


Abstract
Ancestral sequence reconstruction is essential to a variety of evolutionary studies. Here, we present the FastML web server, a user-friendly tool for the reconstruction of ancestral sequences. FastML implements various novel features that differentiate it from existing tools: (i) FastML uses an indel-coding method, in which each gap, possibly spanning multiples sites, is coded as binary data. FastML then reconstructs ancestral indel states assuming a continuous time Markov process. FastML provides the most likely ancestral sequences, integrating both indels and characters; (ii) FastML accounts for uncertainty in ancestral states: it provides not only the posterior probabilities for each character and indel at each sequence position, but also a sample of ancestral sequences from this posterior distribution, and a list of the k-most likely ancestral sequences; (iii) FastML implements a large array of evolutionary models, which makes it generic and applicable for nucleotide, protein and codon sequences; and (iv) a graphical representation of the results is provided, including, for example, a graphical logo of the inferred ancestral sequences. The utility of FastML is demonstrated by reconstructing ancestral sequences of the Env protein from various HIV-1 subtypes. FastML is freely available for all academic users and is available online at http://fastml.tau.ac.il/.
Introduction
Ancestral sequence reconstruction (ASR) methods require as input both a multiple sequence alignment (MSA) of existing sequences and a corresponding phylogenetic tree (either provided or computed from the MSA). They output a statistical inference of the ancestral sequence at any internal node of the phylogenetic tree. ASR is being used in a steadily increasing number of evolutionary studies [reviewed in (1)]. For example, in protein ‘resurrection’ studies, ancestral sequences are synthesized and characterized, thus providing the ability to test evolutionary hypotheses regarding protein evolution. Such an approach was successfully applied to study the evolution of the ancestral archosaur visual pigment rhodopsin (2) and the evolution of the steroid receptor (3) to name a few. ASR was also applied in various other contexts, including protein engineering (4), the study of HIV evolution (5) and the study of variation in DNA turnover due to indels and substitutions among eutherian mammalian lineages (6).
There are two main paradigms for ASR: maximum parsimony (MP) and probabilistic-based reconstruction. The latter includes maximum likelihood (ML) and Bayesian reconstructions, both of which were shown to outperform MP [e.g. (7)]. One advantage of probabilistic-based approaches is that they also provide an estimate of the confidence in each inferred ancestral character, most often expressed as its posterior probability given the data. While MP reconstruction has a time complexity linear in the number of sequences analyzed, efficient algorithms have also been developed for different types of ML-based reconstructions [reviewed in (8)], with a time complexity that is linear with the number of sequence for most algorithms (9,10) and exponential in the worst case scenario for one variant (joint reconstruction with among site rate variation). Notably, all algorithms can be efficiently used for large data sets in most practical cases (11).
Various tools for ASR exist. Most of these tools apply the now standard dynamic programming algorithms to find the most probable sequence at a specific node (9,10). However, they differ in various aspects, among which the most important ones are: (i) the way gaps in the alignment are treated; (ii) the flexibility with respect to the allowed evolutionary models and the number of optional parameters; (iii) the generation of near-optimal solutions (i.e. for a given node of the tree, plausible sequences that only marginally differ from the single most likely ancestral sequence); (iv) the graphical user interface (GUI) and the ease of use. Here we present the FastML web server, which aims to improve upon existing tools with respect to all these aspects, as detailed below. FastML is freely available for use at http://fastml.tau.ac.il/ with no requirement of log-in.
In ASR algorithms, gaps are usually treated as unknown characters (10,12) or as an additional character (13). These approaches are problematic since they ignore dependencies among sites. Furthermore, treating gaps as unknown characters results in ancestral sequences that are longer than all present day sequences, which is unrealistic. One possible way to overcome this limitation is to introduce a heuristic approach that maps indels to a phylogeny so that they maximize some ad hoc scoring function (6). The GASP program (14) also computes probabilities of gaps at internal nodes. However, it uses an unrealistic model in which gaps in adjacent MSA columns are assumed to be independent. Furthermore, the probabilities of gaps at internal nodes are not computed based on a continuous time Markov model, which is used for reconstructing substitution events. As described below, in FastML, we developed a different approach in which we first apply an indel-coding methodology that provides for each indel a presence (‘1’) or absence (‘0’) state in the input sequences. FastML then applies an ML-based reconstruction algorithm for binary data to determine the probability of gap character state in the ancestral sequences.
For protein-coding genes, amino acid-based reconstruction rather than codon-based reconstruction is usually applied [e.g. (2,14)]. This stems from two main reasons: (i) the availability of various empirical amino-acid substitution matrices that were inferred from a large collection of protein sequences; (ii) for more diverged sequences, the synonymous substitutions are often saturated. However, these models ignore the codon structure of coding sequences, and thus they may be less accurate compared to codon models that explicitly account for the chosen codon at each amino acid site. Furthermore, reconstructing ancestral regulatory regions are expected to become more common with the increased availability of fully sequenced genomes. Thus, FastML allows reconstructing ancestral sequences using nucleotide substitution models, amino acid replacement models and codon models.
Simulation studies have shown that at each specific position the most likely ancestral state has a high probability to reflect the ‘true’ one [e.g. (15)]. However, this high accuracy reflects an average over all sites, many of which are conserved sites in which accurate reconstruction is trivial. In practice, the probability of the ‘true’ ancestral sequence to be identical to the reconstructed one across the entire sequence is rather small due to several highly variable sites. Furthermore, it was shown that the most likely reconstructed ancestor might be biased: it tends to favor common amino acids in a particular position over rare variants (15). To account for this problem, most programs not only provide the most likely character at each site, but also give the posterior probabilities of each ancestral character as output. However, correct usage of these probabilities in studies utilizing ancestral sequences is not obvious. In the FastML web server, we do not only report these site-specific probabilities, but additionally we provide the set of the k most likely ancestral sequences at each node. Since ancestral sequences are often used to infer protein variants that are more stable than all current day sequences (15), this set provides a list from which protein engineers may choose to synthesize highly stable proteins. FastML also provides, for each node a list of ancestral proteins sampled from the posterior distribution. In simulations, this set was shown to better represent the amino-acid composition and biochemical properties of the ‘true’ ancestral sequence compared with the most likely ancestral sequences (15). Details on the generation of alternative ancestral states are given in the OVERVIEW section of the web server.
Finally, the web server is tailored for both novice and advanced users. The novice user is provided with a user-friendly interface that requires only an MSA as input. The server further provides a rich graphical output that includes: (i) projection of the ancestral sequences onto the phylogeny; (ii) color-scaled projection of the reconstruction probabilities at the internal nodes of the tree; and (iii) a graphical logo of all possible alternative reconstructions.
MATERIALS AND METHODS
Given an MSA and a phylogenetic tree, the ancestral reconstruction process can be divided into two parts: character reconstruction and indel reconstruction. The results of both reconstructions are integrated to provide the most probable ancestral sequences in each node of the phylogeny. Figure 1 shows a flowchart of the ASR procedure. The minimal input of the web server is an MSA of nucleotide, protein or codon sequences. ASR depends on a tree, which is computed from the MSA using either the neighbor joining algorithm or using the ML tree search procedure as implemented in RAxML (16). Users may also provide their own tree as input. The FastML server then runs two algorithms that together reconstruct the ancestral sequences. The first infers for each indel character whether or not a gap is present in the ancestral sequence. The second algorithm then infers the most likely character states only in the non-gapped positions of the ancestral sequences. A short description of the methodology is provided below and a more detailed one is available at http://fastml.tau.ac.il/ under the OVERVIEW section.
Figure 1.
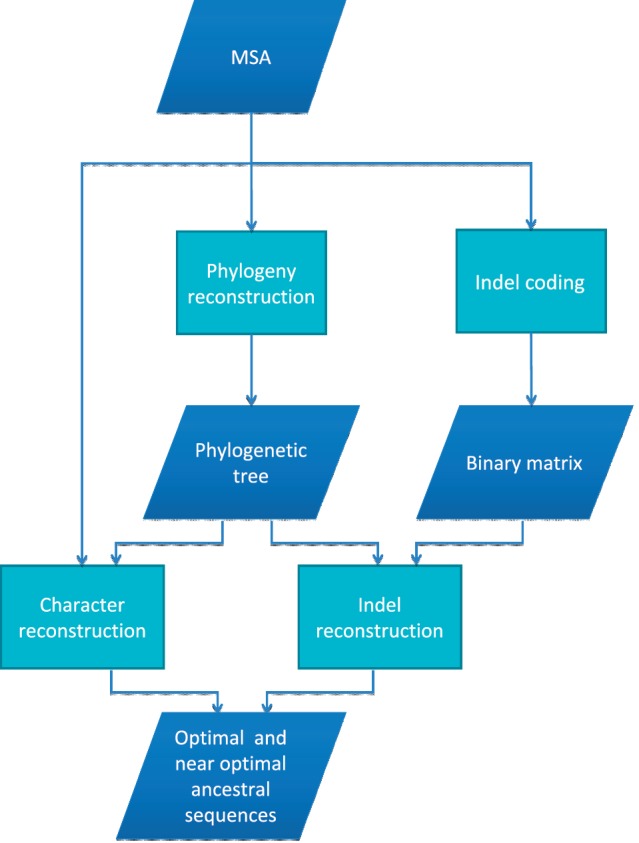
A schematic flow chart of the FastML web server.
To account for the dependence of insertions and deletions among sites, the FastML server first employs the simple indel coding scheme (17) to code all indels in the data as binary (presence\absence) characters, each of which may represent a gap of multiple sites. The binary data matrix is then provided as input to an ML-based ancestral indel reconstruction algorithm. The evolutionary model for this indel reconstruction step allows for variable rates of insertions and deletions among indel sites, similar to the model that we have previously developed for phyletic patterns (18,19). The output of this step is the posterior probability for each indel site at each ancestral node of the phylogeny. Most likely character states in the ancestral nodes are reported only in positions that are inferred to be non-gapped with a probability ≥0.5. Alternatively, users can select to reconstruct the ancestral indel states based on the MP approach (20).
The previously described FastML algorithms are used to infer the most likely ancestral sequences (10,11). Both joint and marginal reconstructions are implemented. Briefly, in joint reconstruction, the most likely set of ancestral states at all the internal nodes is inferred, while in marginal reconstruction the most likely sequence at a specific internal node is inferred, averaging over all possible ancestral states at all other nodes. Rate variation among sites is accounted for by assuming that the rate at each site is sampled from a discrete gamma distribution (21). The user is allowed to choose the evolutionary model that best fits the data analyzed. For amino acids, the server implements the Dayhoff (22), JTT (23), WAG (24), LG (25), mtREV (26) and cpREV (27) replacement matrices; for nucleotides, the JC (28) and the HKY (29) substitution matrices are implemented. For codon characters, the server offers the M5 (30), the empirical codon matrix (31) and the MEC (32) models.
Running time depends on the number of sequences and their length, the evolutionary model, the proportion of gaps and the reconstruction algorithm. Codon models are the most time consuming and nucleotide models are the least. Additionally, accounting for among site rate variation is significantly more complex for the joint reconstruction compared to the marginal reconstruction (11). To aid users with estimating the running time for their data sets, the OVERVIEW section of the server includes detailed information regarding the average running time on simulated data sets of various sizes, using different evolutionary models. In addition, an estimation of the running time is given for each run.
FastML outputs
FastML provides the following outputs:
the posterior probability of each character (or indel) for each site at each ancestral node;
MSAs augmented with the reconstructed ancestral sequences: one MSA according to the joint reconstruction and a second according to the marginal reconstruction;
the reconstructed phylogenetic tree;
the k most probable ancestral sequences for each ancestral node (where k is defined by the user);
a set of l sequences sampled according to the posterior probabilities for each site and each node (where l is defined by the user);
a graphical visualization of the most probable ASR at each node colored according to the posterior probabilities. A graphical logo representing the posterior probabilities of each possible character for each ancestral node is further provided using WebLogo (33); and
a projection of the ancestral sequences onto the phylogeny. Using Jalview (34), the user can view the ASR at each internal node. Furthermore, the user can download the ancestral sequences of specific nodes or the sequences of an entire subtree.
CASE STUDY
Reconstructing the ancestral sequences of HIV-1 is a challenging task due to its fast rate of evolution. Nevertheless, ASR was suggested to be of great value to HIV-1 vaccine design that aims to elicit an immune response against a broad spectrum of contemporary viral strains [e.g. (35)]. Specifically, the envelope protein (Env) exhibits an extraordinary diversity (up to 35% diversity among different HIV-1 subtypes), which is attributed to mutational escape of the virus from the host immune system. The viral high mutation rate is also responsible for the ability of the virus to acquire resistance to drug treatments and is also a major obstacle toward developing an efficient vaccine.
Here, we illustrate the ability of FastML to reconstruct ancestral Env sequences. We run FastML on a sample of HIV-1 group M sequences from subtypes B and C taken from a previous study (36). Our analysis is focused on the marginal reconstruction of the ancestral sequences of subtype C, which is the most prevalent subtype and accounts for nearly half of all infections globally, and subtype B, which is predominant in the western world and accounts for ∼12% of global infections. Sequences were aligned using MAFFT (37). The alignment, running parameters and the results are provided in the web server’s Gallery. Several differences were found between clade B and clade C ancestral sequences, including both different character and indel assignments. Interestingly, some sites were reconstructed with high confidence in subtype C and low confidence in subtype B, and vice versa. Among these sites is position 592 in the MSA, which corresponds to position 414 of gp120, a derived protein of Env. This position is involved in the binding of the co-receptor CCR5. FastML inferred that this site in the ancestral of subtype C was threonine with a high posterior probability (0.997), while the reconstruction of the ancestor of subtype B is arginine with a much lower posterior probability (0.628 only). The different reconstructions are visually presented in Figure 2. The difference in the posterior probability between the ancestors of these two clades in this position may be explained by a previous analysis that suggested that the intensity of selection forces on this position is not constant among the various HIV-1 lineages (36). Specifically, this position is highly conserved in subtype C but is variable in subtype B, which is directly reflected in the posterior probabilities. We further used FastML to provide the 100 most likely ancestral sequences of the ancestral of subtype C. At the abovementioned site, threonine is always inferred, which is in agreement with its high posterior probability. Notably, the difference in log-likelihood between the most likely ancestral sequence at this node and the 100th most likely sequence is only 0.141, indicating that both sequences are almost as likely to reflect the ‘true’ ancestral sequence.
Figure 2.

An example of the FastML output. Graphical representation of the marginal posterior probabilities of the reconstruction of HIV-1 Env ancestral sequences of (A) subtype B ancestor and (B) subtype C ancestor. A red rectangle highlights the different in the reconstruction of position 592 in the MSA, as discussed in the text.
FUNDING
An Israel Science Foundation [878/09]; Edmond J. Safra Bioinformatics Center at Tel-Aviv University (fellowship to O.P., O.C. and H.A.). Funding for open access charge: Israel Science Foundation.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors wish to thank Michael Peeri and Gershon Celniker for their help in developing the web server.
REFERENCES
- 1.Liberles DA. Ancestral Sequence Reconstruction. Oxford: Oxford University Press; 2007. [Google Scholar]
- 2.Chang BS, Jonsson K, Kazmi MA, Donoghue MJ, Sakmar TP. Recreating a functional ancestral archosaur visual pigment. Mol. Biol. Evol. 2002;19:1483–1489. doi: 10.1093/oxfordjournals.molbev.a004211. [DOI] [PubMed] [Google Scholar]
- 3.Thornton JW, Need E, Crews D. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science. 2003;301:1714–1717. doi: 10.1126/science.1086185. [DOI] [PubMed] [Google Scholar]
- 4.Cole MF, Gaucher EA. Utilizing natural diversity to evolve protein function: applications towards thermostability. Curr. Opin. Chem. Biol. 2011;15:399–406. doi: 10.1016/j.cbpa.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaschen B, Taylor J, Yusim K, Foley B, Gao F, Lang D, Novitsky V, Haynes B, Hahn BH, Bhattacharya T, et al. Diversity considerations in HIV-1 vaccine selection. Science. 2002;296:2354–2360. doi: 10.1126/science.1070441. [DOI] [PubMed] [Google Scholar]
- 6.Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Res. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnan NM, Seligmann H, Stewart CB, De Koning AP, Pollock DD. Ancestral sequence reconstruction in primate mitochondrial DNA: compositional bias and effect on functional inference. Mol. Biol. Evol. 2004;21:1871–1883. doi: 10.1093/molbev/msh198. [DOI] [PubMed] [Google Scholar]
- 8.Pupko T, Doron-Faigenboim A, Liberles D, Cannarozzi G. In: Ancestral sequence reconstruction. Liberles D, editor. Oxford: Oxford University Press; 2007. [Google Scholar]
- 9.Koshi JM, Goldstein RA. Probabilistic reconstruction of ancestral protein sequences. J. Mol. Evol. 1996;42:313–320. doi: 10.1007/BF02198858. [DOI] [PubMed] [Google Scholar]
- 10.Pupko T, Pe'er I, Shamir R, Graur D. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol. Biol. Evol. 2000;17:890–896. doi: 10.1093/oxfordjournals.molbev.a026369. [DOI] [PubMed] [Google Scholar]
- 11.Pupko T, Pe'er I, Hasegawa M, Graur D, Friedman N. A branch-and-bound algorithm for the inference of ancestral amino-acid sequences when the replacement rate varies among sites: application to the evolution of five gene families. Bioinformatics. 2002;18:1116–1123. doi: 10.1093/bioinformatics/18.8.1116. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z, Kumar S, Nei M. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics. 1995;141:1641–1650. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menzel P, Stadler PF, Gorodkin J. maxAlike: maximum likelihood-based sequence reconstruction with application to improved primer design for unknown sequences. Bioinformatics. 2011;27:317–325. doi: 10.1093/bioinformatics/btq651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards RJ, Shields DC. GASP: gapped ancestral sequence prediction for proteins. BMC Bioinformatics. 2004;5:123. doi: 10.1186/1471-2105-5-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams PD, Pollock DD, Blackburne BP, Goldstein RA. Assessing the accuracy of ancestral protein reconstruction methods. PLoS Comput. Biol. 2006;2:e69. doi: 10.1371/journal.pcbi.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- 17.Simmons MP, Ochoterena H. Gaps as characters in sequence-based phylogenetic analyses. Syst. Biol. 2000;49:369–381. [PubMed] [Google Scholar]
- 18.Cohen O, Rubinstein ND, Stern A, Gophna U, Pupko T. A likelihood framework to analyse phyletic patterns. Philos. Trans. Roy. Soc. Lond. B. Biol. Sci. 2008;363:3903–3911. doi: 10.1098/rstb.2008.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen O, Pupko T. Inference of gain and loss events from phyletic patterns using stochastic mapping and maximum parsimony—a simulation study. Genome Biol. Evol. 2011;3:1265–1275. doi: 10.1093/gbe/evr101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sankoff D. Minimal mutation trees of sequences. Siam. J. Appl. Math. 1975;28:35–42. [Google Scholar]
- 21.Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- 22.Dayhoff M, Schwartz R, Orcutt B. A model of evolutionary change in proteins. In: Dayhoff M, editor. Atlas of Protein Sequence and Structure. Vol. 5. Washington, DC: National Biomedical Research Foundation; 1978. pp. 345–352. [Google Scholar]
- 23.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 24.Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- 25.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 26.Adachi J, Hasegawa M. Model of amino acid substitution in proteins encoded by mitochondrial DNA. J. Mol. Evol. 1996;42:459–468. doi: 10.1007/BF02498640. [DOI] [PubMed] [Google Scholar]
- 27.Adachi J, Waddell PJ, Martin W, Hasegawa M. Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J. Mol. Evol. 2000;50:348–358. doi: 10.1007/s002399910038. [DOI] [PubMed] [Google Scholar]
- 28.Jukes TH, Cantor CR. In: Mammalian protein metabolism. Munro HN, editor. Academic Press: New York; 1969. pp. 21–123. [Google Scholar]
- 29.Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider A, Cannarozzi GM, Gonnet GH. Empirical codon substitution matrix. BMC Bioinformatics. 2005;6:134. doi: 10.1186/1471-2105-6-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doron-Faigenboim A, Pupko T. A combined empirical and mechanistic codon model. Mol. Biol. Evol. 2007;24:388–397. doi: 10.1093/molbev/msl175. [DOI] [PubMed] [Google Scholar]
- 33.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rolland M, Jensen MA, Nickle DC, Yan J, Learn GH, Heath L, Weiner D, Mullins JI. Reconstruction and function of ancestral center-of-tree human immunodeficiency virus type 1 proteins. J. Virol. 2007;81:8507–8514. doi: 10.1128/JVI.02683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penn O, Stern A, Rubinstein ND, Dutheil J, Bacharach E, Galtier N, Pupko T. Evolutionary modeling of rate shifts reveals specificity determinants in HIV-1 subtypes. PLoS Comput. Biol. 2008;4:e1000214. doi: 10.1371/journal.pcbi.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33:511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]