dependent vasodilation and eNOS function.
Keywords: angiotensin receptor blockers, endothelium, nanosensors, nitric oxide, nitric oxide synthase
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Angiotensin II receptor blockers improve endothelial cell-dependent vasodilation in patients with hypertension through suppression of angiotensin II type 1 receptors but may have additional and differential effects on endothelial nitric oxide synthase (eNOS) function.
WHAT THIS STUDY ADDS
The key finding from this study is that angiotensin II receptor blockers (ARBs) differentially enhanced nitric oxide (NO) release in a manner influenced by certain genetic variants of eNOS. This finding provides new insights into the effects of ARBs on endothelial cell-dependent vasodilation and eNOS function that are of high importance in vascular medicine and clinical pharmacology.
Introduction
The vascular endothelium regulates vascular motion while providing a non-thrombogenic surface and macromolecular barrier between blood elements and the underlying vessel wall. Vascular endothelial cells (ECs) also play an important role in regulating vascular tone, largely through the constitutive release of nitric oxide (NO). Under pathophysiologic conditions, an imbalance occurs between the endothelial release of this powerful vasodilator and the systemic release of vasoconstrictive peptides, such as angiotensin II and endothelin, resulting in increased vascular resistance [1], [2]. Beyond abnormal vasodilation, the loss of NO bioavailability with endothelial dysfunction contributes to various atherogenic processes, including activation of the renin-angiotensin system, thrombus formation and inflammation [3].
In clinical studies, angiotensin II receptor blockers (ARBs) demonstrated a significant improvement in endothelium-dependent vasodilation in patients with hypertension as compared with placebo or other antihypertensive agents [4]–[7]. Losartan significantly improved endothelial function in patients with atherosclerosis and coronary heart disease (CHD) while candesartan improved forearm blood flow in patients with hypercholesterolaemia-associated endothelial dysfunction [8]–[10]. Comparative studies have shown similar effects of ARBs and angiotensin converting enzyme (ACE) inhibitors on endothelial function in patients with hypertension and CHD [9], [11]. The pharmacologic basis for enhanced NO release with ARB treatment is attributed to reduced activity of angiotensin II, similar to the mechanistic effects of ACE inhibitors, leading to anti-oxidant protection and increased NO bioavailability [9], [12]. Unlike ACE inhibitors, ARBs do not enhance concentrations of bradykinin, a stimulus for NO release; but they may enhance eNOS expression [3], [13]–[15].
This study was conducted to compare the effects of multiple ARBs (losartan, olmesartan, telmisartan, valsartan) on stimulated NO release from healthy human ECs with certain endothelial nitric oxide synthase (eNOS) genetic variants as determined by single nucleotide polymorphism (SNP) genotyping analysis. In addition to the normal eNOS genotype, we evaluated the effects of treatment in ECs consisting of two well-characterized eNOS polymorphisms, G894T, a SNP in the exon 7 of the human eNOS gene (located at 7q35-36), which results in a Glu298Asp substitution in the eNOS sequence and T–786C, a SNP at position −786, in the promoter region of the eNOS gene, which results in decreased expression of the eNOS enzyme. These eNOS polymorphisms have been linked, albeit with some inconsistencies, to changes in NO metabolites (nitrites, nitrates), arterial and EC function, as well as susceptibility to hypertension [16]–[21]. While ARBs improve vasodilation in patients with hypertension through suppression of angiotensin II, these agents may have additional effects on eNOS function. To understand these processes, we measured directly the effects of ARBs on NO release in ECs from donors with normal eNOS genotypes as well as those having eNOS polymorphisms linked to enhanced cardiovascular risk.
Methods
Subjects and cell cultures
Human umbilical vein endothelial cells (HUVECs) were isolated into primary cultures from Caucasian female donors (18 subjects) by Clonetics (San Diego, California) and purchased as proliferating cells. HUVECs pooled from the same 18 Caucasian donors were also purchased from Clonetics. All cell culture donors were healthy, with no pregnancy or prenatal complications. Cells were cultured in the recommended complete endothelial cell growth medium and maintained at 37°C in a 5% CO2 humidified incubator. Cells were supplied with fresh medium every other day and propagated using an enzymatic (trypsin) procedure, for a maximum of 16 population doublings.
Preparation of nanosensors for NO detection
Measurements of NO were carried out with an electrochemical nanosensor with a total diameter of 200–500 nm and length of 4–5 µm. Nanosensor design was based on previously developed chemically modified carbon-fibre technology [22], [23]. Each of the nanosensors was made by depositing a sensing material on the tip of a carbon fibre. We used polymeric nickel (II) tetrakis (3-methoxy-4-hydroxyphenyl) porphyrin for the NO sensor.
Measurement of NO
The confluent cells (4 to 5 × 105 cells/35 mm dish) were incubated with ARBs in endothelial basal medium for 6 h. The ARBs were dissolved in 18 µl of dimethy sulfoxide (DMSO) and further diluted in endothelial basal medium (1 ml) to obtain a final concentration of 1 µm. Further experiments were performed to rule out the influence of the small concentration of DMSO on endothelial NO release.
NO release was stimulated by calcium ionophore (CaI), A23187, at a concentration of 1 µm, injected with a nanoinjector that was positioned by a computer-controlled micromanipulator. Four to five measurements of NO were performed per group.
An NO nanosensor with a platinum wire (0.1 mm) counter electrode and saturated calomel reference electrode was applied. Differential pulse voltammetry (DPV) and amperometry were performed using a computer-based Gamry VFP600 multichannel potentiostat. DPV was used to measure the basal NO concentrations and amperometry was used to measure changes in NO concentrations from its basal level with time. The DPV current at the peak potential characteristic for NO oxidation (0.70 V) was directly proportional to the concentrations of this compound in the immediate vicinity of the sensor. Linear calibration curves (current vs. concentration) were constructed for each sensor from 10 nm to 2 µm before and after measurements with aliquots of NO standard solution. The detection limit of the sensors was 1 nm. At a constant distance of the sensors from the surface of an endothelial cell, the reproducibility of measurements was relatively high (5–12%). The position of the nanosensors (x, y, and z coordinates) vs. the ECs was established with the help of a computer controlled micromanipulator. The sensor was moved horizontally (x, y coordinates) and positioned above the surface of a randomly chosen single EC.
Determination of eNOS variants
Maximal generation of NO was correlated with eNOS variants (G894T and T–786C) linked to changes in NO metabolism. These genotypes were determined by polymerase chain reaction (PCR) using a SNP genotyping assay kit from Applied Biosystems Inc. (Foster City, California). Between three and six Caucasian donors with specific eNOS variant combinations were assigned to each group (n = 3–6).
Statistical analyses
In order to control intra-experimental variations, measurements of endothelial NO were performed using a single cell randomly selected from 4–5 different cell culture dishes. The significance of differences between results from independent experimental conditions was tested using either the two-tailed Student's t-test (measurements of NO from various treatments) or one-way analysis of variance (anova) with Student–Newman–Keuls multiple comparisons post hoc analysis. Data were presented as mean ± standard deviation (SD). Mean values were considered significantly different at P < 0.05.
Results
NO release in human endothelial cells with different eNOS gene polymorphisms
We measured NO release from ECs as a function of the G894T and T–786C genotypes. As shown in Figure 1, there was no significant difference in NO release among these variants following CaI stimulation. The average level of NO release (585 ± 34 nm) was consistent with previous studies in ECs from Caucasian donors treated with a similar receptor-independent stimulus like CaI [24].
Figure 1.
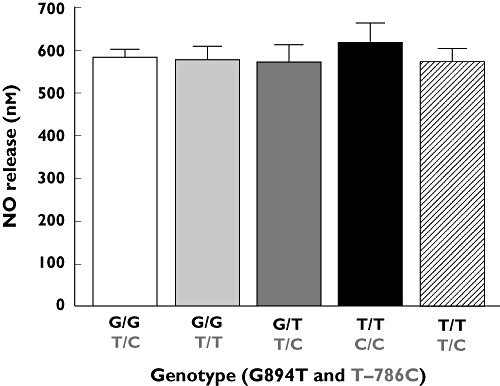
Release of NO from a single HUVEC cell with eNOS gene polymorphisms at positions 894 (G894T) and/or −786 (T–786C). Values are mean ± SD (n = 4–5 measurements of NO per donor, 3–6 donors per group)
Effect of ARBs on NO release from human endothelial cells with different eNOS gene polymorphisms
We compared NO release from HUVECs, pooled from all 18 Caucasian donors carrying all available eNOS variants, in the absence and presence (6 h) of various ARBs (losartan, olmesartan, telmisartan, valsartan) at a concentration of 1 µm (Figure 2). Each ARB caused an increase in NO release as compared with untreated samples (P < 0.01, n = 4–5 measurements of NO per group). The greatest effect was observed with olmesartan, which increased NO release by 30% (from 585 ± 34 nm to 768 ± 98 nm) and was significantly greater (P < 0.01, n = 4–5 measurements of NO per group) than any increases observed with the other ARBs. Olmesartan also increased NO release in most eNOS variant combinations (homozygous and heterozygous) as compared with untreated cells (Figure 3). The most apparent difference in potency between olmesartan and the other ARBs was observed in ECs homozygous for both the 894T and −786C alleles.
Figure 2.
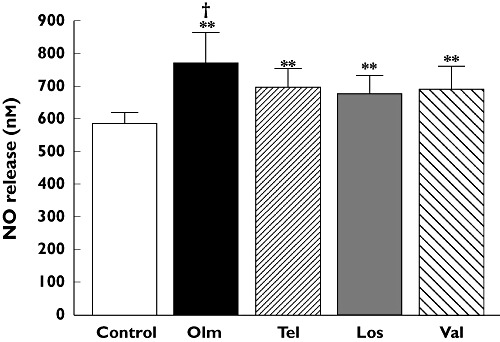
Comparative effects of losartan (Los), olmesartan (Olm), telmisartan (Tel), valsartan (Val) on CaI (1 µm)-induced NO release from HUVECs (pooled from all 18 Caucasian donors carrying all available eNOS variant combinations) following treatment for 6 h at 1 µm. Values are mean ± SD (n = 4–5 measurements of NO per group). **P < 0.01 vs. control; †P < 0.01 vs. all other treatments (Student–Newman–Keuls multiple comparisons test; overall anova: P < 0.0001, F = 17.336)
Figure 3.
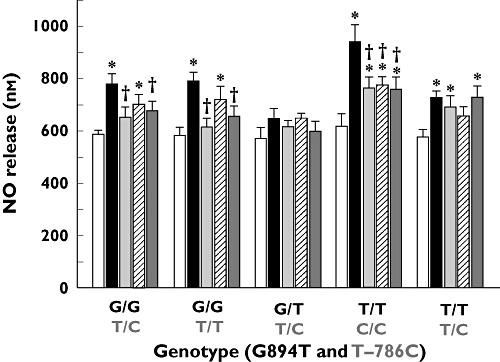
Effect of ARB treatment (1 µm, 6 h treatment) on NO release from HUVECs obtained with eNOS gene polymorphisms at positions 894 (G894T) and/or −786 (T–786C). Values are mean ± SD (n = 4–5 measurements of NO per donor, 3–6 donors per group). *P < 0.05 vs. cognate control; †P < 0.01 vs. cognate olmesartan treatment (Student–Newman–Keuls multiple comparisons test; overall anova: P < 0.0001, F = 18.109). Control ( ); Tel (
); Tel ( ); Val (
); Val ( ); Olm (
); Olm ( ); Los (
); Los ( )
)
Discussion
The key finding from this study is that treatment of human ECs with ARBs differentially enhanced NO release in a manner influenced by certain genetic variants of eNOS. All of the ARBs tested (losartan, olmesartan, telmisartan, valsartan) caused an increased in endothelial NO release compared with untreated cells. Olmesartan had the greatest effect on NO release as it enhanced NO concentrations by 30%. The eNOS polymorphisms were not associated with differences in stimulated NO release except in the presence of the different ARBs. Olmesartan increased NO release in the various eNOS genotypes with the most apparent difference in ECs from donors homozygous for both G894T and T–786C single nucleotide mutations. This finding indicates agent-specific effects of ARBs on eNOS function that are influenced, in turn, by single nucleotide substitutions that are linked to changes in NO metabolism and increased cardiovascular risk, including hypertension [16]–[21]. The explanation for the relative contribution of the different variants to the responses of the ARBs is complex and may require further investigation.
The eNOS genotype may be linked to changes in the homeostatic balance between concentrations of NO and reactive oxygen species, such as superoxide anion, that regulate the cellular redox state essential for normal endothelial function. Endothelial dysfunction and impairment in the capacity of the vessels to dilate are directly linked to enhanced catabolism of NO by superoxide anions [3], [13], [14]. Loss of normal NO bioavailability, coupled with increased oxidative stress, is causally related to various diseases, including atherosclerosis, hypertension and diabetes mellitus [25], [26]. Reductions in NO release have also been associated with eNOS uncoupling, an effect that may be reversed with certain therapies [3], [13], [14]. The differential effects of the ARBs on NO release with these eNOS variants may therefore be linked to changes in the homeostatic balance between NO and free radical generation in these cells.
Tissue angiotensin and its products have a direct role in the pathophysiology of vascular disease through enhanced oxidative stress, inflammation and other mechanisms [27]. Like ACE inhibitors, ARBs suppress the activity of angiotensin II, leading to antioxidant protection that promotes NO bioavailability [9]. Specifically, inhibitors of the renin-angiotensin system reduce NAD(P)H activity, which acts as an important enzymatic source of intracellular superoxide [9], [12]. Although ARBs do not affect bradykinin concentrations, these agents have been shown in bovine pulmonary artery endothelial cells to increase significantly levels of eNOS [15]. Thus, ARBs may promote NO release through the up-regulation of eNOS protein as well as antioxidant protection. Clinical studies have demonstrated that ARBs improve endothelium-dependent vasodilation in patients with hypertension, compared with placebo or comparator agents [4]–[7]. In comparison with ACE inhibitors, ARBs have similar effects on endothelial function in patients with hypertension and with CHD [9], [11].
ARBs inhibit the vasoconstrictor effects of angiotensin II by selectively and reversibly blocking its binding to the AT1 receptor. The basis for observed differences in the endothelial effects of olmesartan may be related to various factors, including its chemical structure and receptor affinity. The covalent structure of olmesartan includes a hydroxyl group and an intermediate-sized alkyl substituent (isopropyl) at the 4-position, which undergoes hydrogen bonding with the carboxylate anion at the 5-position of the imidazole ring, allowing the isopropyl substituent to interact with a hydrophobic region on the AT1 receptor. The other ARBs also interact with a hydrophobic region of the AT1 receptor, so this factor alone is not the basis for uniqueness. The order of potency for the IC50 values (nm) has been reported as follow: olmesartan (0.78), telmisartan (0.88), valsartan (3.1) and losartan (11) [28]. These data indicate that the receptor binding characteristics for olmesartan to the human AT1 receptor is slightly more potent than telmisartan and more potent when compared with the other ARBs, especially losartan. However, the relative differences in the potency of these agents for the AT1 receptor alone failed to predict their complex effects on NO release among the eNOS variants that were tested. Olmesartan may also have a differential effect on eNOS expression, although this mechanism is a less likely contributor to changes observed in this study, given the relatively short treatment periods. The presence of the hydroxyl group may contribute to free radical scavenging properties for olmesartan, including chain-breaking antioxidant activity. In particular, the hydroxyl group may provide proton donation and electron stabilization properties that reduce free radical propagation resulting in loss of NO bioavailability through formation of peroxynitrite ion. Also, it is possible that olmesartan attenuates the activation of NADPH oxidase and decreases superoxide generation in the absence of angiotensin II, leading to an increase in NO bioavailability [29]. These potential effects need to be explored further.
One of the most intriguing findings from this study was the ARB-specific changes in NO release among the various eNOS variants. On the other hand, there were no genotype-specific differences in NO release following stimulation with CaI. This may be due, in part, to the type of stimulus used in these studies. By using a CaI, we fully activated available cellular eNOS in a receptor-independent manner. If these variants influence receptor-dependent pathways related to eNOS activation, then these effects may be missed with the use of the CaI.
This is the first study to compare the direct effects of various ARBs on the NO release capacity of human endothelial cells with different eNOS variants. Additional studies are needed to elucidate the basis for ARB effects on eNOS function, including eNOS protein and co-factor levels, as well as changes in the redox state of the cell, all of which may be linked to their specific chemical structures. These studies also need to be extended to whole animals and human clinical evaluations with well-characterized genotype analyses to understand the relationships between selective AT1 receptor inhibition and endothelial-dependent NO release in patients with cardiovascular risk.
An important limitation of this study was the number of patients with any particular eNOS variant. However, the reproducibility in the genotype-dependent response of these cells to stimulation indicates that this model serves as an important predictor of NO release as a function of treatment with these agents. It will be important to examine the effects of these pharmacologic agents on NO metabolism in a larger numbers of patients having these and other possible eNOS genotypes.
Acknowledgments
This study was supported in part by an investigator-initiated research grant from Daiichi Sankyo to Dr Mason. Also, support from Marvin and Ann Dilley White Endowed Chair (Dr Malinski) is acknowledged.
Competing Interests
Dr Mason has received independent research grants from Astra Zeneca, Forest Laboratories, Pfizer, Sanofi-Aventis and Daiichi Sankyo. All other authors have no conflicts of interest to disclose.
REFERENCES
- 1.Ignarro LJ, Buga GM, Wood KS, Byrnes RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA. 1987;84:9265–9. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rees DD, Palmer RM, Moncada S. The role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci USA. 1989;86:3375–8. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kojda G, Harrison DG. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res. 1999;43:562–71. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 4.Ghiadoni L, Virdis A, Magagna A, Taddei S, Salvetti A. Effect of the angiotensin II type 1 receptor blocker candesartan on endothelial function in patients with essential hypertension. Hypertension. 2000;35(1 Pt 2):501–6. doi: 10.1161/01.hyp.35.1.501. [DOI] [PubMed] [Google Scholar]
- 5.Schiffrin EL, Park JB, Intengan HD, Touyz RM. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation. 2000;101:1653–9. doi: 10.1161/01.cir.101.14.1653. [DOI] [PubMed] [Google Scholar]
- 6.Schiffrin EL, Park JB, Pu Q. Effect of crossing over hypertensive patients from a beta-blocker to an angiotensin receptor antagonist on resistance artery structure and on endothelial function. J Hypertens. 2002;20:71–8. doi: 10.1097/00004872-200201000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Klingbeil AU, John S, Schneider MP, Jacobi J, Handrock R, Schmieder RE. Effect of AT1 receptor blockade on endothelial function in essential hypertension. Am J Hypertens. 2003;16:123–8. doi: 10.1016/s0895-7061(02)03154-0. [DOI] [PubMed] [Google Scholar]
- 8.Prasad A, Tupas-Habib T, Schenke WH, Mincemoyer R, Panza JA, Waclawin MA, Ellahham S, Quyyumi AA. Acute and chronic angiotensin-1 receptor antagonism reverses endothelial dysfunction in atherosclerosis. Circulation. 2000;101:2349–54. doi: 10.1161/01.cir.101.20.2349. [DOI] [PubMed] [Google Scholar]
- 9.Hornig B, Landmesser U, Kohler C, Ahlersmann D, Spiekermann S, Christoph A, Tatge H, Drexler H. Comparative effect of ACE inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dismutase. Circulation. 2001;103:799–805. doi: 10.1161/01.cir.103.6.799. [DOI] [PubMed] [Google Scholar]
- 10.Wassmann S, Hilgers S, Laufs U, Bohm M, Nickenig G. Angiotensin II type 1 receptor antagonism improves hypercholesterolemia-associated endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1208–12. doi: 10.1161/01.atv.0000022847.38083.b6. [DOI] [PubMed] [Google Scholar]
- 11.Leu HB, Charng MJ, Ding PY. A double blind randomized trial to compare the effects of eprosartan and enalapril on blood pressure, platelets, and endothelium function in patients with essential hypertension. Jpn Heart J. 2004;45:623–35. doi: 10.1536/jhj.45.623. [DOI] [PubMed] [Google Scholar]
- 12.Nickenig G, Harrison DG. The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis. Circulation. 2002;105:393–6. doi: 10.1161/hc0302.102618. [DOI] [PubMed] [Google Scholar]
- 13.Drexler H, Hornig B. Endothelial dysfunction in human disease. J Mol Cell Cardiol. 1999;31:51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- 14.Mason RP, Cockcroft JR. Targeting nitric oxide with drug therapy. J Clin Hypertens. 2006;8(Suppl. 4):S40–52. doi: 10.1111/j.1524-6175.2006.06041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thai H, Wollmuth J, Goldman S, Gaballa M. Angiotensin subtype 1 receptor (AT1) blockade improves vasorelaxation in heart failure by up-regulation of endothelial nitric-oxide synthase via activation of the AT2 receptor. J Pharmacol Exp Ther. 2003;307:1171–8. doi: 10.1124/jpet.103.054916. [DOI] [PubMed] [Google Scholar]
- 16.Tsukada T, Yokoyama K, Arai T, Takemoto F, Hara S, Yamada A, Kawaguchi Y, Hosoya T, Igari J. Evidence of association of the ecNOS gene polymorphism with plasma NO metabolite levels in humans. Biochem Biophys Res Commun. 1998;245:190–3. doi: 10.1006/bbrc.1998.8267. [DOI] [PubMed] [Google Scholar]
- 17.Wang XL, Mahaney MC, Sim AS, Wang J, Wang J, Blangero J, Almasy L, Badenhop RB, Wilcken DE. Genetic contribution of the endothelial constitutive nitric oxide synthase gene to plasma nitric oxide levels. Arterioscler Thromb Vasc Biol. 1997;17:3147–53. doi: 10.1161/01.atv.17.11.3147. [DOI] [PubMed] [Google Scholar]
- 18.Bonnardeaux A, Nadaud S, Charru A, Jeunemaitre X, Corvol P, Soubrier F. Lack of evidence for linkage of the endothelial cell nitric oxide synthase gene to essential hypertension. Circulation. 1995;91:96–102. doi: 10.1161/01.cir.91.1.96. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto Y, Saito Y, Kajiyama N, Yoshimura M, Shimasaki Y, Nakayama M, Kamitani S, Harada M, Ishikawa M, Kuwahara K, Ogawa E, Hamanaka I, Takahashi N, Kaneshige T, Teraoka H, Akamizu T, Azuma N, Yoshima Y, Yoshima T, Itoh H, Masuda I, Yasue H, Nakao K. Endothelial nitric oxide synthase gene is positively associated with essential hypertension. Hypertension. 1998;32:3–8. doi: 10.1161/01.hyp.32.1.3. [DOI] [PubMed] [Google Scholar]
- 20.Li R, Lyn D, Lapu-Bula R, Oduwole A, Igho-Pemu P, Lankford B, Morgan J, Nkemdechi S, Liu G, Pack C, Silvestrov N, von Deutsch DA, Song Q, Abukhalaf IK, Ofili E. Relation of endothelial nitric oxide synthase gene to plasma nitric oxide level, endothelial function, and blood pressure in African Americans. Am J Hypertens. 2004;17:560–7. doi: 10.1016/j.amjhyper.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Srinivasan SR, Bond MG, Tang R, Urbina EM, Li S, Boerwinkle E, Berenson GS. Nitric oxide synthase gene polymorphism (G894T) influences arterial stiffness in adults: the Bogalusa Heart Study. Am J Hypertens. 2004;17:553–9. doi: 10.1016/j.amjhyper.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 22.Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676–8. doi: 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- 23.Lvovich V, Scheeline A. Amperometric sensors for simultaneous superoxide and hydrogen peroxide detection. Anal Chem. 1997;69:454–62. doi: 10.1021/ac9606261. [DOI] [PubMed] [Google Scholar]
- 24.Mason RP, Kalinowski L, Jacob RF, Jacoby AM, Malinski T. Nebivolol reduces nitroxidative stress and restores nitric oxide bioavailability in endothelium of black Americans. Circulation. 2005;112:3795–801. doi: 10.1161/CIRCULATIONAHA.105.556233. [DOI] [PubMed] [Google Scholar]
- 25.Drexler H, Hayoz D, Munzel T, Hornig B, Just H, Brunner HR, Zelis R. Endothelial function in chronic congestive heart failure. Am J Cardiol. 1992;69:1596–601. doi: 10.1016/0002-9149(92)90710-g. [DOI] [PubMed] [Google Scholar]
- 26.Gilligan DM, Panza JA, Kilcoyne CM, Waclawiw MS, Casion PR, Quyyumi AA. Contribution of endothelium-derived nitric oxide to exercise-induced vasodilation. Circulation. 1994;90:2853–8. doi: 10.1161/01.cir.90.6.2853. [DOI] [PubMed] [Google Scholar]
- 27.Dzau VJ. Theodore Cooper Lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–52. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- 28.Mizuno M, Sada T, Ikeda M, Fukuda N, Miyamoto M, Yanagisawa H, Koike H. Pharmacology of CS-866, a novel nonpeptide angiotensin II receptor antagonist. Eur J Pharmacol. 1995;285:181–8. doi: 10.1016/0014-2999(95)00401-6. [DOI] [PubMed] [Google Scholar]
- 29.Yatabe J, Sanada H, Yatabe MS, Hashimoto S, Yoneda M, Felder RA, Jose PA, Watanabe T. Angiotensin II type I receptor blocker attenuates the activation of ERK and NADPH oxidase by mechanical strain in mesangial cells in the absence of angiotension II. Am J Physiol Renal Physiol. 2009;296:F1050–62. doi: 10.1152/ajprenal.00580.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]